

Abstract
Distal arthrogryposis (DA) syndromes are the most common of the heritable congenital-contracture disorders, and ∼50% of cases are caused by mutations in genes that encode contractile proteins of skeletal myofibers. DA type 5D (DA5D) is a rare, autosomal-recessive DA previously defined by us and is characterized by congenital contractures of the hands and feet, along with distinctive facial features, including ptosis. We used linkage analysis and whole-genome sequencing of a multiplex consanguineous family to identify in endothelin-converting enzyme-like 1 (ECEL1) mutations that result in DA5D. Evaluation of a total of seven families affected by DA5D revealed in five families ECEL1 mutations that explain ∼70% of cases overall. ECEL1 encodes a neuronal endopeptidase and is expressed in the brain and peripheral nerves. Mice deficient in Ecel1 exhibit perturbed terminal branching of motor neurons to the endplate of skeletal muscles, resulting in poor formation of the neuromuscular junction. Our results distinguish a second developmental pathway that causes congenital-contracture syndromes.
Main Text
Distal arthrogryposis (DA) is a group of at least ten disorders characterized by nonprogressive, congenital contractures that typically affect the hands, feet, wrists, and ankles.1 Over the past decade, mutations causing DA1 (MIM 108120),2,3 DA2A (Freeman-Sheldon syndrome [MIM 193700]),4 DA2B (Sheldon-Hall syndrome [MIM 601680]),2,3 and DA7 (trismus pseudocamptodactyly [MIM 158300])5,6 have been identified in six genes, each of which encodes a component of the contractile apparatus of skeletal myofibers. Of the DA syndromes for which no causative mutation has yet been discovered, DA5 (DA with ophthalmoplegia [MIM 108145])1 is perhaps the most notable because, unlike the other DA disorders, the extraocular muscles of the eye are often involved. Since the original delineation of DA5, the spectrum of ocular defects observed in affected individuals has expanded substantially. Additionally, it has become clear that ophthalmoplegia is sometimes not present in all affected members of a family. Furthermore, a subset of affected individuals develop restrictive lung disease with resultant hypoxemia, hypercarbia, pulmonary hypertension, and early death.7
In an effort to delineate genetically homogeneous subsets of DA5-affected families for gene-discovery studies, we categorized the phenotypic and mapping data from 35 DA5-affected families into one of four different subtypes of DA5 (i.e., DA5A–DA5D). Through this analysis, we delineated seven families affected by a unique phenotype (i.e., DA5D) consisting of severe camptodactyly of the hands, including adducted thumbs and wrists; mild camptodactyly of the toes; clubfoot and/or a calcaneovalgus deformity; extension contractures of the knee; unilateral ptosis or ptosis that was more severe on one side; a round-shaped face; arched eyebrows; a bulbous, upturned nose; and micrognathia (Figure 1, Table 1, and Figures S1 and S2, available online). The presence of ophthalmoplegia was notably absent from this phenotype. Furthermore, unlike in other DA phenotypes described to date, no parent-to-child transmissions were reported, and the fact that families had multiple affected siblings suggested that, unlike all other DA phenotypes described to date, DA5D is an autosomal-recessive disorder.
Figure 1.
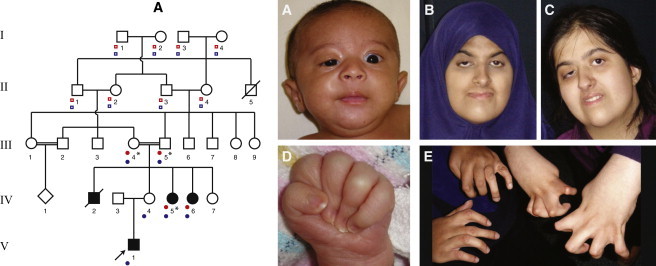
Pedigree and Clinical Features of Family A
Pedigree showing the relationships and affected status of family A. Typical features of the face include unilateral ptosis, a round-shaped face, a small jaw, a bulbous upturned nose, and arched eyebrows. Camptodactyly and severely adducted thumbs are also present. Case identifiers A:V-1 (images A and D), A:IV-6 (images B and E [left]), and A:IV-5 (images C and E [right]) correspond to those in Table 1, where a detailed description of each individual is provided. The proband is indicated with an arrow. Asterisks indicate individuals in whom whole-genome sequencing (WGS) was performed. Red dots indicate individuals from whom genotype data were included in analysis of linkage under a recessive model. Open red squares indicate individuals who were included in the recessive model and from whom genotype data were unavailable. Blue dots indicate individuals from whom genotype data were included in analysis of linkage under a dominant model. Open blue squares indicate individuals who were included in the dominant model and from whom genotype data were unavailable. Larger and more detailed images are presented in Figures S1 and S4.
Table 1.
Mutations and Clinical Findings of Individuals with DA5D
|
Family A |
Family B |
Family D |
Family E |
Family F |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A:V-1 | A:IV-5 | A:IV-6 | B:II-2 | B:II-3 | D:II-2 | D:II-3 | E:II-2 | F:II-3 | |||||||
| Mutation Characteristics | |||||||||||||||
| Exon (ECEL1) | 2 | 2 | 2 | 2 | 4 | 3 | 4 | 3 | 7 | 6 | 7 | 6 | 6 | 2 | 7 |
| Nucleotide (cDNA) | c.716dupA | c.344_355 del | c.716dupA | c.716dupA | c.869A>G | c.797_801delins GCT | c.869A>G | c.797_801delins GCT | c.1252C>A | c.1184+3A>T | c.1252C>A | c.1184+3A>T | c.1184G>A | c.590G>A | c.1252C>T |
| GERP score | NA | NA | NA | NA | 5.51 | NA | 5.51 | NA | 5.18 | 3.07 | 5.18 | 3.07 | 4.84 | 4.12 | 5.18 |
| Protein alteration | p.Tyr239∗ | p.Asn115_Ala118 del | p.Tyr239∗ | p.Tyr239∗ | p.Tyr290Cys | p.Asp266Glyfs∗15 | p.Tyr290Cys | p.Asp266Glyfs∗15 | p.Arg418Ser | − | p.Arg418Ser | − | p.Arg395Gln | p.Gly197Asp | p.Arg418Cys |
| Predicted effect | premature truncation | deletion | premature truncation | premature truncation | missense | frameshift | missense | frameshift | missense | splice site | missense | splice site | missense | missense | missense |
| Genotype | heterozygous | heterozygous | homozygous | homozygous | heterozygous | heterozygous | heterozygous | heterozygous | heterozygous | heterozygous | heterozygous | heterozygous | homozygous | heterozygous | heterozygous |
| Inheritance | maternal | paternal | maternal/paternal | maternal/paternal | maternal | paternal | maternal | paternal | maternal | paternal | maternal | paternal | NA | maternal | paternal |
| Clinical Findings | |||||||||||||||
| Ancestry | Indian and eastern European | Indian | Indian | European American | European American | European American | European American | Indian | European American | ||||||
| Consanguineous parents | no | double first cousins | double first cousins | no | no | no | no | no | no | ||||||
| Foot and/or toe contractures | − | + | + | + | + | + | + | + | + | ||||||
| Ankle contractures | + | + | + | + | + | + | + | + | + | ||||||
| Knee contractures | + flexion | + flexion | + flexion | + extension | + extension | + extension | + extension | + extension | − | ||||||
| Hip contractures | + | + | + | + | + | + | + | + | + | ||||||
| Hand and/or finger contractures | + | + | + | + | + | + | + | + | + | ||||||
| Wrist contractures | + | + | + | + | + | + | + | + | + | ||||||
| Elbow contractures | + | + | + | ND | ND | ND | ND | + | + | ||||||
| Shoulder contractures | + | + | + | + | ND | ND | ND | + | + | ||||||
| Neck contractures | + | + | ND | ND | ND | ND | ND | + | + | ||||||
| Ptosis | R | R | R | BL (mild) | R | R | − | R | BL (severe) | ||||||
| Bulbous nose | + | + | + | + | + | + | + | + | + | ||||||
| Micrognathia | + | + (mild) | + (mild) | + (mild) | + (mild) | + | + | − | + | ||||||
| Pterygia | neck, axillae | − | − | − | − | groin, neck | ND | elbows, axillae, neck | − | ||||||
| Short neck | + | ND | ND | + | + | − | − | − | + | ||||||
| Scoliosis | − | + | + | − | − | − | − | − | − | ||||||
| Other features | reduced facial expression, slightly crouched gait | BL dislocated hips, hip reduction, spinal rods, skin graft on hand | R eyelid lift | glabellar hemangioma, posteriorly rotated ears, rocker bottom (calcaneal valgus) | glabellar hemangioma, wide nasal bridge and upturned nose, cleft palate, ears minimally posteriorly rotated, BL hip dislocation | multiple eyelid surgeries, clubfoot correction, BL hip dysplasia | severe strabismus, glabellar hemangioma | dislocated hips, reduced fetal movements, bushy eyebrows, grooved tongue, high arched palate | cupped ears, L undescended testes, inguinal hernia | ||||||
This table depicts mutation information and associated clinical findings for individuals with DA5D. Plus signs indicate the presence of a finding, and minus signs indicate the absence of a finding. The following abbreviations are used: GERP, genomic evolutionary rate profiling; ND, no data available; NA, not applicable; R, right; L, left; and BL, bilateral.
To identify causative mutations for DA5D, we first screened each proband by Sanger sequencing genes in which mutations are known to cause similar DA disorders; these genes included TPM2 (MIM 190990), TNNT3 (MIM 600692), TNNI2 (MIM 191043), and MYH3 (MIM 160720). We also screened CHRNG (MIM 100730), mutations in which cause congenital contractures and pterygia in Escobar syndrome (MIM 265000).8 Additionally, each proband was screened for copy-number variations (CNVs) by genome-wide array-based comparative genomic hybridization. No pathogenic mutations or CNVs were identified. All studies were approved by the institutional review boards of the University of Washington and Seattle Children’s Hospital, and informed consent was obtained from participants or their parents. Subsequently, we selected three families in which recurrence among siblings was observed (families A, B, and C in Figure S2) for whole-genome sequencing (WGS). To prioritize variants identified via WGS, we genotyped all of the living affected individuals and their parents in family A (Figure 1) by using the HumanCytoSNP-12 DNA Analysis BeadChip, which interrogates ∼300,000 SNPs. Self-reported East Indian ancestry was confirmed with EIGENSTRAT.
In family A (Figure 1), the proband (V-1) had unaffected nonconsanguineous parents, whereas the two affected aunts (IV-5 and IV-6) and a deceased affected uncle (IV-2) were born to parents who were double first cousins. This observation suggested that the father of the proband (IV-3) was either cryptically related to the proband’s mother (IV-4) and carried the same mutation as other family members or that he was a carrier for a different pathogenic variant in the same gene. An alternative explanation was the presence of maternal isodisomy of the region containing the causal variant in proband IV-3. To test whether IV-3 was cryptically related to other family members, we constructed a pedigree that modeled the father of the proband (IV-3) as the great grandchild of I-3 and I-4; this would have permitted cotransmission of the same recessive variant to all affected individuals in the pedigree (Figure S3).
Under this model, we performed parametric linkage analysis in ALLEGRO by using a fully penetrant rare recessive model (f1 = 0; f2 = 1; q = 0.0001) and allele frequencies estimated from unrelated members of the HapMap GIH (Gujarati Indians in Houston, Texas) population on a ∼0.02 cM SNP map. No linkage peaks were observed in this analysis, suggesting that no homozygous genomic regions were inherited identically by descent and shared by both the proband and his affected aunts. This confirmed that either the causal variant in the unaffected carrier father existed on a different haplotype background than that of other family members (e.g., the proband was a compound heterozygote, whereas the affected siblings in the fourth generation were homozygous for the causal variant) or that the common haplotype was too small to be detected on our SNP map.
To test whether IV-3 contributed a different pathogenic variant, we performed parametric linkage analysis as above in only the two affected sibling aunts and their consanguineous parents (Figure 1). This analysis identified approximately 82 Mb of genomic regions showing evidence of linkage with DA5D. To model the additional information present if IV-4 transmitted a single copy of a causal haplotype to V-1, we performed linkage analysis under a dominant model (f0 = 0; f1 = 1; q = 0.0001) in the full pedigree (Figure 1). Comparing results from both analyses (e.g., combining the recessive and dominant modeling of linkage or a “redominant” model) revealed only three genomic regions that overlapped and narrowed the candidate interval to 16.7 Mb.
Next, WGS was performed on each of the selected families. In brief, 1 μm of genomic DNA was subjected to a series of shotgun-library-construction steps, including fragmentation through acoustic sonication (Covaris), end polishing (NEBNext End Repair kit), A-tailing (NEBNext dA Tailing kit), and ligation of 8 bp barcoded sequencing adaptors (Enzymatics Ultrapure T4 Ligase). Libraries were automatically size selected for fragments 350–550 bp in length with the automated PippinPrep cartridge system, which provides for fine control over the insert size and physically isolates each size fraction in separate chambers. Prior to sequencing, the library was amplified via PCR (Kapa HiFi Hotsart). For facilitating optimal flow-cell loading, the library concentration was determined by triplicate quantitative PCR (Kapa Illumina Library Quantification kit), and molecular-weight distributions were verified on the Agilent Bioanalyzer. Massively parallel sequencing by synthesis with fluorescently labeled, reversibly terminating nucleotides was carried out on the HiSeq sequencer. Samples were sequenced to an average depth of at least 30× with paired-end 100 bp reads; a third read sequenced the 8 bp index for confirmation of sample assignments.
Demultiplexed BAM files were aligned to a human reference (hg19) with the Burrows-Wheeler Aligner.9 Read data from a flow-cell lane were treated independently for alignment and quality-control purposes in instances where the merging of data from multiple lanes was required. All aligned read data were subjected to: (1) removal of duplicate reads, (2) indel realignment with the Genome Analysis Toolkit (GATK) IndelRealigner, and (3) base-quality recalibration with GATK TableRecalibration. Variant detection and genotyping were performed with the UnifiedGenotyper tool from GATK (v.1.529). Variant data for each sample were formatted (variant call format [VCF]) as “raw” calls that contained individual genotype data for one or multiple samples and were flagged with the filtration walker (GATK) for marking sites that were of lower quality and potential false positives (e.g., quality scores ≤ 50, allelic imbalance ≥ 0.75, long homopolymer runs > 3, and/or low quality by depth < 5). Variant data were annotated with the SeattleSeq Annotation Server. Variants identified in dbSNP v.134 or in 1,200 exomes from a subset of the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project (ESP) were excluded prior to analysis for all sequenced individuals.
Analysis of annotated WGS variants in family A (Figure 1) revealed in the 82 Mb region 15 homozygous variants shared identically by descent between the affected aunts (IV-5 and IV-6). Limiting the search to the 16.7 Mb haplotype that is homozygous and shared between the affected aunts and heterozygous in the proband (V-1) reduced the number of candidate genes with predicted functional variants to four (ECEL1 [MIM 605896], ALPPL2 [MIM 171810], CHRNG, TRIP11 [MIM 604505]). Examination of each of these four genes for variants in the affected individuals in the two additional families that underwent WGS revealed variants shared in only a single gene, ECEL1 (RefSeq accession number NM_004826.2), in the affected sibling (II-3) of the proband of family B (Figure S2). Specifically, in exon 2, the affected aunt (IV-5) in family A was homozygous for a c.716dupA (p.Tyr239∗) variant predicted to cause a frameshift resulting in an immediate stop codon, whereas the affected sibling in family B (II-3) was a compound heterozygote for a missense mutation (c.869A>G [p.Try290Cys]) in exon 4 and a 5 bp deletion and 3 bp insertion (c.797_801delinsGCT [p.Asp266Glyfs∗15]) predicted to result in a frameshift in exon 3 (Table 1 and Figure 2). Furthermore, Sanger sequencing of ECEL1 in the proband in family A (V-1) found that he inherited the c.716dupA mutation from his mother (IV-4) and additionally inherited a 12 bp in-frame deletion (c.344_355del [p.Asn115_Ala118del]) in exon 2 from his father (IV-3). Accordingly, as suspected from the linkage analysis of family A, DA5D was caused by homozygosity of a c.716dupA variant in ECEL1 in the offspring of the consanguineous mating between III-4 and III-5, and although the proband (V-1) inherited only a single c.716dupA variant from his carrier mother, his father was by chance a carrier for a different pathogenic variant in ECEL1.
Figure 2.
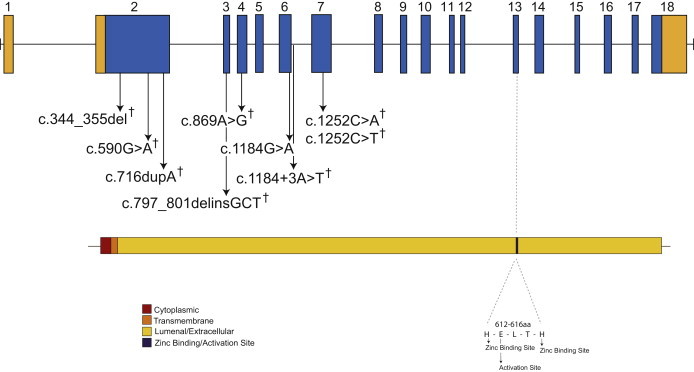
Genomic Structure and Allelic Spectrum of ECEL1 Mutations that Cause DA5D
ECEL1 is composed of 18 exons that encode UTRs (orange) and protein-coding-sequence domains (blue), including cytoplasmic (red), transmembrane (orange), and extracellular (gold) domains and the activation site (black). Arrows indicate the locations of nine different mutations found in five families affected by DA5D. The † symbol indicates mutations that were confirmed to be inherited from the parents.
To determine the extent to which ECEL1 mutations explain cases of DA5D, we used Sanger sequencing to screen four additional unrelated families (with a total of six affected individuals). These families consisted of two trios and two families with two affected siblings each (Figure S2). We identified ECEL1 mutations in three families (D, E, and F in Table 1 and Figure S2). Each of the affected individuals in families D and F was compound heterozygous for predicted functional variants in ECEL1, whereas the affected proband in family E was homozygous for a c.1184G>T (p.Arg395Gln) missense variant. Collectively, between the three families used for discovery and the four families used for validation studies, ECEL1 mutations were found in five out of seven (70%) tested kindreds, which included three of five families with more than one affected individual (60%) and two of two simplex cases (100%). In each of the five families for which DNA was available from one or both parents, heterozygosity for the ECEL1 mutation was found. In total, nine unique mutations, including five missense mutations, two frameshifts, one in-frame deletion, and one splicing mutation, were identified (Table 1 and Figure 2). None of the ECEL1 missense or splice-site mutations identified in the subjects were found in >13,000 chromosomes sequenced as part of the NHLBI ESP. The clinical characteristics of individuals with DA5D caused by ECEL1 mutations were indistinguishable from those of individuals without ECEL1 mutations. Additionally, no heterozygous pathogenic ECEL1 variants were found in 20 tested families affected by dominantly inherited DA5.
ECEL1 encodes endothelin-converting enzyme-like 1 (ECEL1) or damage-induced neuronal endopeptidase, a membrane-bound metalloprotease that, despite its structural similarity to endothelin-converting enzyme (ECE), does not cleave ECE substrates.10 In mice, absence of Ecel1 perturbs the terminal branching of motor neurons to the endplate of skeletal muscles, resulting in poor formation of the neuromuscular junction.11,12 However, the mechanism by which loss of ECEL1 affects distal axonal arborization of skeletal muscle remains unknown. The skeletal muscles affected in Ecel1−/− mice include the diaphragm and intercostal muscles, and Ecel1−/− mice die immediately after birth without taking a breath.11,12 This observation suggests that the restrictive lung disease observed in some individuals with DA5 might be caused by similar, although less severe, effects on respiratory muscles.
Our results suggest that DA5D is genetically heterogeneous. ECEL1 is highly homologous to ECE1 (MIM 600423), and an ECE1 C>T missense mutation resulting in a p.Arg742Cys substitution has previously been reported in an individual with Hirschsprung disease (MIM 142623), dysmorphic facial features, and congenital contractures of the hands.13 In DA5D-affected families in which no ECEL1 mutations were identified, we screened ECE1 but did not identify any pathogenic mutations. Branching defects similar to those observed in Ecel1−/− mice have also been reported in mice deficient in other genes, such as Pea3.14 Similarly, congenital contractures have been reported in individuals with mutations in other genes (e.g., CHRNG) known to contribute to the formation of the neuromuscular junction. Along this line, both of the affected aunts in family A (IV-5 and IV-6) were also homozygous for a c.994C>T (p.Arg332Trp) missense (GERP [genomic evolutionary rate profiling] score of −0.45) variant in CHRNG8 (RefSeq NM_005199.4). We cannot exclude that this CHRNG variant contributed to the phenotype of these two cases, particularly because each affected sibling had features not found in other DA5D cases, such as cutaneous syndactyly of the fingers and scoliosis, which are found in individuals with mutations in CHRNG. However, the c.994C>T variant has not been reported in cases of multiple pterygium syndrome, nor were CHRNG mutations found in other DA5D-affected families.
In summary, we used linkage analysis and WGS of a consanguineous pedigree to discover that mutations in ECEL1 cause DA5D and explain ∼70% of cases in our cohort. This observation suggests that DA5D is genetically heterogeneous. Our findings further support a role for ECEL1 in the development of the neuromuscular junction of human skeletal muscles of both the limbs and the trunk. Finally, our results distinguish a second developmental pathway that causes congenital-contracture syndromes.
Acknowledgments
We thank the families for their participation and support. Our work was supported in part by grants from the National Institutes of Health National Human Genome Research Institute (1U54HG006493 to M.B., D.N., and J.S.; 1RC2HG005608 to M.B., D.N., and J.S.; and 5RO1HG004316 to H.T.), the National Institute of Child Health and Development (HHSN27500503415C to J.M.S and HHSN267200700023C to E.M.F.), the Life Sciences Discovery Fund (2065508 and 0905001), and the Washington Research Foundation.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
FASTX-Toolkit, http://hannonlab.cshl.edu/fastx_toolkit/
Genome Analysis Toolkit (GATK), http://www.broadinstitute.org/gsa/wiki/
HumanCytoSNP-12 DNA Analysis BeadChip Kit, http://www.illumina.com/products/humancytosnp_12_dna_analysis_beadchip_kits.ilmn
Human Genome Variation Society (HGVS), http://www.hgvs.org/mutnomen/
NHLBI Exome Sequencing Project Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Eurexpress, http://www.eurexpress.org
Picard, http://picard.sourceforge.net/
SAMtools, http://samtools.sourceforge.net/
SeattleSeq Variation Annotation, http://gvs.gs.washington.edu/SeattleSeqAnnotation/
References
- 1.Bamshad M., Jorde L.B., Carey J.C. A revised and extended classification of the distal arthrogryposes. Am. J. Med. Genet. 1996;65:277–281. doi: 10.1002/(SICI)1096-8628(19961111)65:4<277::AID-AJMG6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 2.Sung S.S., Brassington A.M., Grannatt K., Rutherford A., Whitby F.G., Krakowiak P.A., Jorde L.B., Carey J.C., Bamshad M. Mutations in genes encoding fast-twitch contractile proteins cause distal arthrogryposis syndromes. Am. J. Hum. Genet. 2003;72:681–690. doi: 10.1086/368294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sung S.S., Brassington A.M., Krakowiak P.A., Carey J.C., Jorde L.B., Bamshad M. Mutations in TNNT3 cause multiple congenital contractures: A second locus for distal arthrogryposis type 2B. Am. J. Hum. Genet. 2003;73:212–214. doi: 10.1086/376418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Toydemir R.M., Rutherford A., Whitby F.G., Jorde L.B., Carey J.C., Bamshad M.J. Mutations in embryonic myosin heavy chain (MYH3) cause Freeman-Sheldon syndrome and Sheldon-Hall syndrome. Nat. Genet. 2006;38:561–565. doi: 10.1038/ng1775. [DOI] [PubMed] [Google Scholar]
- 5.Toydemir R.M., Chen H., Proud V.K., Martin R., van Bokhoven H., Hamel B.C., Tuerlings J.H., Stratakis C.A., Jorde L.B., Bamshad M.J. Trismus-pseudocamptodactyly syndrome is caused by recurrent mutation of MYH8. Am. J. Med. Genet. A. 2006;140:2387–2393. doi: 10.1002/ajmg.a.31495. [DOI] [PubMed] [Google Scholar]
- 6.Veugelers M., Bressan M., McDermott D.A., Weremowicz S., Morton C.C., Mabry C.C., Lefaivre J.F., Zunamon A., Destree A., Chaudron J.M., Basson C.T. Mutation of perinatal myosin heavy chain associated with a Carney complex variant. N. Engl. J. Med. 2004;351:460–469. doi: 10.1056/NEJMoa040584. [DOI] [PubMed] [Google Scholar]
- 7.Williams M.S., Elliott C.G., Bamshad M.J. Pulmonary disease is a component of distal arthrogryposis type 5. Am. J. Med. Genet. A. 2007;143:752–756. doi: 10.1002/ajmg.a.31648. [DOI] [PubMed] [Google Scholar]
- 8.Morgan N.V., Brueton L.A., Cox P., Greally M.T., Tolmie J., Pasha S., Aligianis I.A., van Bokhoven H., Marton T., Al-Gazali L. Mutations in the embryonal subunit of the acetylcholine receptor (CHRNG) cause lethal and Escobar variants of multiple pterygium syndrome. Am. J. Hum. Genet. 2006;79:390–395. doi: 10.1086/506256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shirotani K., Tsubuki S., Iwata N., Takaki Y., Harigaya W., Maruyama K., Kiryu-Seo S., Kiyama H., Iwata H., Tomita T. Neprilysin degrades both amyloid beta peptides 1-40 and 1-42 most rapidly and efficiently among thiorphan- and phosphoramidon-sensitive endopeptidases. J. Biol. Chem. 2001;276:21895–21901. doi: 10.1074/jbc.M008511200. [DOI] [PubMed] [Google Scholar]
- 11.Schweizer A., Valdenaire O., Köster A., Lang Y., Schmitt G., Lenz B., Bluethmann H., Rohrer J. Neonatal lethality in mice deficient in XCE, a novel member of the endothelin-converting enzyme and neutral endopeptidase family. J. Biol. Chem. 1999;274:20450–20456. doi: 10.1074/jbc.274.29.20450. [DOI] [PubMed] [Google Scholar]
- 12.Nagata K., Kiryu-Seo S., Maeda M., Yoshida K., Morita T., Kiyama H. Damage-induced neuronal endopeptidase is critical for presynaptic formation of neuromuscular junctions. J. Neurosci. 2010;30:6954–6962. doi: 10.1523/JNEUROSCI.4521-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hofstra R.M., Valdenaire O., Arch E., Osinga J., Kroes H., Löffler B.M., Hamosh A., Meijers C., Buys C.H. A loss-of-function mutation in the endothelin-converting enzyme 1 (ECE-1) associated with Hirschsprung disease, cardiac defects, and autonomic dysfunction. Am. J. Hum. Genet. 1999;64:304–308. doi: 10.1086/302184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haase G., Dessaud E., Garcès A., de Bovis B., Birling M., Filippi P., Schmalbruch H., Arber S., deLapeyrière O. GDNF acts through PEA3 to regulate cell body positioning and muscle innervation of specific motor neuron pools. Neuron. 2002;35:893–905. doi: 10.1016/s0896-6273(02)00864-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.