

Abstract
Reduced FCGR3B copy number is associated with increased risk of systemic lupus erythematosus (SLE). The five FCGR2/FCGR3 genes are arranged across two highly paralogous genomic segments on chromosome 1q23. Previous studies have suggested mechanisms for structural rearrangements at the FCGR2/FCGR3 locus and have proposed mechanisms whereby altered FCGR3B copy number predisposes to autoimmunity, but the high degree of sequence similarity between paralogous segments has prevented precise definition of the molecular events and their functional consequences. To pursue the genomic pathology associated with FCGR3B copy-number variation, we integrated sequencing data from fosmid and bacterial artificial chromosome clones and sequence-captured DNA from FCGR3B-deleted genomes to establish a detailed map of allelic and paralogous sequence variation across the FCGR2/FCGR3 locus. This analysis identified two highly paralogous 24.5 kb blocks within the FCGR2C/FCGR3B/FCGR2B locus that are devoid of nonpolymorphic paralogous sequence variations and that define the limits of the genomic regions in which nonallelic homologous recombination leads to FCGR2C/FCGR3B copy-number variation. Further, the data showed evidence of swapping of haplotype blocks between these highly paralogous blocks that most likely arose from sequential ancestral recombination events across the region. Functionally, we found by flow cytometry, immunoblotting and cDNA sequencing that individuals with FCGR3B-deleted alleles show ectopic presence of FcγRIIb on natural killer (NK) cells. We conclude that FCGR3B deletion juxtaposes the 5′-regulatory sequences of FCGR2C with the coding sequence of FCGR2B, creating a chimeric gene that results in an ectopic accumulation of FcγRIIb on NK cells and provides an explanation for SLE risk associated with reduced FCGR3B gene copy number.
Introduction
Systemic lupus erythematosus (SLE [MIM 152700]) is a chronic autoimmune disease with effects on many organ systems including skin, lungs, heart, joints, blood, and kidneys. Environmental triggers and genetic factors predispose to development of SLE, but the molecular mechanisms are not well understood.1 However, impaired clearance of immune complexes and dying cells, dysregulation of apoptosis, and presence of autoantibodies to nuclear antigens are believed to be involved.2 There is increased prevalence of disease in non-European populations and women are at ∼10-fold greater risk than men.3
The FCGR locus on chromosome 1q23 is subject to copy-number variation (CNV) with reduced copy number of FCGR3B associating with the immune diseases SLE and rheumatoid arthritis (RA [MIM 180300]) in various populations.4–9 Besides FCGR3B (CD16B [MIM 610665]), the region contains genes for the low-affinity Fc gamma receptors FCGR2A (CD32A [MIM 146790]), FCGR3A (CD16A [MIM 146740]), FCGR2C (CD32C [MIM 612169]), and FCGR2B (CD32B [MIM 604590]), encoding respectively the proteins for FcγRIIa, FcγRIIIa, FcγRIIc, and FcgRIIb. FcγRIIa and FcγRIIc contain immunoreceptor tyrosine-based activatory motifs (ITAMs); FcγRIIIa, like the high affinity activatory FcγRI, signals through a common gamma chain, and FcγRIIIb is membrane-linked by a glycosylphosphatidylinositol (GPI) anchor. These are all activatory receptors unlike FcγRIIb (encoded by FCGR2B), which contains an immunoreceptor tyrosine-based inhibitory motif (ITIM).10 The five FCGR genes are arranged across two ∼85 kb paralogous blocks suggesting an evolutionary origin through tandem duplication (Figure 1A). The centromeric (proximal) block contains FCGR2A, FCGR3A, and the 5′ portion of FCGR2C. The telomeric (distal) block contains the 3′ portion of FCGR2C as well as FCGR3B and FCGR2B.11 Paralog ratio test (PRT) and multiplex ligation-dependent probe amplification assays have demonstrated stable Mendelian inheritance of CNV blocks containing FCGR3A, FCGR2C, and FCGR3B but have not shown CNV at the FCGR2A and FCGR2B loci.7,12 The most common CNV block includes FCGR3B and FCGR2C and it was recently proposed that FCGR3B CNV is the result of nonallelic homologous recombination between proximal and distal FCGR paralogs.13
Figure 1.
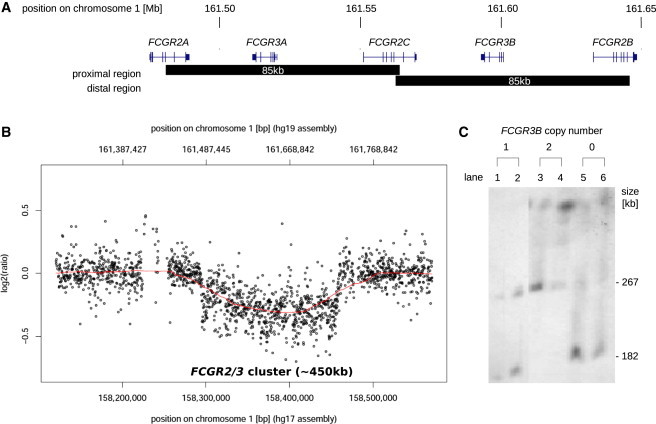
Copy-Number Variation at the FCGR Locus
(A) Arrangement of FCGR genes across the FCGR2/FCGR3 cluster on chromosome one. Black bars below the gene loci indicate proximal and distal regions of paralogy.
(B) CGH array data showing the deletion of FCGR3B and surrounding area. Probe design based on NCBI assembly version 35 (hg17).
(C) Pulsed field gel electrophoresis and Southern blotting with FCGR2 and FCGR3 probes of PmeI digested genomic DNA from individuals with different FCRGR3B copy number. Copy number refers to the number of FCGR3B containing haplotyptes. In two-copy individuals with FCGR3B present on both chromosomes, a band of size 267 kb is detected representing the FCGR3B containing haplotype. The presence of a smaller-size band (182 kb) in individuals with one or both copies of FCGR3B missing, which is not detected in two-copy individuals, indicates the loss of an ∼85 kb region of genomic DNA on FCGR3B-deleted haplotypes.
Previous studies have suggested mechanisms whereby loss of either FCGR3B or FCGR2C may predispose to autoimmunity by reducing binding and clearance of immune complexes. FCGR3B is expressed on neutrophils and a direct positive correlation has been observed between the number of copies of FCGR3B and FcγRIIIb translation and function.7,14 It has previously been suggested that loss of FCGR3B may alter immune complex handling in the tissues by neutrophils4,7,14 although the role of neutrophils in immune complex handling is not well understood. In addition to CNV, FCGR3B exhibits several polymorphisms that affect function of the receptor. Single nucleotide changes at five locations define the two alleles, NA1 and NA2 that produce receptors with similar affinity to IgG1 but FcγRIIIb-NA1 has approximately double the affinity of the NA2 allele for monomeric IgG3.15 Low copy number of FCGR3B, in particular deletion of the NA1 allele is associated with SLE, potentially conferring disease risk by reduced expression due to lower copy number and by reduced binding to the receptor.7
FCGR2B, the only inhibitory Fc receptor, has a polymorphism in the transmembrane domain, Ile232Thr (rs1050501), that alters activity with the 232Thr variant having lower affinity for lipid rafts and thereby a decreased inhibitory effect.16,17 The 232Thr variant is associated with SLE in East Asian and European populations.18 Because FcγRIIb is the only inhibitory FcγR, a reduction in its function could cause increased immune responsiveness and predisposition to autoimmunity. Consistent with this, downregulation of FCGR2B expression has been reported in SLE.19 The known involvement of the various FcγRs on 1q23 in SLE lends support to the hypothesis that FCGR3B CNV may promote autoimmunity by effects other than, or in addition to, those arising directly from the altered expression of the gene encoded within the area of CNV. SLE susceptibility appears to be associated with alleles of lower affinity (FCGR2A-131R, FCGR3A-158F) or loss of higher affinity alleles (FCGR3B-NA1) lending support to the clearance hypothesis.
In this study, to pursue the underlying mechanism of SLE disease association with FCGR3B CNV, we established a detailed map of allelic and paralogous sequence variation (PSV) across the FCGR2/FCGR3 locus. We define the limits of the breakpoint regions associated with structural variation at this locus and further show that deletion of FCGR3B results in creation of a fusion gene with ectopic presence of FcγRIIb on natural killer cells that provides an explanation for SLE susceptibility encoded at this locus.
Material and Methods
Fosmid Clone Sequencing and Assembly
Fosmid clones of ∼40 kb human DNA fragments from eight individuals were obtained from the authors of.20 Based on the mapping of end-sequence pairs (ESPs) to the human reference genome (hg18) provided by the authors, haplotype-assigned fosmid clones21 tiling across the FCGR locus (chr1:161,400,000–161,750,000) were selected.
Fosmid inserts were sequenced in a multiplexed sequencing run on the 454/Roche GS FLX Titanium sequencing platform. Library barcoding was carried out as described previously.22 Sequencing reads were detagged and sorted by fosmid with the untag command line tool (version 0.99.2). Quality and adapter clipped sequences and Phred scaled quality scores were extracted to FASTA formatted files with the sff_extract command line tool (version 2.0). Reads from each fosmid were assembled into contigs with the Roche GS De Novo Assembler Software (version 2.5).
Fosmid End-Sequence Pair Mapping
Fosmid inserts were anchored on the GRCh37 (hg19) assembly of the reference genome by mapping of the ESPs to the reference sequence. To obtain ESP sequences from the NCBI trace archive, we retrieved fosmid-clone-ID-to-NCBI-trace-name mappings from the Human Genome Structural Variation Project website. Trace names were mapped to TI numbers (trace IDs), and the respective trace sequences and quality scores downloaded from the NCBI trace archive by using a Perl script available from the NCBI FTP site. The NCBI project ID required for the query was 29893. Trace sequences were aligned to the GRCh37 (hg19) assembly of human chromosome 1 with the alignment tool BLAT23 (version 3.4). The alignments were subsequently filtered for the highest scoring hits with the pslReps tool distributed with the BLAT software package by using the -singleHit option. Alignments were processed to retain only concordant mappings.
SNP and Indel Calling
To identify SNPs and indels between fosmid inserts and the reference sequence, we mapped the assembled contigs to the reference genome with the BLAST local alignment tool. SNP and indel positions were parsed from the BLAST XML output by using a custom Perl script.
Targeted Sequencing
Genomic DNA was hybridized to custom Roche/NimbleGen capture arrays to enrich a 550 kb region on chromosome 1 (chr1:161,325,000-161,875,000) containing the FCGR2/FCGR3 gene cluster. The array probe design was based on the NCBI 36 (hg18) assembly of the human reference genome. To account for the segmental sequence similarity and to increase probe coverage, we allowed nonunique probes. The captured DNA was sequenced in a multiplexed Roche/454 GS FLX Titanium sequencing run. Ethical approval for the DNA samples was as previously reported.7
CNV-seq Analysis
Sequencing reads were detagged and sorted by sample with the sff_file command line tool of the SFF tools package (version 2.0) provided by Roche/454. From the resulting SFF files sequences and Phred scaled quality scores were extracted to FASTA formatted files with the sff_extract command line tool (version 2.0).
Reads were mapped to the GRCh37 (hg19) assembly of the human genome sequence with BWA.24 Reads with a length ≥200 bp were mapped with BWA-SW for long reads,25 and reads shorter than 200 bp were mapped with BWA for short reads.24
Read depth analysis to identify CNV regions from sequencing data was carried out with the CNV-seq package.26 Read count ratios were calculated across 1.5 kb windows. A p value of 0.05 was chosen as the significance threshold for detected CNV regions defined as four consecutive 1.5 kb windows with a log2 ratio of ≥0.69 (≥1.6-fold difference in read depth).
Flow-Cytometric Analysis
The antibodies used were CD32B-Alexa488 (clone 2B6; Macrogenics), CD19-PE-Cy7 (BD Pharmigen), CD56-APC (Biolegend) Cells. Samples from volunteers were subjected to Ficoll-Hypaque density separation to isolate PBMC. Natural killer (NK) cells were enriched from PBMC by positive selection by using CD56 MACS beads (Miltenyi). B cells were enriched by negative selection by using Dynabeads Untouched Human B Cells.
Nucleic-Acid Isolation
Total RNA was isolated from 107 cells (B or NK cells) by using TRIzol total RNA isolation reagent (Gibco BRL, Grand Island, NY). Four hundred nanograms of total RNA were used to synthesize cDNA with the RevertAid First Strand cDNA Synthesis Kit (Fermentas).
Transcript Analysis
The FCGR2B RT-PCR was performed with 1 μl cDNA, 300 nM each primer (sense primer 5′-GAGAAGGCTGTGACTGCTGT-3′, antisense primer 5′-TACCAGATCTTCCCTCTCTG-3′), 200 μM dNTPs, 1.5 mM MgCl2, and 0.5 units Platinum® Taq DNA polymerase in a 25 μl reaction volume, starting with 94°C for 2 minutes, 35 cycles of denaturing at 94°C for 30 seconds, annealing at 60°C for 30 seconds, extension at 72°C for 1.5 minutes, with a final extension at 72°C for 10 minutes. The product was sequenced by the Sanger method using the primer 5′-CCTCACCTGGAGTTCCAGGAGGGAG-3′ to analyse the FGCR2B SNP rs1050501 (p.Ile232Thr).
Protein Immunoblot
Standard protein immunoblot analysis was performed using FcγRIIb-specific antibody (CD32B[C-20] Santa Cruz Biotechnology).
Results
Assessment of Size of CNV at FCGR2C/FCGR3B
PRT assay27 of ∼2,500 SLE cases, unaffected relatives and controls4,7 identified eight individuals homozygous for loss of FCGR3B, whom we describe as zero-copy individuals. Comparative genomic hybridization (CGH) array analysis was performed on two of these zero-copy individuals. They were arrayed against a known two-copy control. The data indicates that the genomic region around and including FCGR3B is absent in the zero-copy individuals (Figure 1B). However, cross hybridization between the proximal and distal probes prevents this method from identifying the boundaries of the deletion with accuracy.
Pulsed field gel electrophoresis (PFGE) and Southern blotting of PmeI digested gDNA from two of the zero-copy individuals and their heterozygous (one-copy) relatives with FCGR2 and FCGR3 probes confirmed the loss of ∼85 kb in FCGR3B-deleted haplotypes (Figure 1C). These deletions may not be identical in terms of the exact boundaries of the deleted region because the PFGE Southern blot analysis would not detect variations of <10 kb, but the analysis demonstrates that individuals who were shown by PRT to have zero copies (Figure 1C, lanes 5 and 6) or one copy (Figure 1C, lanes 1 and 2) of FCGR3B are missing a similarly sized genomic region. Taken together with the array CGH result, the PFGE data show the approximate size of the area deleted (∼85 kb) in zero- or one-copy individuals but do not identify the precise boundaries of the deletion(s).
Analysis of Sequence Variation at the FCGR Locus
To identify paralogous sequence variants (PSVs) that could be used to map the boundaries of the deleted region in our zero-copy individuals, we aligned the reference sequence (GRCh37/hg19) of the proximal block of the FCGR locus (chr1:161,480,906–161,564,008) to that of the distal block (chr1:161,562,570–161,645,839; Figure 2). Over the alignment length of 85.2 kb, the two blocks showed 93.7% sequence identity. This included a 26.1 kb region of paralogy in FCGR2C and FCGR2B that encompasses the 5′ coding region of 12.8 kb and an upstream region of 13.3 kb, that overall showed 99.2% identity. A total of 1,202 positions showed variation between the proximal and distal 85.2 kb blocks constituting a set of candidate PSVs. However, PCR-based sequence analysis failed to find any PSVs in this region that did not show allelic variation between individuals (data not shown), confounding breakpoint analysis. To identify nonpolymorphic PSVs that could help to identify the location of the breakpoints, we undertook a systematic sequence analysis of the FCGR2/FCGR3 locus in eight unrelated individuals by high-throughput sequencing and de novo assembly of fosmid clones derived from the region.
Figure 2.
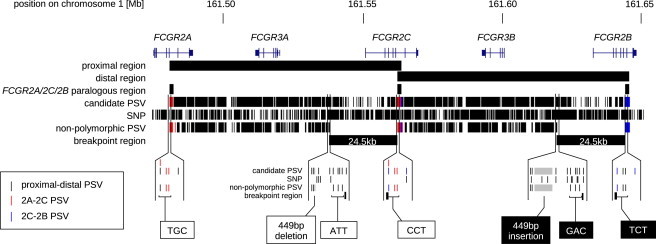
PSV and Breakpoint Analysis at the FCGR Locus
Proximal and distal regions of the reference sequence that were aligned to identify candidate PSVs are indicated below the FCGR gene loci. A region of ∼1.5 kb in the centre of the FCGR2 gene loci is found in all three paralogs (2A-2C-2B paralogous region). The locations of candidate PSVs, SNPs, and PSVs remaining after removal of polymorphic PSVs are shown. Nonpolymorphic PSVs are absent in a 24.5 kb interval starting upstream of FCGR2C/FCGR2B and ending in intron five of FCGR2C/FCGR2B. The blow-ups show the SNPs and PSVs flanking the breakpoint region. The 5′ border is defined by a 449 indel PSV and three single nucleotide PSVs while the 3′ border is defined by a combination of three single nucleotides with the combination CCT identifying the FCGR2C locus and the combination TCT identifying the FCGR2B locus.
A total of 156 fosmids tiling across the FCGR locus between position 161,438,169 and 161,736,847 (see Figure S1 available online) were sequenced on the Roche/454 platform. The average insert size of ∼40 kb, smaller than the size of the duplicated block (∼85 kb), avoided confounding effects caused by sequence of both regions of high similarity being present in a single insert, which could have resulted in chimeric assemblies of proximal and distal sequence.
SNPs were called from alignments of assembled fosmid insert sequences to the reference genome (GRCh37/hg19). The alignments were guided by the anchoring of the fosmids on the genome sequence based on their ESPs facilitating assignment of assembled sequence to proximal or distal block. A total of 1,748 SNPs were detected across the locus. A more accurate pool of potential PSVs was then generated by discarding those PSVs with allelic variation in either the proximal or distal block.
Intersecting the 1,748 SNPs that were detected from the fosmid sequencing with the 1,202 candidate PSVs identified from alignment of the reference sequence (Figure 2; Table S1) revealed that around one third of the PSVs (434) were subject to allelic variation in either the proximal or distal segment and thus unsuitable as paralog-specific markers, leaving 768 potentially informative PSVs. However, nonpolymorphic PSVs were completely absent in a 24.5 kb interval that is contained within the 26.1 kb region of 99.2% identity defined by the reference sequence alignment and includes the upstream region and first five exons/introns of FCGR2C/FCGR2B (Figure 2).
Interestingly, two haplotype blocks at either end of the 24.5 kb PSV-free regions were found to have swapped between the proximal and distal region in some of the sequenced individuals (Figure 3). Comparison of paralogous sequence variation (Figure 3A) and allelic sequence variation (Figure 3B) showed that in some cases the pattern of allelic variation in the proximal block was similar or identical to the PSV pattern of the distal block, and vice versa, suggesting that ancestral sequence exchange has occurred between the two regions. Because the ESPs of the fosmids that contained the swapped regions map to sequence that is specific to either the proximal or the distal block, incorrect anchoring to the reference could be excluded as a cause for the observed haplotype swapping.
Figure 3.
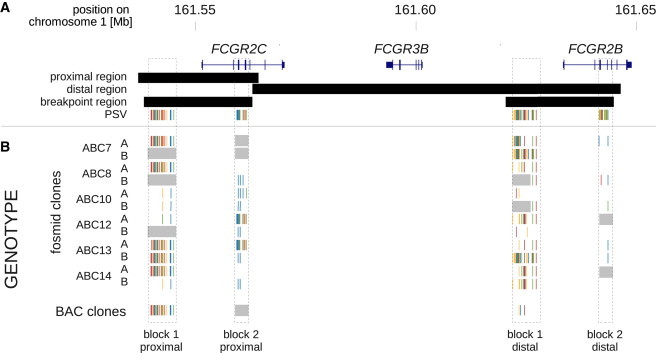
Swapping of Proximal and Distal Haplotype Blocks
Comparison of SNP patterns to candidate PSVs derived from the reference sequence shows that two haplotype blocks located at the outer boundaries of the breakpoint region (dashed boxes) appear to be swapped between proximal and distal region on a number of chromosomes.
(A) Paralogous sequence variation within swapped haplotype blocks. PSVs were derived by global alignment of proximal and distal region of paralogy. Only candidate PSVs within the two swapped blocks are shown with the color coding representing the PSV “allele” at the paralogous site.
(B) Allelic sequence variation within swapped haplotype blocks. SNPs were identified by sequencing of fosmid clones from six individuals (ABC7, ABC8, ABC10, ABC12, ABC13, ABC14) and hydatidiform mole BAC clones from one individual. SNP alleles at the respective positions are shown below for each individual (ABCx) and chromosome (A/B). Grey shading indicates missing haplotype information. High similarity between SNP and PSV patterns suggest that sequence between the two paralogous regions has been swapped.
To exclude the possibility that structural inconsistencies in the reference assembly as described recently for other loci that are subject to structural variation28 could underlie the observed phenomenon and could potentially hamper our analyses, we sought to confirm the structural arrangement of the FCGR2/FCGR3 locus in the reference assembly. Five clones from a haploid hydatidiform mole bacterial artificial chromosome (BAC) library (Table S2) tiling across the FCGR2/FCGR3 locus (see Figure S2) with an average insert size of 200 kb were sequenced by capillary sequencing. Alignment of the assembled BAC sequences to the reference sequence confirmed the overall organization of the locus. The only discrepancy observed was discordance between the expected insert size of two clones (CH17-318H14, CH17-135E1) and the region they aligned to in the reference assembly. However, this was explained by compression of a tandem repeat region upstream of the FCGR2/FCGR3 cluster in the reference. This inconsistency had already become evident from mapping of sequence capture data generated across the locus (see Figure S2), which we discuss further below. Furthermore, the BAC clone sequencing also confirmed the haplotype swapping we had observed in the fosmid data as clone CH17-135E1 also contained a swapped haplotype (see Figure 3) which explained why the 3′ end of the clone had initially been wrongly anchored at the FCGR2B locus by ESP mapping (Figure S3).
Mapping of FCGR3B CNV Breakpoints
Of the 768 potentially informative PSVs that remained after analysis of the fosmid sequence data, we specifically examined those close to the approximate breakpoint position estimated from PFGE and CGH arrays. The selected PSVs (Table 1) were further substantiated by Sanger sequencing in 12 individuals with normal FCGR3B diploid copy number (two copies, one on each chromosome) to confirm that a proximal and distal variant (parallele) was always present (data not shown).
Table 1.
Chromosomal Positions, as Annotated in the GRCh37/hg19 Assembly, of Nonpolymorphic PSVs Used to Distinguish Proximal and Distal Sequence at the FCGR2C/FCGR2B Loci in Proximity of the FCGR3B CNV Breakpoint Region
|
Proximal Region |
Distal Region |
||
|---|---|---|---|
| Position | PSV “allele” | Position | PSV “allele” |
| 161,537,464 | 449 bp deletion | 161,618,848–161,619,296 | 449 bp insertion |
| 161,537,898 | A | 161,619,727 | G |
| 161,537,999 | T | 161,619,829 | A |
| 161,538,110 | T | 161,619,940 | C |
| 24.5 kb interval without PSVs | |||
| {161,562,577;161,562,582;161,562,584} | {C;C;T}a | {161,644,408;161,644,413;161,644,415} | {T;C;T}a |
PSVs flanking the 3′ end of the 24.5 kb interval have to be interrogated in combination because individual PSVs do not identify FCGR2C/FCGR2B sequence unambiguously as a result of FCGR2A/FCGR2C/FCGR2B paralogy (see Figure 2).
The verified PSVs were used to map the region containing the breakpoint in our eight individuals homozygous for the deleted haplotype (FCGR3B zero-copy). For each individual, PSVs were typed by PCR and sequencing to identify the presence of proximal or distal paralleles. Absence of either the proximal or distal parallele was used to map the boundaries of the absent genomic segment containing FCGR3B. A 449 bp indel PSV plus three single nucleotide PSVs upstream of FCGR2C/FCGR2B mark the 5′ end of the breakpoint-containing region and a combination of three single nucleotide PSVs in intron five of FCGR2C/FCGR2B mark the 3′ boundary in all eight of our zero-copy individuals (Figure 2; Table 1). This leaves a ∼24.5 kb region bounded by FCGR2C upstream sequence and FCGR2B intron five in the deleted individuals within which the breakpoint(s) must lie.
To derive further supporting data of the location of the breakpoint, FCGR loci of two trios and two single parent families containing FCGR3B zero-copy individuals (five one-copy and five of our eight zero-copy individuals) were sequenced on the Roche/454 Titanium platform. Genomic DNA from these individuals was hybridized to custom Roche/NimbleGen capture arrays to enrich a 550 kb region (chr1:161,325,000–161,875,000) containing the FCGR2/FCGR3 locus and then sequenced.
Copy-number-variable regions in zero-copy offspring were detected by read depth analysis (CNV-seq) by using one-copy parents as control samples. Significant copy-number variation was detected in the region containing FCGR3B and surrounding sequence in the deleted haplotype individuals. This confirmed deletion of FCGR3B and gave an indication of the extent of the deletion from the middle of FCGR2C to immediately 5′ of FCGR2B (see Figure S4). However, the high sequence similarity between the 5′ regions of FCGR2C and FCGR2B resulted in cross-mapping of sequencing reads derived from these regions. This hampered detection of CNV from read depth in the region upstream of FCGR2B.
Reads from these individuals were also used to verify the 449 bp indel PSV upstream of exon one and the four PSVs located in intron five of FCGR2C and FCGR2B. The sequencing reads from this region on 15 deleted haplotypes (the five zero-copy individuals and their five one-copy relatives) were searched for evidence of an unbalanced breakpoint, as evidenced by chimeric sequence in which verified proximal and distal PSVs were juxtaposed in a single read. However, our analysis failed to find a single such chimeric sequence. Furthermore, the reads captured from zero-copy individuals could be assembled de novo into contigs (data not shown), which, when aligned to the reference, like the mapping analysis, did not show any evidence of an unbalanced breakpoint. Figure 4A shows the organization of the FCGR locus in an FCGR3B zero-copy individual derived from the de novo assembly of captured reads.
Figure 4.
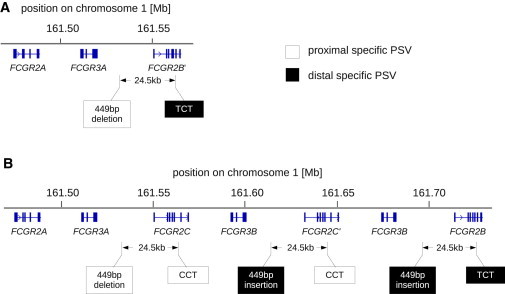
Organization of the FCGR2/FCGR3 Locus in FCGR3B Copy-Number-Variable Individuals
(A) Organization of the FCGR2/FCGR3 locus in an FCGR3B zero-copy individual obtained by de novo assembly of sequence capture data. The PSV alleles (see Figure 2) confirm that the FCGR2B′ locus is a chimera of 5′ proximal sequence and 3′ distal sequence.
(B) Organization of the FCGR2/FCGR3 locus in an individual with an FCGR3B duplication obtained by de novo assembly of fosmid clones. The organization of the locus in both individuals with an FCGR3B duplication and individuals with an FCGR3B deletion suggests that the FCGR3B CNV breakpoint is balanced.
By using PRT, we genotyped the eight HapMap individuals, whose genome had been cloned in the fosmid libraries, for copy-number polymorphism at FCGR3B. Two of these individuals (libraries ABC9 and ABC11) carry a duplicated FCGR3B haplotype. We were able to identify fosmids from these libraries where the 5′ end of the fosmid discordantly mapped to the FCGR3B and the 3′ end discordantly to the FCGR2C locus of the reference assembly (Figure S5A). Thus these fosmid inserts must contain the duplication breakpoint, which is likely to be similar to the deletion breakpoint. No hybrid reads from these fosmids were found, confirming that the duplication breakpoint also lies in a region of high similarity between proximal and distal region. Figure 4B shows the organization of the FCGR locus on a haplotype carrying an FCGR3B duplication derived from de novo assembly of fosmid inserts tiling across the locus (Figure S5C).
The absence of nonpolymorphic PSVs in the 24.5 kb region of paralogy together with the absence of chimeric reads in sequence data derived from zero-copy individuals indicate that all breakpoints in our data set lie within the 24.5 kb regions of 100% paralogy between proximal and distal region with proximal sequence running seamlessly into identical distal sequence. The breakpoint can be located anywhere between the PSVs flanking the PSV-free 24.5 kb block.
Given our evidence that the breakpoint in deleted and duplicated haplotypes is symmetrical, we believe that FCGR3B deletion is a balanced recombination event that creates a chimeric FCGR2C/FCGR2B gene that we designate FCGR2B′ (Figure 4A). FCGR2B′ consists of upstream elements and a 5′ coding region that derive from FCGR2C and a 3′ coding region that derives from FCGR2B. The coding sequence of FCGR2B′ is therefore identical to FCGR2B, but FCGR2B′ would be expected to be under the control of 5′ flanking sequence derived from FCGR2C.
Functional Analysis of FCGR2B′ Expression
FCGR2C transcripts have been reported in both NK cells (CD56+ lymphocytes) and in B lymphocytes (CD19+ lymphocytes), whereas FCGR2B transcripts are only present in B lymphocytes.29,30 This tissue specificity led us to postulate that because of the approximation of the FCGR2C regulatory sequence with FCGR2B coding sequence in FCGR2B′, FCGR2B′ expression would be detectable in NK cells of individuals carrying an FCGR3B zero-copy haplotype but would be undetectable in individuals not carrying an FCGR3B zero-copy haplotype.
To test this hypothesis, we analyzed blood samples from individuals with zero, one, and two copies (one on each chromosome of the diploid genome) of FCGR3B by flow cytometry employing anti-CD56 and anti-CD19 and the specific anti-FcγRIIb antibody 2B6.31 Each sample contained a CD19+ 2B6+ population (Figure 5A, top left, center, and right plots), corresponding to B cells on which FcγRIIb is present. Individuals with zero or one copy of FCGR3B present two 2B6+ cell populations (2B6low and 2B6bright, Figure 5A, top center and right plots), whereas those with two copies of FCGR3B, not carrying a deleted haplotype, show only one 2B6+ population, which corresponds to 2B6bright (Figure 5A, top-left plot). The detection of this 2B6low population suggested that there might be ectopic FcγRIIb on cells that did not express CD19. By using CD56 expression as a marker for NK cells, we showed that those without an FCGR3B-deleted haplotype (two-copy individuals) exhibited a negligible (0.66%) proportion of CD56+ 2B6+ cells as expected (Figure 5A, bottom-left plot). In marked contrast, a significant proportion (>70%) of CD56+ cells from individuals with zero or one copy of FCGR3B, bearing a deleted haplotype (Figure 5A, bottom-center and -right plots) showed expression of FCGR2B in their NK cells. Irrespective of FCGR3B CNV, all individuals show the two CD56+ populations (CD56low and CD56bright, Figure 5A, bottom-left, -centre and -right plots) described previously.32,33 In FCGR3B zero- and one-copy individuals, the CD56low population is predominantly 2B6+, which demonstrates that the individuals who carry a deleted FCGR3B haplotype translate FcγRIIb in this subset of NK cells (Figure 5A, bottom-center and -right plots). These CD56low cells produce low levels of NK-derived cytokines, but they are potent mediators of antibody-dependent cell mediated cytotoxicity (ADCC).32 In the samples analyzed from individuals with zero-, one-, or two copies of FCGR3B, in >95% of CD19+ cells FcγRIIb is detected. Similar proportions, 67% and 74%, respectively, of CD56+ (NK) cells from individuals who carry an FCGR3B-deleted haplotype (FCGR3B one- and zero-copy) translate FcγRIIb (Figure 5B). A comparison between the mean fluorescence of 2B6 in B and NK cells shows that accumulation of FcγRIIb is approximately 8-fold higher in B cells than in NK cells in zero- and one-copy FCGR3B individuals (Table S3). There is no statistically significant difference (independent samples Kruskal-Wallis test significance: 0.117) in the magnitude of FcγRIIb accumulation on B cells between unrelated zero-copy, one-copy, and two-copy donors, although we acknowledge that the cohort size is small.
Figure 5.
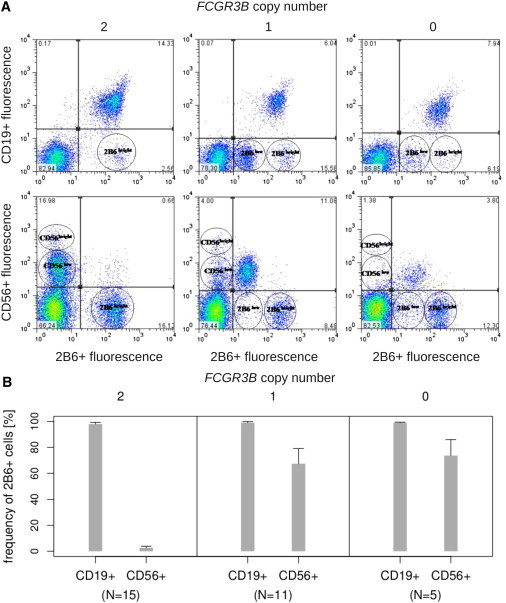
Flow Cytometry Analysis of FcγRIIb Accumulation in B and NK Lymphocytes in Relation to FCGR3B Copy Number
(A) FcγRIIb accumulation in B and NK cells of individuals with different FCGR3B copy number. The top plots show accumulation of FcγRIIb (2B6+) in B cells (CD19+) of FCGR3B two-copy (not carrying an FCGR3B-deleted haplotype), one-copy (carrying one deleted haplotype) and zero-copy (carrying two deleted haplotypes) individuals, respectively. The two 2B6+ populations (2B6low, 2B6bright) are indicated. The bottom plots show accumulation of FcγRIIb (2B6+) in NK cells (CD56+) of FCGR3B two-, one-, and zero-copy individuals. The two populations of CD56+ (CD56low, CD56bright) and 2B6+ (2B6low, 2B6bright) are indicated. The seemingly more pronounced CD56+ 2B6+ signal in one-copy individuals compared to zero-copy individuals is a result of the difference in the total cell count in the two samples. The quantitative flow cytometry data (Table S3) summarized in (B) shows that the percentages of NK cells on which FcγRIIb is detected (2B6+) are similar in one- and zero-copy individuals.
(B) Relative frequency of B and NK cells on which FcγRIIb is detected (2B6+) in individuals with different FCGR3B copy number. Shown is the mean percentage of CD19+ (B) and CD56+ (NK) cells on which FcγRIIb is detected (2B6+) in FCGR3B two-, one-, and zero-copy individuals, respectively. The number of individuals n analyzed is given below each plot. Whiskers indicate SD.
Immunoblotting of B and NK cell preparations with a second antibody specific to an intracellular FcγRIIb C-terminal epitope (C-20) confirms the ectopic presence on NK cells from individuals with a deleted FCGR3B haplotype and corroborates the flow cytometry data obtained with the 2B6 antibody (Figure 6).
Figure 6.
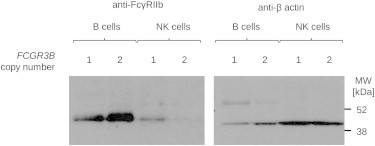
Protein Immunoblot Analysis of FcγRIIb Accumulation in B and NK Lymphocytes in Relation to the FCGR3B Copy Number
FcγRIIb was detected with a goat polyclonal antibody (CD32B [C-20]) (left panel). Anti-β actin was used for quantification (right panel).
Haplotypic Origin of Ectopic FcγRIIb
We designed a reverse transcription (RT)-PCR assay to characterize the FcγRIIb-encoding transcripts present in NK cells in an individual carrying a deleted FCGR3B haplotype and also heterozygous for the FGCR2B-I232T SNP rs1050501 (Figure 7A), enabling measurement of the allelic expression of FCGR2B from the two parental chromosomes. We isolated RNA from highly purified (>95%) B and NK cell preparations from the same individual and sequenced the cDNA. The presence of two peaks at the SNP site in the transcript sequence from purified B cells (Figure 7B) is evidence that the transcript is generated from both alleles. The presence of only one peak in the sequence from NK cells (Figure 7C) indicates that the transcript is produced only from one allele and that the ectopically expressed FCGR2B RNA (and therefore protein) originates from the FCGR2B′ gene on the zero-copy haplotype.
Figure 7.
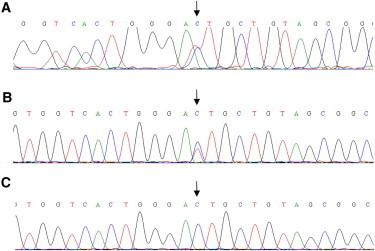
Sequence Analysis of FCGR2B Transcripts Present in NK Cells of a an FCGR3B One-Copy Individual
FCGR2B sequence around SNP rs1050501 (arrow) amplified from (A) genomic DNA, (B) mRNA isolated from purified B cells, (C) mRNA isolated from purified NK cells.
Discussion
Intensive genetic analysis of SLE over several decades, including a number of recent genome-wide association studies, has revealed more than 30 robust susceptibility loci for the disease.34 Studies in rodent models of SLE have also yielded insights, with clear evidence that Fc gamma receptors have an influence on SLE susceptibility phenotypes.35 Amongst these, the finding of Fcgr3 copy-number variation in rats and humans led to replicated reports of association between FCGR3B copy-number variation and SLE in humans.4,7,14,18,36
The studies presented here aim to understand in more depth the relationship between FCGR3B copy-number variation and susceptibility to SLE. We identify and designate a chimeric gene, FCGR2B′, that occurs as a consequence of FCGR3B deletion on FCGR3B zero-copy haplotypes, that (1) is defined by the juxtaposition of the regulatory region of FCGR2C and the coding region of FCGR2B, (2) is expressed ectopically in NK cells, and (3) provides a previously unexpected explanation for the mechanism underlying association of low FCGR3B copy number and susceptibility to SLE.
We carried out extensive sequence analysis of the FCGR2/FCGR3 locus to establish the extent of interindividual variation in FCGR2/FCGR3 haplotype structure and the location of breakpoints leading to FCGR3B copy-number variation. The high segmental sequence similarity found across the FCGR locus makes the study of the genomic pathology of FCGR3B CNV particularly challenging. By using a combination of in silico analyses and high-throughput sequencing of fosmid clones, we identified a set of informative PSVs that allowed us to narrow the potential breakpoint region to a 24.5 kb region of paralogy between the two ancestral duplicated blocks. However, the complete absence of nonpolymorphic PSVs in the 24.5 kb region prevented more precise localization of breakpoints in FCGR3B-deleted or FCGR3B-duplicated haplotypes. The absence of nonpolymorphic PSVs across the duplicated 24.5 kb region makes it likely that the breakpoint region cannot be narrowed down any further with current sequencing technologies.
A recent study reported mapping of FCGR3B CNV breakpoints based on PSVs identified from alignment of the reference sequence of the proximal and distal blocks, assuming that the vast majority of differences between the two blocks are true PSVs.13 Our fosmid sequencing, however, shows that 36% of candidate PSVs identified by sequence alignment are subject to allelic variation, invalidating their use as unambiguous PSVs, which is likely to have compromised the breakpoint analysis presented in that report. Our fosmid analysis also showed evidence of swapping of sequence haplotypes between the proximal and distal ancestral blocks in the duplicated 24.5 kb region of near identity. The haplotype swapping that we observe strongly supports the hypothesis that FCGR3B CNV breakpoints most likely reside within the two 24.5 kb regions of near identity in the proximal and distal blocks.
An illustrative schema of the proposed mechanism through which swapping of haplotype sequences between distal and proximal blocks could occur is shown in Figure 8. Continuous ancestral shuffling of proximal and distal sequence by nonallelic homologous recombination that resulted in a homogenization of paralogous sequence variation across the region would explain why every PSV position in the two 24.5 kb regions is also a SNP position. Recurrent recombination has been proposed previously as a mechanism leading to the homogenization of long stretches of homologous sequence and as a confounding factor in breakpoint analyses.37 The absence of PSVs as well as the observed phenomenon of haplotype swapping between proximal and distal regions gives reason to speculate that a defined breakpoint does not exist but that recombination can take place across the duplicated 24.5 kb region. This, together with the observation of different swapping patterns in the analyzed individuals suggests that FCGR3-deleted haplotypes have not originated from a single ancestral recombination event (identity by descent) but have arisen on multiple occasions through recurrent recombinations (identity by state).
Figure 8.
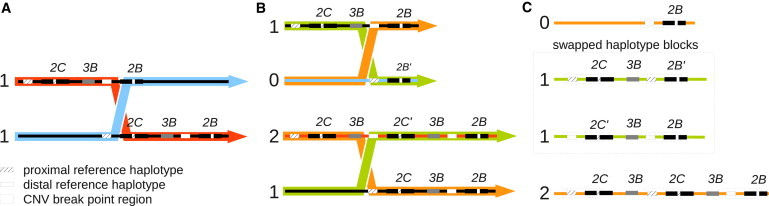
Proposed Mechanism of Proximal and Distal Haplotype Swapping within the FCGR2C/FCGR3B/FCGR2B CNV Breakpoint Region
Scenario of sequential recombination events between FCGR3B normal copy, duplicated and deleted haplotypes occurring at different positions across the CNV breakpoint region that could explain swapping of sequence between proximal and distal region of the locus. Numbers to the left of the haplotypes indicate FCGR3B copy number. (A) Nonhomologous recombination between FCGR2C and FCGR2B, occurring between the two haplotype blocks bordering the 24.5 kb PSV-free regions, results in (B) a deleted and a duplicated FCGR3B locus and a juxtaposition of proximal and distal haplotype blocks in the breakpoint region. The resulting FCGR2B′ and FCGR2C′ loci are in fact FCGR2C/FCGR2B and FCGR2B/FCGR2C chimera, respectively. However, as a result of the high sequence similarity across the breakpoint region, these chimeric genes are indistinguishable from the bona fide FGCR2B and FCGR2C genes.
(C) Subsequent recombination with an FCGR3B normal copy locus occurring at the 5′ end of the breakpoint region results in loci with proximal sequence swapped to the distal region and vice versa.
By using the obtained PSV information, we showed that in FCGR3B zero-copy haplotypes, the regulatory sequence located upstream of FCGR2C and the coding sequence of FCGR2B are juxtaposed as a consequence of the deletion of FCGR3B. Thus the deletion of FCGR3B may be considered as a mechanism that generates a hybrid FCGR2C/FCGR2B gene that we designate FCGR2B′ because its coding sequence is indistinguishable from that of FCGR2B. On account of the different tissue distribution for the expression of FCGR2B and FCGR2C, we sought evidence of aberrant expression of FCGR2B transcripts that arose from FCGR2B′ on zero-copy haplotypes. We showed that in individuals with FCGR3B-deleted haplotypes, FcγRIIb is present on NK cells. Furthermore, we showed that in an individual with a single FCGR3B-deleted haplotype, the NK cell FCGR2B expression was derived from a single chromosome.
The presence of FcγRIIb on B cells does not appear to be affected by the deletion indicating that FCGR2C upstream regulatory regions are sufficient for expression in both B and NK cells. Despite the high identity between the near upstream regions of FCGR2C and FCGR2B, it is likely that there is a PSV (or a set of PSVs) between these two loci that confers expression in NK cells. Its identification will be of considerable value in future research that aims to target expression to this cell type. In this sense, the PSV analysis shows candidate regions that could alter the expression within lymphocyte subsets.
The genomic data directly implicate both FCGR3B and FCGR2C in the genomic perturbation caused by copy-number polymorphism at this locus and this may throw new light on the mechanisms by which deletion at this locus increases the risk of SLE and RA. It is unlikely that the loss of FCGR2C exerts a major impact on SLE risk because the FCGR2C SNP rs10917661 (p.Gln57∗) in exon three produces a stop codon in the majority of alleles, truncating the protein product in the healthy population, and no association between this SNP and SLE has been observed.38 We7 and others14 have shown that the reduced FCGR3B copy number correlates with reduced protein accumulation and FcγRIIIb function on neutrophils, and reduced expression in neutrophils has been shown to impair IgG-mediated opsonization.39 Studies using transgenic mice suggest that neutrophil FcγRIIIb may mediate a less inflammatory effect on immune complex binding compared with FcγRIIa.40 However, the importance of the role of the neutrophil in immune complex clearance is not clear. This prompted us to consider alternative, possibly additional mechanisms, by which copy-number polymorphism at the FCGR locus might impact on immune function.
The role of NK cells in autoimmunity is not fully understood. However, there is an increasing body of literature, dating back more than 30 years, demonstrating clearly that NK cell function is impaired in SLE41–43 and RA:44,45 this includes reduced NK cell numbers as well as NK cell mediated cytotoxicity. The predominant Fc receptor that mediates cytotoxicity on NK cells is FcγRIIIa. This receptor exhibits common polymorphism, and the lower functioning 158F allele of FCGR3A shows an independent genetic association with increased SLE risk.46 Ectopic presence of FcγRIIb on NK cells may inhibit the function of other Fc receptors, particularly FcγRIIIa, because the two receptors are likely to colocalize on the cell surface in lipid rafts.17,47 Our data indicate that the ectopic expression is most marked in CD56dim NK cells, a subset that is implicated in antibody-dependent cytotoxicity. Thus, we speculate that the ectopic presence of FcγRIIb contributes to impaired NK cell cytotoxic function. This hypothesis is given further support by the observation made by two groups33,48 that a subset of NK cells could express FCGR2B. The NK cells expressing FCGR2B were shown to exhibit weaker antibody-dependent degranulation, which could be reversed on blocking FcγRIIb. These data indicate that FcγRIIb on NK cells exerts an inhibitory effect on NK cell function. Recently, NK cells (specifically CD56dim NK cells) have been shown to promote interferon-alpha production by plasmacytoid dendritic cells: a capacity that is impaired in NK cells from SLE patients.49 This is an alternative SLE-associated NK cell function alteration to which ectopic FcγRIIb might contribute.
Our data point to an additive mechanism for predisposition to SLE in people with zero or one copy of FCGR3B: reduced CNV at the FCGR locus, in addition to removing FCGR3B, also generates a hybrid FCGR2C/FCGR2B gene (FCGR2B′) leading to functional, ectopic presence of the only inhibitory FcγR. Both of these effects may contribute additively to SLE risk, with reduction of FcγRIIIb levels on neutrophils decreasing Ig binding and clearance of immune complexes and appearance of FcγRIIb on NK cells, which in turn inhibits the activity of these cells. Impaired NK cell function is recognized as a characteristic immunological feature of SLE. Our data suggest that CNV at a given locus may exert multiple functional effects such as gene dosage effects and aberrant regulation of transcription, important factors when exploring the relationship between CNV and associated phenotypes.
Acknowledgements
We acknowledge support from the Wellcome Trust to H.T.C., T.J.V. and T.J.A., from the MRC to T.J.A., and from the Imperial NIHR-funded Biomedical Research Centre to T.J.A. and T.J.V.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
UCSC Genome Browser, http://genome.ucsc.edu
NCBI Trace Archive, http://www.ncbi.nlm.nih.gov/Traces/home
Human Genome Structural Variation Project, http://hgsv.washington.edu
Fosmid-clone-ID-to-NCBI-trace-name mapping, http://hgsv.washington.edu/general/download/clone_mapping/
The URLs scripts and software tools that were obtained are as follows:
untag command line tool, http://bioinf.eva.mpg.de/pts
sff_extract command line tool, http://bioinf.comav.upv.es/sff_extract
query_tracedb Perl script, ftp://ftp.ncbi.nih.gov/pub/TraceDB/misc/query_tracedb
BLAT software package, http://hgwdev.cse.ucsc.edu/∼kent/src/blatSrc34.zip
CNV-seq R package, http://tiger.dbs.nus.edu.sg/cnv-seq/cnv-seq.tar.gz
Accession Numbers
The fosmid clone sequence data reported in this paper have been deposited at the European Nucleotide Archive, which is hosted at the European Bioinformatics Institute, under accession number ERP001881. The sequence capture data has been deposited at the European Genome-Phenome Archive, which is hosted at the European Bioinformatics Institute, under accession number EGAS00001000376. The Genbank accession numbers for the hydatidiform mole BAC clone sequences reported in this paper are AC243898, AC243440, AC243509, AC243424, AC243499.
References
- 1.Deng Y., Tsao B.P. Genetic susceptibility to systemic lupus erythematosus in the genomic era. Nat Rev Rheumatol. 2010;6:683–692. doi: 10.1038/nrrheum.2010.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muñoz L.E., Lauber K., Schiller M., Manfredi A.A., Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol. 2010;6:280–289. doi: 10.1038/nrrheum.2010.46. [DOI] [PubMed] [Google Scholar]
- 3.Johnson A.E., Gordon C., Palmer R.G., Bacon P.A. The prevalence and incidence of systemic lupus erythematosus in Birmingham, England. Relationship to ethnicity and country of birth. Arthritis Rheum. 1995;38:551–558. doi: 10.1002/art.1780380415. [DOI] [PubMed] [Google Scholar]
- 4.Aitman T.J., Dong R., Vyse T.J., Norsworthy P.J., Johnson M.D., Smith J., Mangion J., Roberton-Lowe C., Marshall A.J., Petretto E. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439:851–855. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- 5.Fanciulli M., Vyse T.J., Aitman T.J. Copy number variation of Fc gamma receptor genes and disease predisposition. Cytogenet. Genome Res. 2008;123:161–168. doi: 10.1159/000184704. [DOI] [PubMed] [Google Scholar]
- 6.Willcocks L.C., Smith K.G.C., Clatworthy M.R. Low-affinity Fcgamma receptors, autoimmunity and infection. Expert Rev. Mol. Med. 2009;11:e24. doi: 10.1017/S1462399409001161. [DOI] [PubMed] [Google Scholar]
- 7.Morris D.L., Roberts A.L., Witherden A.S., Tarzi R., Barros P., Whittaker J.C., Cook T.H., Aitman T.J., Vyse T.J. Evidence for both copy number and allelic (NA1/NA2) risk at the FCGR3B locus in systemic lupus erythematosus. Eur. J. Hum. Genet. 2010;18:1027–1031. doi: 10.1038/ejhg.2010.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKinney C., Fanciulli M., Merriman M.E., Phipps-Green A., Alizadeh B.Z., Koeleman B.P.C., Dalbeth N., Gow P.J., Harrison A.A., Highton J. Association of variation in Fcgamma receptor 3B gene copy number with rheumatoid arthritis in Caucasian samples. Ann. Rheum. Dis. 2010;69:1711–1716. doi: 10.1136/ard.2009.123588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinson J.I., Carr I.M., Cooper D.L., Rashid L.H., Martin S.G., Emery P., Isaacs J.D., Barton A., Wilson A.G., Barrett J.H., Morgan A.W., BRAGGSS Confirmation of association of FCGR3B but not FCGR3A copy number with susceptibility to autoantibody positive rheumatoid arthritis. Hum. Mutat. 2012;33:741–749. doi: 10.1002/humu.22031. [DOI] [PubMed] [Google Scholar]
- 10.Ravetch J.V., Lanier L.L. Immune inhibitory receptors. Science. 2000;290:84–89. doi: 10.1126/science.290.5489.84. [DOI] [PubMed] [Google Scholar]
- 11.Warmerdam P.A., Nabben N.M., van de Graaf S.A., van de Winkel J.G., Capel P.J. The human low affinity immunoglobulin G Fc receptor IIC gene is a result of an unequal crossover event. J. Biol. Chem. 1993;268:7346–7349. [PubMed] [Google Scholar]
- 12.Breunis W.B., van Mirre E., Geissler J., Laddach N., Wolbink G., van der Schoot E., de Haas M., de Boer M., Roos D., Kuijpers T.W. Copy number variation at the FCGR locus includes FCGR3A, FCGR2C and FCGR3B but not FCGR2A and FCGR2B. Hum. Mutat. 2009;30:E640–E650. doi: 10.1002/humu.20997. [DOI] [PubMed] [Google Scholar]
- 13.Machado L.R., Hardwick R.J., Bowdrey J., Bogle H., Knowles T.J., Sironi M., Hollox E.J. Evolutionary history of copy-number-variable locus for the low-affinity Fcγ receptor: mutation rate, autoimmune disease, and the legacy of helminth infection. Am. J. Hum. Genet. 2012;90:973–985. doi: 10.1016/j.ajhg.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Willcocks L.C., Lyons P.A., Clatworthy M.R., Robinson J.I., Yang W., Newland S.A., Plagnol V., McGovern N.N., Condliffe A.M., Chilvers E.R. Copy number of FCGR3B, which is associated with systemic lupus erythematosus, correlates with protein expression and immune complex uptake. J. Exp. Med. 2008;205:1573–1582. doi: 10.1084/jem.20072413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nagarajan S., Chesla S., Cobern L., Anderson P., Zhu C., Selvaraj P. Ligand binding and phagocytosis by CD16 (Fc gamma receptor III) isoforms. Phagocytic signaling by associated zeta and gamma subunits in Chinese hamster ovary cells. J. Biol. Chem. 1995;270:25762–25770. doi: 10.1074/jbc.270.43.25762. [DOI] [PubMed] [Google Scholar]
- 16.Kono H., Kyogoku C., Suzuki T., Tsuchiya N., Honda H., Yamamoto K., Tokunaga K., Honda Z.-I. FcgammaRIIB Ile232Thr transmembrane polymorphism associated with human systemic lupus erythematosus decreases affinity to lipid rafts and attenuates inhibitory effects on B cell receptor signaling. Hum. Mol. Genet. 2005;14:2881–2892. doi: 10.1093/hmg/ddi320. [DOI] [PubMed] [Google Scholar]
- 17.Floto R.A., Clatworthy M.R., Heilbronn K.R., Rosner D.R., MacAry P.A., Rankin A., Lehner P.J., Ouwehand W.H., Allen J.M., Watkins N.A., Smith K.G. Loss of function of a lupus-associated FcgammaRIIb polymorphism through exclusion from lipid rafts. Nat. Med. 2005;11:1056–1058. doi: 10.1038/nm1288. [DOI] [PubMed] [Google Scholar]
- 18.Niederer H.A., Willcocks L.C., Rayner T.F., Yang W., Lau Y.L., Williams T.N., Scott J.A.G., Urban B.C., Peshu N., Dunstan S.J. Copy number, linkage disequilibrium and disease association in the FCGR locus. Hum. Mol. Genet. 2010;19:3282–3294. doi: 10.1093/hmg/ddq216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Su K., Yang H., Li X., Li X., Gibson A.W., Cafardi J.M., Zhou T., Edberg J.C., Kimberly R.P. Expression profile of FcgammaRIIb on leukocytes and its dysregulation in systemic lupus erythematosus. J. Immunol. 2007;178:3272–3280. doi: 10.4049/jimmunol.178.5.3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kidd J.M., Cooper G.M., Donahue W.F., Hayden H.S., Sampas N., Graves T., Hansen N., Teague B., Alkan C., Antonacci F. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kidd J.M., Cheng Z., Graves T., Fulton B., Wilson R.K., Eichler E.E. Haplotype sorting using human fosmid clone end-sequence pairs. Genome Res. 2008;18:2016–2023. doi: 10.1101/gr.081786.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meyer M., Stenzel U., Hofreiter M. Parallel tagged sequencing on the 454 platform. Nat. Protoc. 2008;3:267–278. doi: 10.1038/nprot.2007.520. [DOI] [PubMed] [Google Scholar]
- 23.Kent W.J. BLAT—the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H., Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie C., Tammi M.T. CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinformatics. 2009;10:80. doi: 10.1186/1471-2105-10-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hollox E.J., Detering J.-C., Dehnugara T. An integrated approach for measuring copy number variation at the FCGR3 (CD16) locus. Hum. Mutat. 2009;30:477–484. doi: 10.1002/humu.20911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Antonacci F., Kidd J.M., Marques-Bonet T., Teague B., Ventura M., Girirajan S., Alkan C., Campbell C.D., Vives L., Malig M. A large and complex structural polymorphism at 16p12.1 underlies microdeletion disease risk. Nat. Genet. 2010;42:745–750. doi: 10.1038/ng.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cassel D.L., Keller M.A., Surrey S., Schwartz E., Schreiber A.D., Rappaport E.F., McKenzie S.E. Differential expression of Fc gamma RIIA, Fc gamma RIIB and Fc gamma RIIC in hematopoietic cells: analysis of transcripts. Mol. Immunol. 1993;30:451–460. doi: 10.1016/0161-5890(93)90113-p. [DOI] [PubMed] [Google Scholar]
- 30.Metes D., Ernst L.K., Chambers W.H., Sulica A., Herberman R.B., Morel P.A. Expression of functional CD32 molecules on human NK cells is determined by an allelic polymorphism of the FcgammaRIIC gene. Blood. 1998;91:2369–2380. [PubMed] [Google Scholar]
- 31.Veri M.-C., Gorlatov S., Li H., Burke S., Johnson S., Stavenhagen J., Stein K.E., Bonvini E., Koenig S. Monoclonal antibodies capable of discriminating the human inhibitory Fcgamma-receptor IIB (CD32B) from the activating Fcgamma-receptor IIA (CD32A): biochemical, biological and functional characterization. Immunology. 2007;121:392–404. doi: 10.1111/j.1365-2567.2007.02588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cooper M.A., Fehniger T.A., Turner S.C., Chen K.S., Ghaheri B.A., Ghayur T., Carson W.E., Caligiuri M.A. Human natural killer cells: a unique innate immunoregulatory role for the CD56(bright) subset. Blood. 2001;97:3146–3151. doi: 10.1182/blood.v97.10.3146. [DOI] [PubMed] [Google Scholar]
- 33.Dutertre C.-A., Bonnin-Gélizé E., Pulford K., Bourel D., Fridman W.-H., Teillaud J.-L. A novel subset of NK cells expressing high levels of inhibitory FcgammaRIIB modulating antibody-dependent function. J. Leukoc. Biol. 2008;84:1511–1520. doi: 10.1189/jlb.0608343. [DOI] [PubMed] [Google Scholar]
- 34.Hindorff, L., MacArthur, J., Wise, A., Junkins, H., Hall, P., Klemm, K., and Manolio, T. (2012). A Catalog of Published Genome-Wide Association Studies, http://www.genome.gov/gwastudies.
- 35.Lin Q., Xiu Y., Jiang Y., Tsurui H., Nakamura K., Kodera S., Ohtsuji M., Ohtsuji N., Shiroiwa W., Tsukamoto K. Genetic dissection of the effects of stimulatory and inhibitory IgG Fc receptors on murine lupus. J. Immunol. 2006;177:1646–1654. doi: 10.4049/jimmunol.177.3.1646. [DOI] [PubMed] [Google Scholar]
- 36.Fanciulli M., Norsworthy P.J., Petretto E., Dong R., Harper L., Kamesh L., Heward J.M., Gough S.C.L., de Smith A., Blakemore A.I.F. FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nat. Genet. 2007;39:721–723. doi: 10.1038/ng2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blanco P., Shlumukova M., Sargent C.A., Jobling M.A., Affara N., Hurles M.E. Divergent outcomes of intrachromosomal recombination on the human Y chromosome: male infertility and recurrent polymorphism. J. Med. Genet. 2000;37:752–758. doi: 10.1136/jmg.37.10.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Su K., Wu J., Edberg J.C., McKenzie S.E., Kimberly R.P. Genomic organization of classical human low-affinity Fcgamma receptor genes. Genes Immun. 2002;3(Suppl 1):S51–S56. doi: 10.1038/sj.gene.6363879. [DOI] [PubMed] [Google Scholar]
- 39.Fossati G., Moots R.J., Bucknall R.C., Edwards S.W. Differential role of neutrophil Fcgamma receptor IIIB (CD16) in phagocytosis, bacterial killing, and responses to immune complexes. Arthritis Rheum. 2002;46:1351–1361. doi: 10.1002/art.10230. [DOI] [PubMed] [Google Scholar]
- 40.Tsuboi N., Asano K., Lauterbach M., Mayadas T.N. Human neutrophil Fcgamma receptors initiate and play specialized nonredundant roles in antibody-mediated inflammatory diseases. Immunity. 2008;28:833–846. doi: 10.1016/j.immuni.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsokos G.C., Rook A.H., Djeu J.Y., Balow J.E. Natural killer cells and interferon responses in patients with systemic lupus erythematosus. Clin. Exp. Immunol. 1982;50:239–245. [PMC free article] [PubMed] [Google Scholar]
- 42.Green M.R.J., Kennell A.S.M., Larche M.J., Seifert M.H., Isenberg D.A., Salaman M.R. Natural killer cell activity in families of patients with systemic lupus erythematosus: demonstration of a killing defect in patients. Clin. Exp. Immunol. 2005;141:165–173. doi: 10.1111/j.1365-2249.2005.02822.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park Y.-W., Kee S.-J., Cho Y.-N., Lee E.-H., Lee H.-Y., Kim E.-M., Shin M.-H., Park J.-J., Kim T.-J., Lee S.-S. Impaired differentiation and cytotoxicity of natural killer cells in systemic lupus erythematosus. Arthritis Rheum. 2009;60:1753–1763. doi: 10.1002/art.24556. [DOI] [PubMed] [Google Scholar]
- 44.Hendrich C., Kuipers J.G., Kolanus W., Hammer M., Schmidt R.E. Activation of CD16+ effector cells by rheumatoid factor complex. Role of natural killer cells in rheumatoid arthritis. Arthritis Rheum. 1991;34:423–431. doi: 10.1002/art.1780340407. [DOI] [PubMed] [Google Scholar]
- 45.Conigliaro P., Scrivo R., Valesini G., Perricone R. Emerging role for NK cells in the pathogenesis of inflammatory arthropathies. Autoimmun. Rev. 2011;10:577–581. doi: 10.1016/j.autrev.2011.04.017. [DOI] [PubMed] [Google Scholar]
- 46.Wu J., Edberg J.C., Redecha P.B., Bansal V., Guyre P.M., Coleman K., Salmon J.E., Kimberly R.P. A novel polymorphism of FcgammaRIIIa (CD16) alters receptor function and predisposes to autoimmune disease. J. Clin. Invest. 1997;100:1059–1070. doi: 10.1172/JCI119616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kondadasula S.V., Roda J.M., Parihar R., Yu J., Lehman A., Caligiuri M.A., Tridandapani S., Burry R.W., Carson W.E., 3rd Colocalization of the IL-12 receptor and FcgammaRIIIa to natural killer cell lipid rafts leads to activation of ERK and enhanced production of interferon-gamma. Blood. 2008;111:4173–4183. doi: 10.1182/blood-2007-01-068908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van der Heijden J., Breunis W.B., Geissler J., de Boer M., van den Berg T.K., Kuijpers T.W. Phenotypic variation in IgG receptors by nonclassical FCGR2C alleles. J. Immunol. 2012;188:1318–1324. doi: 10.4049/jimmunol.1003945. [DOI] [PubMed] [Google Scholar]
- 49.Hagberg N., Berggren O., Leonard D., Weber G., Bryceson Y.T., Alm G.V., Eloranta M.-L., Rönnblom L. IFN-α production by plasmacytoid dendritic cells stimulated with RNA-containing immune complexes is promoted by NK cells via MIP-1β and LFA-1. J. Immunol. 2011;186:5085–5094. doi: 10.4049/jimmunol.1003349. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.