

Abstract
The risk of Crohn disease (CD) has a large genetic component. A recent meta-analysis of 6 genome-wide association studies reported 71 chromosomal intervals but does not account for all of the known genetic contribution. Here, we refine localization of the previously reported intervals and also identify additional CD susceptibility genes using a mapping approach that localizes causal variants based on genetic maps in linkage disequilibrium units (LDU maps). Using 2 of the 6 cohorts, 66 of the 71 previously reported loci are confirmed and more precise location estimates for these intervals are given. We identify 78 additional gene regions that pass genome-wide significance, providing strong evidence for 144 genes. Additionally, 56 nominally significant signals, but with more stringent and precise colocalization, are identified. In total, we provide evidence for 200 gene regions confirming that CD is truly multifactorial and complex in nature. Many identified genes have functions that are compatible with involvement in immune/inflammatory processes and seem to have a large effect in individuals with extra ileal as well as ileal inflammation. The precise locations and the evidence that some genes reflect phenotypic subgroups will help identify functional variants and will lead to greater insight of CD etiology.
Main Text
Crohn Disease (CD) is, like ulcerative colitis (UC), a major subtype of Inflammatory Bowel Disease (IBD [MIM 266600]). Although CD differs from UC in several respects (e.g., clinical manifestations, cytokine profile), they both involve chronic intestinal inflammation but with variable and somewhat overlapping manifestations. CD principally involves the ileum, but there is variation in disease phenotype. There are inflammatory, stricturing, or penetrating types that may affect different sites in the gastrointestinal tract (GI) and also differences in the age of onset. This variability results from the interaction of environmental factors, including gut microbiota and the immune and inflammatory mechanisms of the genetically susceptible host. CD is a polygenic disorder with some high-penetrance genes and an estimated individual sibling recurrence risk ratio (λs) ranging from 15 to 30.1 Because of this high λs, CD is one of the most widely studied common multifactorial diseases. The genetic contribution was first explored by linkage analysis using families, which led to the important identification of the role of NOD2 on chromosome 16q (MIM 605956). Further progress was subsequently made through genome-wide association studies (GWASs).2–11 A recent meta-analysis explored 6 GWASs and identified a total of 71 signals of association with CD using more than 6,000 cases and 15,000 controls.5 The authors estimated that these loci explain less than one quarter5 of the reported heritability in liability. A GWAS utilizes hundreds of thousands of SNP markers across the genome. There is a lack of power to detect genome-wide association partly because of (1) the highly stringent significance thresholds required as a result of multiple testing, and (2) the unrealistic assumption that by testing each SNP one at time, one of the markers in the genotyping platform used will either be the causal or in almost complete linkage disequilibrium (LD) with the causal variant. An additional problem is that in order to achieve sufficient power, studies are often combined with the aim of increasing sample size. As a result, data sets often include cases with variable disease onset, variable phenotype definition, and sample collection in different geographic regions. Taken together, this not only means that rare variants of large effect are likely to be missed but that common variants with small effect or that apply only to a particular subgroup could also be missed.
Our recent study of the chromosome 16q linkage region using two of these GWASs illustrated how a multimarker approach based on high-resolution LD maps can provide additional study power. We were also able to explain heterogeneity in a genomic region first identified by linkage analysis.12 By using UK data provided by the Wellcome Trust Case Control Consortium (WTCCC), we identified, on this chromosome arm, with high statistical confidence, several new gene regions that are involved in inflammation and immune dysregulation. These distinct signals were replicated with high precision of location using independent North American data provided by the National Institute of Diabetes and Digestive and Kidney Diseases IBD Genetics Consortium (NIDDK). The study highlighted the importance of genetic heterogeneity (i.e., the involvement of different risk genes in different individuals) within the extensively studied NOD2 gene region and demonstrated the independent involvement of the nearby CYLD (MIM 605018).12 We also illustrated the importance of accurate and detailed phenotype definition in revealing gene association. By examining the group of NIDDK cases that had involvement of nonileal intestinal regions, as well as the ileum, we were able to replicate association with CDH1 (encoding E-cadherin [MIM 192090]), which is located coincident with a previously described linkage peak in cases without NOD2 mutations.
The purpose of this study is to analyze the whole genome using a gene-mapping approach based on LDU maps in order to identify additional genes for CD and provide the basis for new etiological insights into the disease. In order to accomplish this aim, we used 2 GWA data sets. From the UK WTCCC GWAS data2 based on the Affymetrix 500K Array, we analyzed a total of 1,698 cases of CD and 2,948 controls. The CD cases consisted of individuals with any subtype of CD. The GWAS replication data consisted of 813 North American CD cases and 947 ethnically matched controls, and all cases exhibit inflammation of the ileum. These GWAS data are available from the NIDDK IBDGC with the genotype data derived from the Illumina HumanHap300 Array. This array consists of a much lower SNP resolution and a set of SNPs that only partially overlap with the WTCCC 500K Affymetrix Array. Although a smaller data set, the NIDDK data contains a richer phenotypic description of cases and includes the various additional locations of intestinal inflammation for each individual. Further details about the GWAS data sets are available in other reports.2,11
The mapping approach in this study uses genetic locations derived from a high-resolution LD map with distances in LD units (LDU).13 High-density LDU maps were constructed using data from the HapMap Phase II Project. They can be visualized by plotting marker location in LDU against the marker distances in kb. Plotting these LDU maps reveals the nonlinear relationship between physical distance and the underlying LD together with the “block-step” structure of the region in question (see Figure 1). “Blocks” of LD represent areas of low haplotype diversity whereas “steps” define LD breakdown mainly caused by recombination because crossover profiles agree with LD patterns.14
Figure 1.
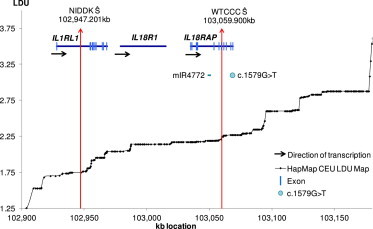
Localization within the IL1RL1/IL18RAP Region
The LD map of the region is shown by plotting LDU (y axis) against kb (x axis). The red vertical arrows are the estimated locations Ŝ for the WTCCC and NIDDK data sets. The WTCCC Ŝ is within an intron and between a miRNA (mIR4772) and coding rare variant associated with CD within IL18RAP (NM_003853.2) c.1579G>T (p.Val527Leu).
The construction of the LDU maps is based on the Malécot Model, which describes the decline of pairwise SNP associations as a function of physical distance in kilobases (kb).13 For the association mapping analyses, all the SNPs in the WTCCC and NIDDK data sets were assigned an LDU location from the HapMap LDU maps. We then divided each chromosome arm into nonoverlapping analytical windows, which were constrained to 10 LDU and a minimum of 60 SNPs. The windows were identical for both data sets. For each window along the whole genome, a composite-likelihood test15 was used, in which all the SNPs within a window are simultaneously tested. Therefore, the method avoids the need for imputation. Because each window is a test, then the total number of tests performed is substantially lower than single SNP analysis. This has important implications in multiple testing. Each test is also based on the same Malécot model used to construct the LDU maps, although in this case, we model the decline of affection-status-by-SNP association as a function of genetic distance in LDU. The model uses the locations from the LDU map in order to estimate the most likely location (Ŝ) of the causal agent. The method returns a p value for association of the window together with an estimated Ŝ for each window. In windows where Ŝ mapped to the end of a window-boundary, that window was extended in that direction to capture information on the far side to improve the accuracy of location. The construction of the LDU maps using the HapMap Phase II data has been described in detail elsewhere16 and the association mapping methodology presented here using the LDU maps is given by Maniatis et al.15 This method has been shown to increase power and localization over kb maps.12,15
Our recent work12 showed that stratifying the analysis according to the occurrence of extraileal intestinal involvement, as opposed to ileal inflammation alone (a subdivision that appears to accompany genetic stratification17), further increases the power of detecting association. We stratified the NIDDK data based on detailed information regarding the location of individuals’ intestinal inflammation. For all the analytical windows that showed nominal statistical evidence of association using the WTCCC data, we analyzed the same windows using the entire NIDDK data and also using a subset of the NIDDK data containing CD cases that reported extraileal in addition to ileal inflammation. Unlike the controls, the vast majority of cases in this subdivision were non-Jewish. We therefore analyzed only non-Jewish cases with extraileal inflammation and non-Jewish controls. This subdivision reduced the sample size from 813 to 277 CD cases and from 947 to 515 controls.
In an initial analysis, we looked for signals within the previously published intervals from the recent meta-analysis5 and present p values for the relevant data sets. Because the majority of these intervals were more than 200 kb in length (defined around the most significant SNP using LD information5), it was difficult to meta-analyze the results obtained using this mapping procedure. We therefore present the p values obtained from WTCCC and NIDDK separately. For the additional genes we identified, we present a meta-analysis p value for each analytical window that shows (1) evidence of nominal statistical significance for each data sample and (2) that the Ŝ estimates for both samples are within 150 kb of one another. These two criteria were used in order to reduce the possibility of heterogeneity and thus facilitate the meta-analysis of shared locations. In order to account for multiple testing, we used a Bonferroni p value threshold of 1 × 10−5, which corresponds to the 4,993 tests, to the final meta p value. This number of tests included the total number of analytical windows across the genome (4,399 windows with identical boundaries for both data sets) plus the additional analyses performed for the NIDDK data using the extraileal subgroup.
In order to capture replications of slightly lower significance but with more precisely agreeing Ŝ values, we used a second threshold in which the Ŝ for both data sets had to lie within 80 kb of each other to be considered a replication and with a meta p value within the range of 1 × 10−3 and 1 × 10−5.
For nominally significant windows of each data set, the closest gene of the estimated location Ŝ was retrieved and then listed in the tables. Using BioMart, the Gene Ontology (GO) annotations attributed to each gene were retrieved and are listed. Using the Cytoscape plugin BiNGO,18 we also checked for overrepresentation of GO terms from the GO Slim GOA database using a hypergeometric test.
Our first goal was to determine whether the LDU mapping method could detect the 71 intervals previously identified from the most recent large meta-analysis of six independent data sets. Table 1 shows some examples and Table S1 presents the statistical evidence and location estimates, for significant signals within each of these 71 intervals.5 Our analyses identified a total 66 out of the 71 intervals (loci) in one or both of these data sets using an uncorrected significance threshold of p < 0.05. Only five intervals showed no evidence of association for either WTCCC or NIDDK using the Malécot model. Notably, the first published results by the WTCCC2 reported just nine loci, but Table S1 (available online) shows 28 signals for the WTCCC alone that passed our genome-wide significance threshold (1 × 10−5). The majority of the 66 intervals (88%) were identified with the larger WTCCC data with the remaining eight intervals (12%) showing nominal evidence of association for the smaller NIDDK data set alone. Interestingly, these eight include regions (e.g., 10q22, 16p11) that have previously been implicated for pediatric IBD cases7 (Table S1). This is consistent with the fact that the NIDDK data set probably includes a larger number of individuals with early onset CD (∼37%) than WTCCC, where this information is not recorded. It is worth noting that one of the six contributory data sets to the most recent meta-analysis included entirely pediatric IBD cases.5
Table 1.
Examples Taken from the Previously Reported Intervals, Showing the Whole-Genome Association Statistics and Closest Gene to the Estimated Location of the Causal Agent Ŝ
| Chr | Reported LD Interval (Mb) | Interval Span (Mb) | WTCCC Ŝ | WTCCC p Value | NIDDK Ŝ | NIDDK p Value | Gene (WTCCC/NIDDK If different) |
|---|---|---|---|---|---|---|---|
| 1p31.3 | 67.36 – 67.77 | 0.41 | 67,684.8 | 2.1 × 10−11 | 67,707.3 | 3.0 × 10−10 | IL23Ra |
| 2q12.1 | 102.80 – 103.30 | 0.50 | 103,059.8 | 1.8 × 10−08 | 102,951.7 | 1.7 × 10−05 | IL18RAPa/IL1RL1a |
| 2q37.1 | 234.15 – 234.57 | 0.42 | 234,144.8 | 5.6 × 10−25 | 234,171.9 | 2.2 × 10−09 | ATG16L1a |
| 5q31.1 | 129.38 – 132.02 | 2.64 | 131,748.2 | 2.7 × 10−22 | 131,631.4 | 8.9 × 10−07 | C5orf56a/SLC22A4a |
| 16q12.1 | 50.46 – 50.85 | 0.39 | 50,803.2 | 2.6 × 10−15 | 50,803.2 | 1.1 × 10−09 | NOD2a |
| 50,846.3 | 1.6 × 10−13 | 50,846.5 | 4.1 × 10−04 | CYLD |
At least one of the estimated locations Ŝ is located within the identified gene. All locations in kb are in NCBI 37. Full details on the entire list of 71 LD intervals are given in Table S1.
Unlike the recent meta-analysis, where a particular gene or genes within these intervals was identified through a series of in silico analyses, we present the genes closest to the location estimates we have obtained using this method (Table 1; Table S1). In several instances, these agree with the previously identified genes (e.g., STAT3 [MIM 102582], IL23R [MIM 607562]; Table S1), but in other instances there are differences. For several regions, we obtain two different estimates of localization (Ŝ) for WTCCC and NIDDK, both within the same interval. Heterogeneity in some regions is expected and location estimates can help identify this. For example, for the interleukin-rich interval (2q12.1), the WTCCC data set yielded an estimate within IL18RAP (MIM 604509) but for NIDDK the Ŝ is within IL1RL1 (MIM 601203; Table 1). Both genes have been previously suggested as candidates within the 2q12.1 interval,5 but an independent fine mapping study has shown strong association to the IL18RAP rs917997 SNP for both CD and UC,19 which is approximately 10 kb away from WTCCC Ŝ (Table 1; Figure 1). A recent study20 using deep sequencing identified a rare coding variant of possible functional significance within IL18RAP (NM_003853.2:c.1579G>T [p.Val527Leu]), which is only 8 kb away from the WTCCC location, though it is probable that other functional variants are also involved. As far as the second gene is concerned, the importance of IL1RL1 to inflammatory processes has also been documented21 for a variety of human pathologies including celiac disease.22 The LDU map clearly shows that there are several recombination hot spots between IL18RAP and IL1RL1 (Figure 1) making it likely that these two signals are independent and that there is genetic heterogeneity within these previously identified intervals. This result is similar to that previously noted in the case of NOD2 and CYLD12 (Table 1).
As well as our 66 localization estimates, we identified 134 additional signals, of which 78 passed our genome-wide significance threshold (1 × 10−5; Table 2; Table S2). Of these 78 signals, 64 were replicated using the complete NIDDK data set, of which half (32) were also replicated in the smaller subset of the data (ileal and extraileal). However, notably, in half of these (16) the association showed higher significance in the smaller subset despite the substantial decrease in the number of cases and controls (see entries in bold marked with b in Table 2). Furthermore, the remaining 14 (22%) of the WTCCC signals were only replicated using this subset of the NIDDK data.
Table 2.
Whole-Genome Association Statistics and the Closest Gene to the Estimated Location of the Causal Agent for the 78 Gene-Regions that Passed Our Genome-Wide Significance Threshold 1 × 10−5 with Ŝ Locations of 150 kb or Less between the Two Data Sets
| Chr | Gene region (WTCCC/NIDDK if different) |
|---|---|
| 1 | NFIAb, PGM1a/EFCAB7a,b, RORCa/THEM4, KCNK1a,b, LGALS8a, ACTN2a/HEATR1 |
| 2 | LOC400940, FAM49A, DNAJC27-AS1a/ADCY3a, DCTN1a/RTKNa,b, intergenic (2q14.1), STK39a, ZNF804Aa, ERBB4a |
| 3 | STAC/ARPP21,ITGA9a, EPHA6b, COL8A1 |
| 4 | TECa/SLAIN2, LNX1/RPL21P44b, PDGFRAb, COL25A1a,b, SNX25a |
| 5 | MCCa,b, MEGF10/PRRC1b |
| 6 | GPLD1a, GABBR1a, EYSa, FNDC1a, Tb, |
| 7 | MACC1/TMEM196, NPYb, INHBA-AS1/GLI3, SEMA3Ca, TSPAN12/KCND2a, intergenicb(7q31.33), PODXLb |
| 8 | MTUS1a, LOC100507632, STMN2b, intergenic (8q21.13), DCAF13/FZD6b, intergenicb(8q23.3) |
| 9 | DOCK8a,b |
| 10 | GPR158a,b, FAM13Ca, ARID5B/C10orf107, PLAUb, KCNMA1a, XPNPEP1, NRAPa |
| 11 | OR52N4/OR52N2b, BDNF, OR9G4, KIRREL3a |
| 12 | CCDC91b, TMEM117a,b, C12orf74b, ANO4a |
| 13 | MIR4703 |
| 14 | PNN/GEMIN2a,b, NRXN3a,b |
| 15 | OTUD7Aa, PGBD4/CHRM5a |
| 16 | RBFOX1b, RBFOX1a, BRD7,HSD11B2b, IRF8 |
| 17 | GRB2a/MIR3678 |
| 18 | GATA6, RIT2a,b, IER3IP1b |
| 20 | HAO1a,b, LOC339568, CD40/SLC12A5a,b |
| 22 | CSF2RBa/LOC1005006241 |
| X | FMR1NB |
bold denotes that the signal is replicated using the data stratified according to non-Jewish ileal and extraileal inflammation.
At least one of the estimated locations Ŝ is located within the identified gene.
The signal is significant in the pooled NIDDK data as well as the stratified non-Jewish ileal and extraileal data set. All locations in kb are in NCBI 37. Full details (p values and estimates of Ŝ) are given in Table S2.
For the majority (74%) of the 78 signals, the location estimates from both data sets point to the same gene, with approximately half of the signals being intragenic and half intergenic locations (gene with “a” indicates that at least one of the signals is within the gene; Table 2). In all signals, including the ∼50% that reside outside of genes, the closest gene is considered as a candidate. It should however be noted that in some instances, functional variation in cis-elements that regulate genes far away from the location determined here may be involved.23
Several of the additional genes we identified have been reported to be involved in inflammatory and/or immune dysregulation conditions (e.g., DOCK8 [MIM 611432], ITGA9 [MIM 603963]). In other instances, the genes have been previously implicated in colonic inflammation or epithelial cell morphology based on functional studies. By way of illustration, Figure 2 presents an example of the estimated locations (Ŝ) for the genomic region that harbors ERBB4 ([MIM 600543], 2q34 window). For both data sets, this window was significantly associated with CD with a meta p value of 3 × 10−6 (Table 2). Figure 2 also shows the LDU map starting from the 3′ region and spanning halfway along the gene, which stretches across more than 1 megabase (Mb). The map shows numerous short LD blocks across the region. However, despite the large LD breakdown in the area, both estimates of Ŝ are close to each other and within the same intron. Interestingly, ERBB4 (which encodes Receptor protein-tyrosine kinase erbB-4) is expressed at high levels in the inflamed colonic mucosa of CD cases.24 Using adult mouse colon, it was also shown that ERBB4 is an important regulator in the epithelial response to inflammation and injury.24 ERBB4 expression has been linked to a number of cellular processes such as cell survival, proliferation, and tumorigenesis in different tissues.25,26 A recent study suggested that this elevated ERBB4 expression could lead to colitis-associated development of colorectal tumors.27 ERBB4 has also been implicated, together with E-cadherin (encoded by CDH1 on 16q22.1, previously identified as a risk gene for CD12 and ulcerative colitis28), in the suppression of anoikis29 (programmed cell death as a result of cell detachment from the extracellular matrix), suggesting that there may be interaction between these two genes. Despite the fact that ERBB4 is a very large gene (>1 Mb), the estimated locations of the causal agent for both data sets (WTCCC and NIDDK) are very close and within the same intron, making it an excellent target for follow-up fine mapping studies and resequencing.
Figure 2.

Localization within the ERBB4 Region
The LD map of the region is shown by plotting LDU (y axis) against kb (x axis). The red vertical arrows are the estimated locations Ŝ for the WTCCC and NIDDK data sets.
Table 3 shows the list of 56 of the 134 additional signals that showed association with CD with p values that ranged between 1 × 10−3 and 1 × 10−5. These p values were below the Bonferroni threshold, but the windows were selected on more stringent criteria for colocalization (i.e., estimates of localization in the two data sets were not more than 80 kb apart from each other). Detailed information (p values, location estimates) for the all the genes listed in Table 3 are given in the Table S3. As in Table 2, most of the signals in Table 3 give estimates of Ŝ that point to the same gene. Several of these genes are also very interesting. For example, the window harboring BTLA (MIM 607925) gave location estimates that are just 2 kb apart. A study using a mouse model of colitis has shown functional evidence of BTLA involvement in colitis.30
Table 3.
List of Genes for the 56 Signals with p Values 10−3−10−5 and with Ŝ Locations of 80 kb or Less between the Two Data Sets
| Chr | Gene region (WTCCC/NIDDK if different) |
|---|---|
| 1 | LOC400752, DAB1a, GADD45A, intergenic (1p31.1), intergenicb (1q31.3), RENa, DNAH14a, H3F3Ab |
| 2 | ETAA1b, IL1R2a, LRP2a, NYAP2 |
| 3 | CACNA2D3, ROBO2a, BTLAa, AGTR1a |
| 4 | HSP90AB2b, intergenic (4q13.2), PRDM5a |
| 5 | CDH12, intergenic (5q21.1), FAM71B/ITKa,b |
| 6 | SOX4, BEND3a, MARCKS, STLb, IFNGR1, intergenic (6q25.3), SNX9a,b |
| 7 | MACC1, NPSR1a, IQUBb, KELa |
| 8 | SLAa |
| 9 | C9orf85/C9orf57, GNA14a,b, intergenic (9q33.1) |
| 10 | FASb |
| 11 | MOB2a, MICALCLa |
| 12 | CAND1, KITLGb, LOC100128554b |
| 13 | GPR12, NBEAa, intergenic (13q31.3), intergenic (13q31.3) |
| 14 | intergenicb(14q31.1) |
| 15 | ATP8B4a |
| 16 | CDH8a, CNTNAP4, WWOXa,b |
| 20 | MACROD2a |
| 21 | DSCAMa |
| 22 | ISXb |
| X | intergenic (Xq23) |
bold denotes that the signal is replicated using the data stratified according to non-Jewish ileal and extraileal inflammation.
At least one of the estimated locations Ŝ is located within the identified gene.
The signal is significant in the Pooled NIDDK data as well as the stratified non-Jewish ileal and extraileal data set. All locations in kb are in NCBI 37. Full details (p values and estimates of Ŝ) are given in Table S3.
In summary, using a powerful multimarker association mapping approach based on LDU maps, we confirm 66 of the 71 previously reported loci from meta-analysis and 134 additional gene regions that are associated with Crohn disease, providing evidence for 200 gene regions that include CD susceptibility loci. We provide more precise location estimates for the 66 previously published intervals, as well as locations for the 134 additional signals. This is a major step forward in identifying the relevant genes and functional variants and thus elucidating the genetics of CD etiology. The very large numbers of genes listed in Tables 2 and 3 confirm that CD is truly polygenic and complex in nature. Many genes show functions that are compatible with involvement in immune and/or inflammatory processes as well as integrity of the intestinal epithelium and differentiation. The groups of genes were enriched for GO terms such as signal transducer activity, receptor activity, and binding. These functions are important in inflammatory processes (see Supplemental Information; Tables S4 and S5).
Gene mapping by linkage has a long history, but association mapping was in its infancy when high-throughput SNP genotyping was developed. There is currently some skepticism regarding the success of genome-wide association mapping, but here we demonstrate its great potential in dissecting the genetics of common disease—at least for Crohn, for which relatively highly penetrant genes are known to exist (as indicated by high λS estimates). We also demonstrate that the use of smaller data sets and detailed phenotypic information can detect a great proportion of the genes involved, as well as genetic heterogeneity. Here we get a glimpse of the way in which different genes are likely to be involved in different but overlapping pathways. Many of the replicated signals were more significant using a subset of the North American data (NIDDK), which contained individuals with ileal CD who had involvement of at least one extraileal intestinal location. Despite the substantial decrease in sample size, analysis of this subphenotype using our mapping method yielded much higher significance than the analysis of the full data. This result shows that accurate and detailed phenotype information is extremely important in genetic studies especially when assessing a trait that shows clinical variation.
The majority of the 200 signals unambiguously identify single genes. Their confirmation in independent cohorts makes it likely that most have an effect (in many instances small, though sometimes large) on the risk of CD. The genomic locations we provide are estimates of the positions of the variants of functional significance assuming a monophylogenetic origin (i.e., no substantial allelic heterogeneity). We have previously shown the greater power of LDU compared to physical maps in detecting association and refining location using the same multimarker methodology.31 The close distance between the location estimates in the two different cohorts (10 kb or less in 30% of the additional signals) suggests that these allelic variants will often be the same, but in some instances there are clear indications of independent allelic variants. In either case, the localization offers much greater precision than other methods for future in depth analysis using bioinformatics and resequencing.
The identified genes will facilitate more powerful experimental research designs including a range of functional studies and analytical pathway analyses for CD. Given the high-resolution estimates, the results presented here will allow more targeted, intelligently designed resequencing studies, where carefully characterized, homogeneous subgroups of cases can be more thoroughly investigated. Such research strategies should move us closer to more effective, personalized medical treatment for individuals with Crohn disease.
Acknowledgments
We are very grateful to the Wellcome Trust, the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and the International HapMap Project for making their invaluable data available to the scientific community. The data from the Wellcome Trust Case-Control Consortium (WTCCC)2 was funded by the Wellcome Trust. A full list of the investigators who contributed to the generation of the data is available on the WTCCC website. The NIDDK IBD Genetics Consortium Crohn Disease GWAS was conducted by Judy H. Cho, Yale University; Steven Brant, Johns Hopkins University; Richard Duerr, University of Pittsburgh; Huiying Yang, Cedars-Sinai Medical Center; John Rioux, University of Montreal; and Mark Silverberg, University of Toronto, with support from the NIDDK. This manuscript was not prepared in collaboration with the laboratories of any of the investigators responsible for generating the data and does not necessarily reflect the views or opinions of these investigators or the NIDDK. The data sets used were obtained from the database of Genotypes and Phenotypes (dbGaP) at accession number phs000130. We thank Toby Andrew for his useful comments and suggestions and the Annals of Human Genetics for the financial support for H.E.’s PhD degree.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Gene Ontology, http://www.geneontology.org/
International HapMap Project, http://hapmap.ncbi.nlm.nih.gov/
National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), www.niddk.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
UCSC Genome Browser, http://genome.ucsc.edu/
Wellcome Trust Case Control Consortium (WTCCC), http://www.wtccc.org.uk/
References
- 1.Schreiber S., Rosenstiel P., Albrecht M., Hampe J., Krawczak M. Genetics of Crohn disease, an archetypal inflammatory barrier disease. Nat. Rev. Genet. 2005;6:376–388. doi: 10.1038/nrg1607. [DOI] [PubMed] [Google Scholar]
- 2.Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barrett J.C., Hansoul S., Nicolae D.L., Cho J.H., Duerr R.H., Rioux J.D., Brant S.R., Silverberg M.S., Taylor K.D., Barmada M.M., NIDDK IBD Genetics Consortium. Belgian-French IBD Consortium. Wellcome Trust Case Control Consortium Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat. Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duerr R.H., Taylor K.D., Brant S.R., Rioux J.D., Silverberg M.S., Daly M.J., Steinhart A.H., Abraham C., Regueiro M., Griffiths A. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Franke A., McGovern D.P., Barrett J.C., Wang K., Radford-Smith G.L., Ahmad T., Lees C.W., Balschun T., Lee J., Roberts R. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hampe J., Franke A., Rosenstiel P., Till A., Teuber M., Huse K., Albrecht M., Mayr G., De La Vega F.M., Briggs J. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 2007;39:207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 7.Imielinski M., Baldassano R.N., Griffiths A., Russell R.K., Annese V., Dubinsky M., Kugathasan S., Bradfield J.P., Walters T.D., Sleiman P., Western Regional Alliance for Pediatric IBD. International IBD Genetics Consortium. NIDDK IBD Genetics Consortium. Belgian-French IBD Consortium. Wellcome Trust Case Control Consortium Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat. Genet. 2009;41:1335–1340. doi: 10.1038/ng.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Libioulle C., Louis E., Hansoul S., Sandor C., Farnir F., Franchimont D., Vermeire S., Dewit O., de Vos M., Dixon A. Novel Crohn disease locus identified by genome-wide association maps to a gene desert on 5p13.1 and modulates expression of PTGER4. PLoS Genet. 2007;3:e58. doi: 10.1371/journal.pgen.0030058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mathew C.G. New links to the pathogenesis of Crohn disease provided by genome-wide association scans. Nat. Rev. Genet. 2008;9:9–14. doi: 10.1038/nrg2203. [DOI] [PubMed] [Google Scholar]
- 10.McGovern D.P., Jones M.R., Taylor K.D., Marciante K., Yan X., Dubinsky M., Ippoliti A., Vasiliauskas E., Berel D., Derkowski C., International IBD Genetics Consortium Fucosyltransferase 2 (FUT2) non-secretor status is associated with Crohn’s disease. Hum. Mol. Genet. 2010;19:3468–3476. doi: 10.1093/hmg/ddq248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rioux J.D., Xavier R.J., Taylor K.D., Silverberg M.S., Goyette P., Huett A., Green T., Kuballa P., Barmada M.M., Datta L.W. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elding H., Lau W., Swallow D.M., Maniatis N. Dissecting the genetics of complex inheritance: linkage disequilibrium mapping provides insight into Crohn disease. Am. J. Hum. Genet. 2011;89:798–805. doi: 10.1016/j.ajhg.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maniatis N., Collins A., Xu C.F., McCarthy L.C., Hewett D.R., Tapper W., Ennis S., Ke X., Morton N.E. The first linkage disequilibrium (LD) maps: delineation of hot and cold blocks by diplotype analysis. Proc. Natl. Acad. Sci. USA. 2002;99:2228–2233. doi: 10.1073/pnas.042680999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Webb A.J., Berg I.L., Jeffreys A. Sperm cross-over activity in regions of the human genome showing extreme breakdown of marker association. Proc. Natl. Acad. Sci. USA. 2008;105:10471–10476. doi: 10.1073/pnas.0804933105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maniatis N., Collins A., Morton N.E. Effects of single SNPs, haplotypes, and whole-genome LD maps on accuracy of association mapping. Genet. Epidemiol. 2007;31:179–188. doi: 10.1002/gepi.20199. [DOI] [PubMed] [Google Scholar]
- 16.Lau W., Kuo T.Y., Tapper W., Cox S., Collins A. Exploiting large scale computing to construct high resolution linkage disequilibrium maps of the human genome. Bioinformatics. 2007;23:517–519. doi: 10.1093/bioinformatics/btl615. [DOI] [PubMed] [Google Scholar]
- 17.Ahmad T., Armuzzi A., Bunce M., Mulcahy-Hawes K., Marshall S.E., Orchard T.R., Crawshaw J., Large O., de Silva A., Cook J.T. The molecular classification of the clinical manifestations of Crohn’s disease. Gastroenterology. 2002;122:854–866. doi: 10.1053/gast.2002.32413. [DOI] [PubMed] [Google Scholar]
- 18.Maere S., Heymans K., Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- 19.Zhernakova A., Festen E.M., Franke L., Trynka G., van Diemen C.C., Monsuur A.J., Bevova M., Nijmeijer R.M., van ’t Slot R., Heijmans R. Genetic analysis of innate immunity in Crohn’s disease and ulcerative colitis identifies two susceptibility loci harboring CARD9 and IL18RAP. Am. J. Hum. Genet. 2008;82:1202–1210. doi: 10.1016/j.ajhg.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rivas M.A., Beaudoin M., Gardet A., Stevens C., Sharma Y., Zhang C.K., Boucher G., Ripke S., Ellinghaus D., Burtt N., National Institute of Diabetes and Digestive Kidney Diseases Inflammatory Bowel Disease Genetics Consortium (NIDDK IBDGC); United Kingdom Inflammatory Bowel Disease Genetics Consortium. International Inflammatory Bowel Disease Genetics Consortium Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat. Genet. 2011;43:1066–1073. doi: 10.1038/ng.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akhabir L., Sandford A.J. Genome-wide association studies for discovery of genes involved in asthma. Respirology. 2011;16:396–406. doi: 10.1111/j.1440-1843.2011.01939.x. [DOI] [PubMed] [Google Scholar]
- 22.Amundsen S.S., Rundberg J., Adamovic S., Gudjónsdóttir A.H., Ascher H., Ek J., Nilsson S., Lie B.A., Naluai A.T., Sollid L.M. Four novel coeliac disease regions replicated in an association study of a Swedish-Norwegian family cohort. Genes Immun. 2010;11:79–86. doi: 10.1038/gene.2009.67. [DOI] [PubMed] [Google Scholar]
- 23.Jones B., Swallow D. The impact of cis-acting polymorphisms on the human phenotype. The HUGO Journal. 2011;5:13–23. doi: 10.1007/s11568-011-9155-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frey M.R., Edelblum K.L., Mullane M.T., Liang D., Polk D.B. The ErbB4 growth factor receptor is required for colon epithelial cell survival in the presence of TNF. Gastroenterology. 2009;136:217–226. doi: 10.1053/j.gastro.2008.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Erlich S., Goldshmit Y., Lupowitz Z., Pinkas-Kramarski R. ErbB-4 activation inhibits apoptosis in PC12 cells. Neuroscience. 2001;107:353–362. doi: 10.1016/s0306-4522(01)00350-5. [DOI] [PubMed] [Google Scholar]
- 26.Starr A., Greif J., Vexler A., Ashkenazy-Voghera M., Gladesh V., Rubin C., Kerber G., Marmor S., Lev-Ari S., Inbar M. ErbB4 increases the proliferation potential of human lung cancer cells and its blockage can be used as a target for anti-cancer therapy. International Journal of Cancer. 2006;119:269–274. doi: 10.1002/ijc.21818. [DOI] [PubMed] [Google Scholar]
- 27.Frey M.R., Hilliard V.C., Mullane M.T., Polk D.B. ErbB4 promotes cyclooxygenase-2 expression and cell survival in colon epithelial cells. Laboratory Investigation; a journal of technical methods and pathology. 2010;90:1415–1424. doi: 10.1038/labinvest.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anderson C.A., Boucher G., Lees C.W., Franke A., D’Amato M., Taylor K.D., Lee J.C., Goyette P., Imielinski M., Latiano A. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 2011;43:246–252. doi: 10.1038/ng.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang H.G., Jenabi J.M., Zhang J., Keshelava N., Shimada H., May W.A., Ng T., Reynolds C.P., Triche T.J., Sorensen P.H. E-cadherin cell-cell adhesion in ewing tumor cells mediates suppression of anoikis through activation of the ErbB4 tyrosine kinase. Cancer Res. 2007;67:3094–3105. doi: 10.1158/0008-5472.CAN-06-3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steinberg M.W., Turovskaya O., Shaikh R.B., Kim G., McCole D.F., Pfeffer K., Murphy K.M., Ware C.F., Kronenberg M. A crucial role for HVEM and BTLA in preventing intestinal inflammation. J. Exp. Med. 2008;205:1463–1476. doi: 10.1084/jem.20071160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andrew T., Maniatis N., Carbonaro F., Liew S.H.M., Lau W., Spector T.D., Hammond C.J. Identification and replication of three novel myopia common susceptibility gene loci on chromosome 3q26 using linkage and linkage disequilibrium mapping. PLoS Genet. 2008;4:e1000220. doi: 10.1371/journal.pgen.1000220. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.