

Abstract
Opsismodysplasia (OPS) is a severe autosomal-recessive chondrodysplasia characterized by pre- and postnatal micromelia with extremely short hands and feet. The main radiological features are severe platyspondyly, squared metacarpals, delayed skeletal ossification, and metaphyseal cupping. In order to identify mutations causing OPS, a total of 16 cases (7 terminated pregnancies and 9 postnatal cases) from 10 unrelated families were included in this study. We performed exome sequencing in three cases from three unrelated families and only one gene was found to harbor mutations in all three cases: inositol polyphosphate phosphatase-like 1 (INPPL1). Screening INPPL1 in the remaining cases identified a total of 12 distinct INPPL1 mutations in the 10 families, present at the homozygote state in 7 consanguinous families and at the compound heterozygote state in the 3 remaining families. Most mutations (6/12) resulted in premature stop codons, 2/12 were splice site, and 4/12 were missense mutations located in the catalytic domain, 5-phosphatase. INPPL1 belongs to the inositol-1,4,5-trisphosphate 5-phosphatase family, a family of signal-modulating enzymes that govern a plethora of cellular functions by regulating the levels of specific phosphoinositides. Our finding of INPPL1 mutations in OPS, a severe spondylodysplastic dysplasia with major growth plate disorganization, supports a key and specific role of this enzyme in endochondral ossification.
Main Text
Opsismodysplasia (OPS [MIM 258480]) is a rare chondrodysplasia, first described in 1977 by Zonana et al.1 and coined as “opsismodysplasia” (from “opsismos,” Greek for “late”) by Maroteaux et al. in 1984.2,3 To date, 30 cases have been reported and recurrence in sibs and/or consanguinity have suggested an autosomal-recessive mode of inheritance.4 The disorder is characterized by pre- and postnatal micromelia with extremely short hands and feet. The main radiological features are severe platyspondyly, squared metacarpals, major delay in skeletal ossification, and metaphyseal cupping. The outcome is more variable than initially thought, ranging from severe prenatal findings to late survival.4
In the international nosology for skeletal dysplasias,5 OPS belongs to the group of severe spondylodysplastic dysplasias (group 14). This group also includes (1) achondrogenesis type 1A (ACG1A [MIM 200600]) due to TRIPP11 mutations (MIM 604505) and distinct by poor ossification of vertebral bodies and skull, (2) Schneckenbecken dysplasia (MIM 296250) due to SLC35D1 mutations (MIM 610804) and characterized by a snail-like appearance of the ilia, (3) spondylometaphyseal dysplasia (SMD) Sedaghatian type (MIM 250220), a less severe condition, characterized by laciness of the iliac wings, and finally (4) fibrochondrogenesis (FCG [MIM 228520]), the molecular basis of which remains unknown.
In order to identify the mutations causing OPS, a total of 16 cases from 10 unrelated families were included in this study. Among them, seven were terminated pregnancies (14–29 weeks of gestation) and nine were postnatal cases (birth to 19 years). Recurrency was observed in 5/10 families and consanguinity in 7/10. Inclusion criteria were (1) major delay in epiphyseal ossification, (2) platyspondyly, (3) metaphyseal cupping, and (4) very short metacarpals and phalanges (Figure 1). The clinical details are summarized in Table 1. Histological study of the femoral growth plate performed in the three cases from family 5 and in the prenatal case from family 3 (Table 1) showed similar disorganization of the growth plate with absence of columnar arrangement of proliferative cells and reduced hypertrophic zone with small number of hypertrophic chondrocytes (Figure 1A, III and IV). In two postnatal cases (Table 1, families 3 and 7), the phosphocalcic work up was normal (including blood levels of creatinine, calcium, phosphorus, thyroxin, thyrotropin, 25-hydroxyvitaminD, 1,25-dihydroxyvitaminD, parathyroid hormone, and urinary levels of creatinine, calcium, and phosphorus).
Figure 1.
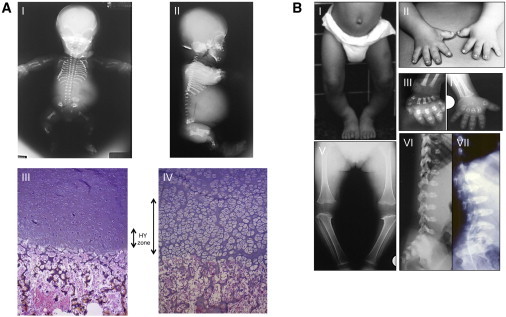
Clinical, Radiological, and Histological Features of OPS Cases
(A) Antenatal radiological and histological features in two OPS cases from family 5. Skeleton X-rays (I, II) in case 1 showing hypoplastic vertebral bodies, very short metacarpals, horizontal acetabular roof, and metaphyseal irregularities. Growth plate study at the femoral head in sib 2 (15 WG, III) showing nearly absent columnar organization and reduced hypertrophic zone (HY zone) with small reduced number of hypertrophic chondrocytes compared to wild-type growth plate (15 WG, IV).
(B) Clinical and radiological findings in case 1 (family 3) at 4 years (I, II, IV, V, VII) and 2 years (III, VI) showing lower limb valgus deformity (I), extremely short hands (II), short metacarpal with metaphyseal cupping and dysplastic carpal ossification (III, IV), severe epiphyseal delay and metaphyseal cupping around the knee (V), and severe platyspondyly (VI, VII).
Table 1.
Clinical Features of the Ten OPS Families
| Ethnic Origin | CS | Prenatal Findings | Parameters at Birth | Facial Features | Respiratory Insufficiency | Short Hand, Feet | Other | Outcome | |
|---|---|---|---|---|---|---|---|---|---|
| Family 1, Sib 1 | Arab Moslem | 1st cousin | short long bones (25 WG) | W 2,700 g; L 41.5 cm; HC 32.5 cm | hypertelorism, high forehead, short nose, long philtrum, large fontanelle | – | yes | – | died at 15 months |
| Family 1, Sib 2 | Arab Moslem | 1st cousin | short long bones | ? | ? | – | yes | – | termination of pregnancy |
| Family 2 (male) | Somalian | no | ? | ? | coarse face, hypertelorism, high forehead, short nose, large fontanelle | lung infections | yes | – | 4 years old; L, W ≤5 SD, HC P3 |
| Family 3, Sib 1 (male) | French/Algerian | no | ureteral dilatation | W 3,400 g; L 49 cm; HC 36 cm | coarse face, high forehead, short nose, long philtrum, large fontanelle | – | yes | paraplegia after surgery for scoliosis | 19 years old; H 140 cm; W 36 kg |
| Family 3, Sib 2 (male) | French/Algerian | no | polyhydramnios, short femora (22 WG), narrow thorax, short hands, feet | W 440 g; L 19.5 cm (22 WG) | high forehead, short nose | lung hypoplasia | yes | – | termination of pregnancy (22 WG) |
| Family 4 (male) | Portuguese | 1st cousin | short limbs | W 3,300 g; L 45 cm; HC 38 cm | hypertelorism, frontal bossing, short nose, long philtrum, macrostomia, wide fontanelle | yes; narrow thorax | yes | cervical kyphosis, scoliosis | 19 years |
| Family 5, Sib 1 (male) | French | no | short limbs (24 WG), short hands, narrow thorax | W 1,280 g; L 33 cm; HC 29 cm (29 WG) | coarse face, high forehead, short nose, long philtrum | pulmonary hypoplasia | yes | – | termination of pregnancy (29 WG) |
| Family 5, Sib 2 (female) | French | no | hygroma, short limbs (14 WG), narrow thorax | W 30 g; L 9.5 cm (15 WG) | – | – | – | – | termination of pregnancy (15 WG) |
| Family 5, Sib 3 (female) | French | no | hygroma, narrow thorax, short limbs | W 40 g; L 8.5 cm (14 WG) | – | – | – | – | termination of pregnancy (14 WG) |
| Family 6 (male) | Arab (Saudi) | 1st cousin | short limbs | ? | hypertelorism, midface hypoplasia, wide fontanelle, broad forehead | chronic lung disease | yes | tricuspide and mitral valve prolapse | 3.5 years old |
| Family 7 (male) | Algerian | 2nd cousin | – | W 3120 g; L 34 cm; HC 34 cm | high forehead, short nose with anteverted nares | – | yes | scoliosis, lower limb varus deformity | 9 years old; H 115 cm (≤4 SD); W 24.9 kg; HC 55.5 cm |
| Family 8, Sib 1 (female) | French | no | short femora (24 W), short hands, narrow thorax | W 1,400 g; L 29 cm; HC 31 cm | coarse face, high forehead, short nose | pulmonary hypoplasia | yes | – | termination of pregnancy (29 WG) |
| Family 8, Sib2 (female) | French | no | hygroma, IUGR | L 9 cm (15 WG) | – | – | yes | – | termination of pregnancy (15 WG) |
| Family 9 (male) | Brazil | 1st cousin | short limbs, polyhydramnios | W 1,455 g; L 32 cm; HC 29 cm | brachycephaly, midface hypoplasia, broad forehead, short nose, long philtrum, retrognathia, low-set ears | pulmonary hypoplasia | yes | short neck, narrow thorax, prominent calcaneus | stillborn (30 WG) |
| Family 10, Sib 1 (male) | Brazil | 7th cousin | short limbs, narrow thorax, polyhydramnios | W 2,620 g; L 40 cm; (35 WG) | brachycephaly, midface hypoplasia, broad forehead, short nose, low-set ears | yes | yes | absence right kidney (prenatal US) | died at 5 days |
| Family 10, Sib 2 | Brazil | 7th cousin | short limbs, narrow thorax, platspondyly | W 2,420 g; L 35.5 cm; HC 36 cm (36 WG) | brachycephaly, midface hypoplasia, broad forehead, short nose, low-set ears | yes | yes | – | died at 30 min |
Abbreviations are as follows: CS, consanguinity; WG, weeks of gestation; L, length; W, weight; HC, head circumference; IUGR, intrauterine growth retardation; ?, unknown; –, no.
Families 3–5 have been previously published in Cormier-Daire et al.4 and family 4 was previously published in Santos et al.17
Informed consent for participation and sample collection were obtained by protocols approved by the Necker Hospital ethics board committee. We first excluded the genes involved in the lethal spondylodysplastic group by direct sequencing, namely SBDS (MIM 607444) involved in some cases of spondylometaphyseal dysplasia Sedhagatian type,6 SLC35D1,7 and TRIP11.8 We then decided to undertake an exome capture-sequencing in three OPS cases from families 1–3.
Exome capture was performed at the French National Sequencing Institute (CNG) with the SureSelect Human All Exon kit (Agilent Technologies).9 Single-end sequencing was performed on an Illumina Genome Analyzer IIx (Illumina), generating 72-base reads. For sequence alignment, variant calling, and annotation, the sequences were aligned to the human genome reference sequence (hg18 build), via BWA aligner.10 Downstream processing was carried out with the Genome Analysis Toolkit (GATK),11 SAMtools,12 and Picard Tools. Substitution calls were made with GATK Unified Genotyper, whereas indel calls were made with a GATK IndelGenotyperV2. All calls with a read coverage ≤2× and a Phred-scaled SNP quality of ≤20 were filtered out. All the variants were annotated with an in-house developed annotation software system. We first focused our analyses on nonsynonymous variants, splice acceptor and donor site mutations, and coding indels, anticipating that synonymous variants were far less likely to be disease causing (Table S1 available online). We also defined variants as previously unidentified if they were absent from both control populations and data sets including dbSNP129, the 1000 Genomes Project, and in-house exome data.
Based on the recessive mode of inheritance of OPS, only one gene was found to harbor mutations in all three cases (Table S1) and was therefore selected. This gene, inositol polyphosphate phosphatase-like 1 (INPPL1), is also referred to as SHIP2, for SH2 (Src homology 2)-domain-containing inositol phosphatase (MIM 600829). Indeed, exome analysis detected five INPPL1 mutations in the three individuals and was present at the homozygote state in case 1 and compound heterozygote state in cases 2 and 3. These results were confirmed by Sanger sequencing. INPPL1 (RefSeq accession number NM_001567.3) is composed of 28 coding exons and encodes a protein of 1,258 amino acids characterized by a N-terminal SH2 domain, a conserved catalytic 5-phosphatase domain, a C-terminal proline-rich region with consensus sites for SH3 domain interactions, a ubiquitin interacting motif, and a sterile alpha motif (SAM) (Figure 2). Subsequent screening of the 28 INPPL1 coding exons in the remaining cases led to identification of seven additional mutations. Altogether, we identified a total of 12 distinct INPPL1 mutations in the 10 families (Figure 2, Table 2). Among them, 2/12 were nonsense mutations (c.2845C>T [p.Arg949∗], c.2719C>T [p.Arg907∗]) and 4/12 were frameshift mutations (c.276_280del [p.Gln93Profs∗3], c.1845dupT [p.Ile616Tyrfs∗14], c.94_121del [p.Glu32Metfs∗77], c.1328delinsTA [p.Thr443Ilefs∗23]) located in regions encoding the SH3 binding, SH2, or 5-phosphatase domains. They were expected to result in a truncated protein with no prolin-rich and SAM domains, which are crucial for protein-protein interaction. In addition, 2/12 were splice-site mutations (c.519−3A>G, c.1951+1G>A). Alamut Splicing Predictions, via bioinformatic analysis SSF, MaxEnt, NNSPLICE, and HSF, predicted a new acceptor site in c.519−2, generating a frameshift of two additional bases followed by a premature stop codon, and the suppression of the donor site in c.1951, generating a premature stop codon. Finally, 4/12 were missense mutations (c.1975C>T [p.Pro659Ser], c.1201C>T [p.Arg401Trp], c.2164T>A [p.Phe722Ile], c.2064G>T [p.Trp688Lys]) located in the 5-phosphatase domain. These mutations cosegregated with the disease, were present at the heterozygote state in the parents, were considered as pathogenic in the PolyPhen and Sift database, and were absent from alleles in 200 ethnicity-matched controls.
Figure 2.
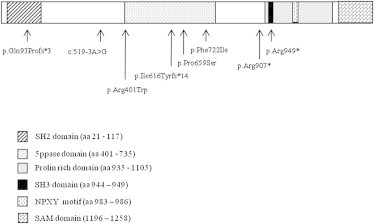
Localization of INPPL1 Mutations Identified in OPS Individuals
Table 2.
INPPL1 Mutations in Families in Opsismodyplasia
| Family | Ethnic Origin | CS | No. of Affected Children | Nucleotide Change | Status | Amino Acid Change | Location | Domain |
|---|---|---|---|---|---|---|---|---|
| 1 | Arab Moslem | yes | 2 | c.2845C>T/ | ho | p.Arg949∗ | Ex25 | SH3 binding domain |
| 2 | Somalian | no | 1 | c.276_280del | he | p.Gln93Profs∗3 | Ex3 | SH2 domain |
| c.1975C>T | he | p.Pro659Ser | Ex17 | 5phophatase domain | ||||
| 3 | French/Algerian | no | 2 | c.1201C>T | he | p.Arg401Trp | Ex11 | 5phophatase domain |
| c.2164T>A | he | p.Phe722Ile | Ex19 | 5phophatase domain | ||||
| 4 | Portuguese | yes | 1 | c.2719C>T | ho | p.Arg907∗ | Ex24 | / |
| 5 | French | yes | 3 | c.1845dupT | ho | p.Ile616Tyrfs∗14 | Ex15 | 5phophatase domain |
| 6 | Arab (Saudi) | yes | 1 | c.519-3A>G | ho | ? | In4 | / |
| 7 | Algerian | yes | 1 | c.1951+1G>A | ho | ? | In16 | / |
| 8 | French | no | 2 | c.1328delinsTA c.2064G>T |
he he |
p.Thr443Ilefs∗23 p.Trp688Lys |
Ex12 Ex18 |
5phophatase domain 5phophatase domain |
| 9 | Brazil | yes | 1 | c.94_121del | ho | p.Glu32Metfs∗77 | Ex1 | SH2 domain |
| 10 | Brazil | yes | 2 | c.94_121del | ho | p.Glu32Metfs∗77 | Ex1 | SH2 domain |
Abbreviations are as follows: Cs, consanguinity; ho, homozygote; he, heterozygote; /, mutation not localized in a known protein domain.
Here, we report INPPL1 mutations in ten unrelated families of opsismodysplasia. All cases clearly fulfilled the diagnostic criteria for OPS but were variable in severity. Indeed, prenatal findings detected in four families led to early termination of pregnancies (especially in recurrent sibs) in 7/16 cases and hygroma, short long bones, short extremities, and narrow thorax were consistently observed. Four children died early (stillborn at 30 weeks of gestation to 15 months of age). The five remaining cases ranged in age from 3 to 19 years old and had normal cognitive development, severe short stature (<4 SDS), lower limb deformity, and severe scoliosis with atlanto axial instability (at least in one case).
Most mutations (6/12) resulted in premature stop codons, 2/12 were splice-site mutations, and 4/12 were missense mutations located in the catalytic domain, 5-phosphatase, presumably responsible for impaired catalytic activity.
INPPL1 belongs to the inositol-1,4,5-trisphosphate 5-phosphatase family, a family of signal-modulating enzymes that govern a plethora of cellular functions by regulating the levels of specific phosphoinositides. Growth factor or insulin stimulation induces a canonical cascade resulting in the transient phosphorylation of phosphatidylinositol (PtdIns) (4,5)P(2) by PI3K (phosphoinositide 3-kinase) to form PtdIns(3,4,5)P(3), which is rapidly dephosphorylated either by phosphatase and tensin homolog (PTEN) back to PtdIns(4,5)P(2) or by the inositol polyphosphate 5-phosphatases (5-ptases) generating PtdIns(3,4)P(2). Ten mammalian 5-ptases have been identified. Their gene-targeted deletion in mice has revealed that these enzymes regulate haemopoietic cell proliferation, synaptic vesicle recycling, insulin signaling, endocytosis, vesicular trafficking, and actin polymerization.13 More specifically, INPPL1 has been implicated in the negative regulation of insulin signaling and glucose homeostasis in specific tissues.14 SNP analysis in the Japanese population have suggested that INPPL1 polymorphisms are associated with a predisposition to insulin resistance.15 Moreover, animal models lacking Inppl1 had increased glucose intolerance and insulin sensitivity. However, Inppl1−/− mice were viable and had normal glucose and insulin levels but were highly resistant to weight gain, suggesting that Inppl1 mediates obesity resistance.16 In the four survivors from our series, no insulin resistance was reported and length and weight were both ≤4 SD.
Recent studies have suggested additional noncatalytic properties of INPPL1 that may act as a docking protein for a large number of proteins including cytoskeletal, focal adhesion, or scaffold proteins, phosphatases, and tyrosine kinase-associated receptors, like EGF receptor. Moreover, loss of INPPL1 in zebrafish led to an increased and expanded expression of outputs of FGF-mediated signaling.13
The finding of INPPL1 mutations in OPS, a severe spondylodysplastic dysplasia with major growth plate disorganization, supports a key and specific role of this enzyme in the endochondral ossification process, through either its role in postranslational modifications (phosphorylation or ubiquitination) or its interaction with specific protein network. We conclude that INPPL1 mutations are responsible for OPS. Ongoing studies will hopefully lead to an understanding of the specific role of this enzyme in the ossification process.
Acknowledgments
We thank all families for their contribution to this work. We thank the GIS Maladies Rares for the funding of the exome project.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Picard, http://picard.sourceforge.net/
UniProt, http://www.uniprot.org/
References
- 1.Zonana J., Rimoin D.L., Lachman R.S., Cohen A.H. A unique chondrodysplasia secondary to a defect in chondroosseous transformation. Birth Defects Orig. Artic. Ser. 1977;13(3D):155–163. [PubMed] [Google Scholar]
- 2.Maroteaux P., Stanescu V., Stanescu R. Four recently described osteochondrodysplasias. Prog. Clin. Biol. Res. 1982;104:345–350. [PubMed] [Google Scholar]
- 3.Maroteaux P., Stanescu V., Stanescu R., Le Marec B., Moraine C., Lejarraga H. Opsismodysplasia: a new type of chondrodysplasia with predominant involvement of the bones of the hand and the vertebrae. Am. J. Med. Genet. 1984;19:171–182. doi: 10.1002/ajmg.1320190117. [DOI] [PubMed] [Google Scholar]
- 4.Cormier-Daire V., Delezoide A.L., Philip N., Marcorelles P., Casas K., Hillion Y., Faivre L., Rimoin D.L., Munnich A., Maroteaux P., Le Merrer M. Clinical, radiological, and chondro-osseous findings in opsismodysplasia: survey of a series of 12 unreported cases. J. Med. Genet. 2003;40:195–200. doi: 10.1136/jmg.40.3.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Warman M.L., Cormier-Daire V., Hall C., Krakow D., Lachman R., LeMerrer M., Mortier G., Mundlos S., Nishimura G., Rimoin D.L. Nosology and classification of genetic skeletal disorders: 2010 revision. Am. J. Med. Genet. A. 2011;155A:943–968. doi: 10.1002/ajmg.a.33909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boocock G.R.B., Morrison J.A., Popovic M., Richards N., Ellis L., Durie P.R., Rommens J.M. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat. Genet. 2003;33:97–101. doi: 10.1038/ng1062. [DOI] [PubMed] [Google Scholar]
- 7.Hiraoka S., Furuichi T., Nishimura G., Shibata S., Yanagishita M., Rimoin D.L., Superti-Furga A., Nikkels P.G., Ogawa M., Katsuyama K. Nucleotide-sugar transporter SLC35D1 is critical to chondroitin sulfate synthesis in cartilage and skeletal development in mouse and human. Nat. Med. 2007;13:1363–1367. doi: 10.1038/nm1655. [DOI] [PubMed] [Google Scholar]
- 8.Smits P., Bolton A.D., Funari V., Hong M., Boyden E.D., Lu L., Manning D.K., Dwyer N.D., Moran J.L., Prysak M. Lethal skeletal dysplasia in mice and humans lacking the golgin GMAP-210. N. Engl. J. Med. 2010;362:206–216. doi: 10.1056/NEJMoa0900158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Byun M., Abhyankar A., Lelarge V., Plancoulaine S., Palanduz A., Telhan L., Boisson B., Picard C., Dewell S., Zhao C. Whole-exome sequencing-based discovery of STIM1 deficiency in a child with fatal classic Kaposi sarcoma. J. Exp. Med. 2010;207:2307–2312. doi: 10.1084/jem.20101597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ooms L.M., Horan K.A., Rahman P., Seaton G., Gurung R., Kethesparan D.S., Mitchell C.A. The role of the inositol polyphosphate 5-phosphatases in cellular function and human disease. Biochem. J. 2009;419:29–49. doi: 10.1042/BJ20081673. [DOI] [PubMed] [Google Scholar]
- 14.Dyson J.M., Kong A.M., Wiradjaja F., Astle M.V., Gurung R., Mitchell C.A. The SH2 domain containing inositol polyphosphate 5-phosphatase-2: SHIP2. Int. J. Biochem. Cell Biol. 2005;37:2260–2265. doi: 10.1016/j.biocel.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 15.Kagawa S., Sasaoka T., Yaguchi S., Ishihara H., Tsuneki H., Murakami S., Fukui K., Wada T., Kobayashi S., Kimura I., Kobayashi M. Impact of SRC homology 2-containing inositol 5′-phosphatase 2 gene polymorphisms detected in a Japanese population on insulin signaling. J. Clin. Endocrinol. Metab. 2005;90:2911–2919. doi: 10.1210/jc.2004-1724. [DOI] [PubMed] [Google Scholar]
- 16.Sleeman M.W., Wortley K.E., Lai K.-M.V., Gowen L.C., Kintner J., Kline W.O., Garcia K., Stitt T.N., Yancopoulos G.D., Wiegand S.J., Glass D.J. Absence of the lipid phosphatase SHIP2 confers resistance to dietary obesity. Nat. Med. 2005;11:199–205. doi: 10.1038/nm1178. [DOI] [PubMed] [Google Scholar]
- 17.Santos H.G., Saraiva J.M. Opsismodysplasia: another case and literature review. Clin. Dysmorphol. 1995;4:222–226. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.