

Abstract
Klippel-Feil syndrome (KFS) is a segmentation malformation of the cervical spine; clinically, it manifests as a short neck with reduced mobility and a low posterior hairline. Several genes have been proposed as candidates for KFS when it is present with other associated anomalies, but the genetics of isolated KFS have been difficult to study because of the syndrome’s mostly sporadic occurrence. We describe a multiplex consanguineous family in which isolated KFS maps to a single 17q21.31 locus that harbors a homozygous frameshift deletion in MEOX1; this deletion results in complete instability of the transcript. Direct sequencing of this gene in two siblings from another consanguineous family affected by isolated KFS uncovered another homozygous truncating (nonsense) MEOX1 mutation that also leads to complete degradation of the transcript. This gene encodes a transcription factor with a well-established and nonredundant role in somite development, and homozygous null alleles of Meox1 in mice have a cervical skeletal defect that is remarkably similar to the one we observe in human individuals with MEOX1 mutations. Our data strongly suggest that KFS is the human phenotypic equivalent of the sclerotome polarity defect that results from Meox1 deficiency in mice.
Main Text
The vertebral column is a major anatomical structure that distinguishes an entire subphylum of the animal kingdom. Its conspicuous segmented pattern as individual vertebrae joined together by various joints and ligaments provides many mechanical advantages that facilitate movements in various axes.1 This segmentation is the result of a highly orchestrated and complex developmental process that is marked by the formation of somites, mesoderm-derived compartments that are mostly mesenchymal but are encapsulated by epithelium, and that proceeds in caudal-rostral direction.1 Extensive patterning takes place during somitogenesis and involves polarization, differentiation, and segmentation, the end result of which is that individual somites divide into three different compartments: sclerotome, myotome, and dermatome. These compartments each give rise to a distinct group of tissues: vertebral bodies, vertebral muscles, and dorsal skin dermis, respectively.1–3
Several somite segmentation defects are known to give rise to recognizable clinical phenotypes in humans. Those affecting the cervical spine are particularly disabling because of its high range of mobility. Klippel-Feil syndrome (KFS [MIM 214300]) is a prototypic segmentation defect in the cervical spine; it manifests clinically as a short neck with painless restriction in mobility and a low posterior hairline.4 Radiologically, KFS is characterized by a variable degree of neck vertebral fusion, which is often associated with additional skeletal defects such as Sprengel’s deformity of the scapula (the scapula is positioned higher than normal) and omovertebral bone (extra bone forms between the medial aspect of the scapula and the vertebral body).5 KFS affects 1:40,000 individuals and is often sporadic.6 However, familial occurrence and the observation of KFS in association with various chromosomal aberrations support a genetic origin of KFS, although this has been difficult to study.7 PAX1 (MIM 167411), GDF6 (MIM 601147), and GDF3 (MIM 606522) play roles in somite development and have been the focus of sequencing projects as candidate genes. Several missense variants have been identified in these genes in a small percentage of KFS-affected individuals with various other manifestations.8–10 Interestingly, some of these presumed pathogenic variants are now listed in Exome Variant Server (EVS) as population variants with a frequency much higher than that of KFS, raising doubts about the original proposed link. The best available evidence for a Mendelian form of KFS comes from previously reported consanguineous multiplex pedigrees in which KFS appears to follow an autosomal-recessive mode of inheritance.11 However, no mapping data are available for any of these families, so the gene or genes that underlie KFS in these families remain unknown. Here, we describe an autosomal-recessive form of KFS that segregates with truncation mutations in MEOX1 (MIM 600147), encoding a transcription factor with a well-established role in somite development, in two multiplex consanguineous families.
The index is a 10-year-old Saudi girl who was referred by orthopedics because of KFS. She was born to healthy first-cousin parents who had previously had a healthy male child (Figure 1). Respiratory distress was diagnosed shortly after her term delivery, but she only had a brief requirement for supplemental oxygen. A short neck was noted on neonatal examination, and radiological examination revealed a cervical segmentation defect consistent with KFS. Her development has been normal, and there was no history suggestive of the involvement of other systems. Physical examination revealed normal weight and head circumference but borderline short stature (fifth centile for age). Her neck was short, and she had a low posterior hairline (Figure 1). She was unable to flex, extend, or rotate her neck or lift her right hand to touch the back of her head. The rest of the physical examination was normal. A full skeletal survey confirmed the C2-C3 fusion and Sprengel’s deformity but revealed no additional skeletal defects (Figure 1). The family history and photographs revealed the presence of several similarly affected relatives (Figure 1).
Figure 1.
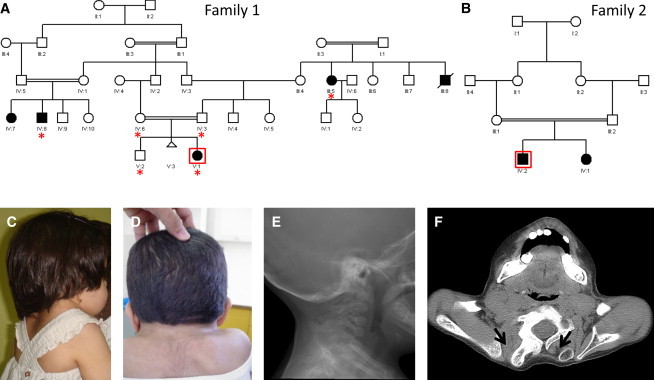
Identification of Two Families with Autosomal-Recessive KFS
(A) Pedigree of family 1 (red asterisks denote individuals whose DNA was available for analysis).
(B) Pedigree of family 2.
(C) Clinical photograph of the index individual in family 1; note a short neck and elevated scapulae.
(D) Clinical photograph of the index individual in family 2; note a short neck, a low posterior hair line, and elevated scapulae.
(E) Cervical-spine radiogram showing a segmentation defect.
(F) Computed-tomography image of the shoulder of the index individual in family 2. Note that the bilateral omovertebral bone (black arrow) is much more developed on the right, where it is fused with the right posterior segment of the C7 vertebral body.
The family pedigree was highly suggestive of an autosomal-recessive mode of inheritance, so we enrolled available surviving affected members and relevant unaffected relatives (denoted by red asterisks in Figure 1) in a King Faisal Specialist Hospital and Research Center (KFHSRC) IRB-approved protocol after obtaining written informed consent from the participants (RAC# 2080006). DNA was extracted from whole blood, and genotyping on an Axiom platform was then performed according to the Manufacturer’s instructions (Affymetrix, Santa Clara, CA, USA). Autozygosity mapping was performed on the resulting genotypes with autoSNPa.12 Interestingly, only one run of homozygosity (ROH), chr17: 29,323,850–45,448,560 (hg18), was exclusively shared by the affected members and was subsequently confirmed by linkage analysis with easyLINKAGE13 (Figure 2). This interval contains 477 genes, but close examination for genes with relevant phenotypes in the mouse knockout highlighted MEOX1 as the most compelling candidate given the mouse phenotype and its established role in somite formation, especially in the cervical spine (see below). Therefore, we proceeded with direct sequencing of MEOX1 and uncovered a 1 bp deletion, c.94delG (RefSeq accession number NM_004527.3), that results in complete loss of the mutant transcript, most likely as a result of nonsense-mediated decay (NMD) (Figure 3). The mutation predicts frameshift and premature truncation of the resulting protein (p.Ala32Profs∗165), so even if a mutant protein is made, it will completely lack the DNA-binding homeobox and is thus likely to be null (Figure 3). This mutation fully segregated with the phenotype and was absent in 210 Saudi controls and in the Exome Variant Server (EVS; see Web Resources) database.
Figure 2.
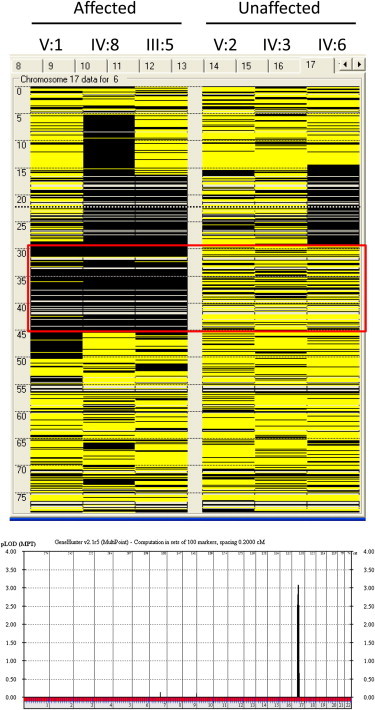
Identification of an Autosomal-Recessive KFS Locus
Upper panel: Homozygosity mapping of family 1 via autoSNPa. Each column represents the homozygosity profile of one individual, and each row represents the call of an individual SNP on chromosome 17 (yellow, heterozygous; black, homozygous). Note the large run of homozygosity that is exclusively shared by the affected members (boxed in red) in family 1 (only three members were available for analysis).
Lower panel: Linkage analysis confirmed the critical run of homozygosity, evidenced by the presence of a single significant linkage peak corresponding to the same interval.
Figure 3.
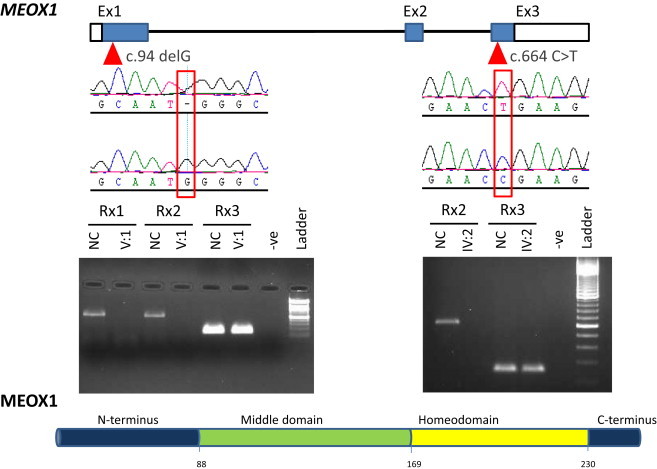
Identification of Two Null Alleles in MEOX1
The location of the two mutations is shown above the schematic depiction of MEOX1 along with sequence chromatograms of the affected individual and normal control. An RT-PCR experiment for the mutation in family 1 and family 2 is also shown; Rxn1 refers to one segment of exon 1, Rxn2 refers to another segment of exon 1, and Rxn3 is the GAPDH control. Note the complete absence of the mutant transcript in the affected individuals compared to the control. The source of RNA was lymphoblasts from individual V:1 and whole blood from individual IV:2.
In the course of this study, we were able to recruit another individual with typical KFS. He was diagnosed with Pierre-Robin sequence (MIM 261800) at birth (he had micrognathia and the typical U-shaped cleft palate, which was later repaired surgically). Congenital ptosis was diagnosed during follow up. His physical examination revealed a short neck with severely limited mobility, a low posterior hairline, scoliosis, and ptosis. His skeletal survey revealed, in addition to a cervical segmentation defect, the presence of omovertebral bones, Sprengel’s deformity, and dextroscoliotic deformity of the thoracic spine between T2 and T11(Figure 1). His family history revealed that his parents are first cousins and that he has an affected sister. Subsequent evaluation of the sister revealed identical physical findings except that the cleft palate was absent. We sequenced the entire coding region of MEOX1 and uncovered a homozygous transition, c.664C>T, that created a premature stop codon, i.e., nonsense mutation p.Arg222∗ (Figure 3). Although this mutation affects the last exon and so is unlikely to be subject to NMD, RT-PCR with blood-derived RNA consistently showed complete absence of the mutant transcript (Figure 3). NMD caused by introduction of premature termination codon in the last exon has been described before but very rarely.14,15 Our results indicate that both mutations are null, which is consistent with the similarity in clinical phenotype between the two families.
Meox1 and its paralogue, Meox2, encode homeobox proteins that are localized to many mesodermal structures.16,17 Their expression patterns are both overlapping and distinct.16–19 The focus has mostly been on their roles in somite development, which also appear to be both overlapping and distinct. Meox1 appears to have a non-redundant role in the development of the sclerotome whereas Meox2 appears to have a nonredundant role in the development of the myotome such that in Meox1/Meox2 double homozygous knockout mice there is severe deficiency of the sclerotome- and myotome-derived structures, whereas the presence of one copy of Meox1 or Meox2 rescues sclerotome or myotome development, respectively.20 Two independent null alleles in Meox1 have been described, and although heterozygotes are phenotypically normal, homozygotes have an identical segmentation defect of the cervical spine but normal muscle development. This pattern is remarkably similar to the phenotype we observe in individuals with MEOX1-truncating mutations.21
The mechanism by which Meox1 controls normal somite development is incompletely understood. One proposed mechanism is that Meox1 maintains the appropriate expression of downstream genes known to regulate somite development; namely, these genes are Bapx1, Tbx18, and Uncx.21,22 This proposal is supported by the fact that Meox1 physically binds the promoter of these genes and that its deficiency perturbs their expression domain. One conspicuous effect of Meox1 deficiency is the diminution of the normal caudal/rostral polarity of somites at very early stages of development, and although differential cell proliferation has been proposed as a mechanism, the exact mechanism remains unknown.21 Interestingly, the authors who describe the distinct skeletal profile in Meox1−/− mice speculated that the equivalent human phenotype might be a spondylocostal dysostosis syndrome, and our data strongly support the idea that KFS is indeed the human phenotypic equivalent. Of note, a feedback loop between Meox1 and Hoxa2 has been demonstrated in the development of the second branchial arch, and although Meox1 was proposed to have a redundant role in this process, we note that the Pierre-Robin sequence seen here in one of the two human individuals with a MEOX1 mutation suggests that this role may not be completely redundant in humans.23
In summary, we show that homozygous truncating mutations in the gene encoding mesenchyme homeobox 1 cause an autosomal-recessive form of KFS. The mode of inheritance, the predilection of the segmentation defect to the cervical spine, and the sparing of myotome-derived structures are remarkably conserved features between humans and mice deficient in this gene. It remains to be seen what the contribution of MEOX1 is, if any, to sporadic forms of KFS.
Acknowledgments
We thank the families for their enthusiastic participation. We thank the Genotyping and Sequencing Core Facilities at the King Faisal Specialist Hospital and Research Center for their technical help. This work was supported by King Abdulaziz City for Science and Technology grant 09-MED941-20 (F.S.A.) and Dubai-Harvard Foundation for Medical Research collaborative research grant (F.S.A.).
Web Resources
The URLs for data presented herein are as follows:
Exome Variant Server (EVS), http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Eckalbar W.L., Fisher R.E., Rawls A., Kusumi K. Scoliosis and segmentation defects of the vertebrae. Wiley Interdisciplinary Reviews: Developmental Biology. 2012;1:401–423. doi: 10.1002/wdev.34. [DOI] [PubMed] [Google Scholar]
- 2.Gridley T. The long and short of it: Somite formation in mice. Developmental Dynamics: An Official Publication of the American Association of Anatomists. 2006;235:2330–2336. doi: 10.1002/dvdy.20850. [DOI] [PubMed] [Google Scholar]
- 3.Takahashi Y., Sato Y. Somitogenesis as a model to study the formation of morphological boundaries and cell epithelialization. Dev. Growth Differ. 2008;50(Suppl 1):S149–S155. doi: 10.1111/j.1440-169X.2008.01018.x. [DOI] [PubMed] [Google Scholar]
- 4.Tracy M.R., Dormans J.P., Kusumi K. Klippel-Feil syndrome: Clinical features and current understanding of etiology. Clin. Orthop. Relat. Res. 2004;424:183–190. [PubMed] [Google Scholar]
- 5.McBride W.Z. Klippel-Feil syndrome. Am. Fam. Physician. 1992;45:633–635. [PubMed] [Google Scholar]
- 6.Cooper J.C. The Klippel-Feil syndrome. A rare cause of cervico-facial deformity. Br. Dent. J. 1976;140:264–268. doi: 10.1038/sj.bdj.4803748. [DOI] [PubMed] [Google Scholar]
- 7.Gunderson C.H., Greenspan R.H., Glaser G.H., Lubs H.A. The Klippel-Feil syndrome: Genetic and clinical reevaluation of cervical fusion. Medicine (Baltimore) 1967;46:491–512. doi: 10.1097/00005792-196711000-00003. [DOI] [PubMed] [Google Scholar]
- 8.McGaughran J.M., Oates A., Donnai D., Read A.P., Tassabehji M. Mutations in PAX1 may be associated with Klippel-Feil syndrome. Eur. J. Hum. Genet. 2003;11:468–474. doi: 10.1038/sj.ejhg.5200987. [DOI] [PubMed] [Google Scholar]
- 9.Tassabehji M., Fang Z.M., Hilton E.N., McGaughran J., Zhao Z., de Bock C.E., Howard E., Malass M., Donnai D., Diwan A. Mutations in GDF6 are associated with vertebral segmentation defects in Klippel-Feil syndrome. Hum. Mutat. 2008;29:1017–1027. doi: 10.1002/humu.20741. [DOI] [PubMed] [Google Scholar]
- 10.Ye M., Berry-Wynne K.M., Asai-Coakwell M., Sundaresan P., Footz T., French C.R., Abitbol M., Fleisch V.C., Corbett N., Allison W.T. Mutation of the bone morphogenetic protein GDF3 causes ocular and skeletal anomalies. Hum. Mol. Genet. 2010;19:287–298. doi: 10.1093/hmg/ddp496. [DOI] [PubMed] [Google Scholar]
- 11.Da Silva E.O. Autosomal recessive Klippel-Feil syndrome. J. Med. Genet. 1982;19:130–134. doi: 10.1136/jmg.19.2.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carr I.M., Flintoff K.J., Taylor G.R., Markham A.F., Bonthron D.T. Interactive visual analysis of SNP data for rapid autozygosity mapping in consanguineous families. Hum. Mutat. 2006;27:1041–1046. doi: 10.1002/humu.20383. [DOI] [PubMed] [Google Scholar]
- 13.Hoffmann K., Lindner T.H. easyLINKAGE-Plus—Automated linkage analyses using large-scale SNP data. Bioinformatics. 2005;21:3565–3567. doi: 10.1093/bioinformatics/bti571. [DOI] [PubMed] [Google Scholar]
- 14.Natsuga K., Nishie W., Shinkuma S., Arita K., Nakamura H., Ohyama M., Osaka H., Kambara T., Hirako Y., Shimizu H. Plectin deficiency leads to both muscular dystrophy and pyloric atresia in epidermolysis bullosa simplex. Hum. Mutat. 2010;31:E1687–E1698. doi: 10.1002/humu.21330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan D., Weng Y.M., Graham H.K., Sillence D.O., Bateman J.F. A nonsense mutation in the carboxyl-terminal domain of type X collagen causes haploinsufficiency in schmid metaphyseal chondrodysplasia. J. Clin. Invest. 1998;101:1490–1499. doi: 10.1172/JCI1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Candia A.F., Hu J., Crosby J., Lalley P.A., Noden D., Nadeau J.H., Wright C.V. Mox-1 and Mox-2 define a novel homeobox gene subfamily and are differentially expressed during early mesodermal patterning in mouse embryos. Development. 1992;116:1123–1136. doi: 10.1242/dev.116.4.1123. [DOI] [PubMed] [Google Scholar]
- 17.Candia A.F., Wright C.V. Differential localization of Mox-1 and Mox-2 proteins indicates distinct roles during development. Int. J. Dev. Biol. 1996;40:1179–1184. [PubMed] [Google Scholar]
- 18.Candia A.F., Wright C.V. The expression pattern of Xenopus Mox-2 implies a role in initial mesodermal differentiation. Mech. Dev. 1995;52:27–36. doi: 10.1016/0925-4773(95)00384-d. [DOI] [PubMed] [Google Scholar]
- 19.Grigoriou M., Kastrinaki M.C., Modi W.S., Theodorakis K., Mankoo B., Pachnis V., Karagogeos D. Isolation of the human MOX2 homeobox gene and localization to chromosome 7p22.1-p21.3. Genomics. 1995;26:550–555. doi: 10.1016/0888-7543(95)80174-k. [DOI] [PubMed] [Google Scholar]
- 20.Mankoo B.S., Skuntz S., Harrigan I., Grigorieva E., Candia A., Wright C.V., Arnheiter H., Pachnis V. The concerted action of Meox homeobox genes is required upstream of genetic pathways essential for the formation, patterning and differentiation of somites. Development. 2003;130:4655–4664. doi: 10.1242/dev.00687. [DOI] [PubMed] [Google Scholar]
- 21.Skuntz S., Mankoo B., Nguyen M.T., Hustert E., Nakayama A., Tournier-Lasserve E., Wright C.V., Pachnis V., Bharti K., Arnheiter H. Lack of the mesodermal homeodomain protein MEOX1 disrupts sclerotome polarity and leads to a remodeling of the cranio-cervical joints of the axial skeleton. Dev. Biol. 2009;332:383–395. doi: 10.1016/j.ydbio.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodrigo I., Bovolenta P., Mankoo B.S., Imai K. Meox homeodomain proteins are required for Bapx1 expression in the sclerotome and activate its transcription by direct binding to its promoter. Mol. Cell. Biol. 2004;24:2757–2766. doi: 10.1128/MCB.24.7.2757-2766.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kirilenko P., He G., Mankoo B.S., Mallo M., Jones R., Bobola N. Transient activation of meox1 is an early component of the gene regulatory network downstream of hoxa2. Mol. Cell. Biol. 2011;31:1301–1308. doi: 10.1128/MCB.00705-10. [DOI] [PMC free article] [PubMed] [Google Scholar]