

Abstract
Development of post-GWAS (genome-wide association study) methods are greatly needed for characterizing the function of trait-associated SNPs. Strategies integrating various biological data sets with GWAS results will provide insights into the mechanistic role of associated SNPs. Here, we present a method that integrates RNA sequencing (RNA-seq) and allele-specific expression data with GWAS data to further characterize SNPs associated with follicular lymphoma (FL). We investigated the influence on gene expression of three established FL-associated loci—rs10484561, rs2647012, and rs6457327—by measuring their correlation with human-leukocyte-antigen (HLA) expression levels obtained from publicly available RNA-seq expression data sets from lymphoblastoid cell lines. Our results suggest that SNPs linked to the protective variant rs2647012 exert their effect by a cis-regulatory mechanism involving modulation of HLA-DQB1 expression. In contrast, no effect on HLA expression was observed for the colocalized risk variant rs10484561. The application of integrative methods, such as those presented here, to other post-GWAS investigations will help identify causal disease variants and enhance our understanding of biological disease mechanisms.
Main Text
Follicular lymphoma (FL [MIM 613024]), a common subtype of non-Hodgkin lymphoma (NHL [MIM 605027]), is a heterogeneous malignancy of the lymphoid system. Genome-wide association studies (GWASs) have identified several major susceptibility loci for FL in the human-leukocyte-antigen (HLA) region; these loci include SNPs in HLA class II (rs10484561 [p = 1.12 × 10−29]1 and rs2647012 [p = 2 × 10−21]2) and class I (rs6457327 [p = 4.7 × 10−11]3) regions. However, despite additional work that uncovered possible biological relevance of these associated variants,4 their function remains to be established. Recent advances in genetic studies on gene expression provide new opportunities for connecting trait-predisposing variants to cellular mechanisms.5,6 Such studies have already refined candidate-gene selection in numerous GWASs by identifying expression quantitative trait loci (eQTLs) that coordinately influence study traits and gene expression.7–9 In this study, we used existing GWAS data to investigate the influence on gene expression of rs2647012, rs6457327, and rs10484561 by measuring their correlation with RNA-sequencing (RNA-seq) data, and we made use of allele-specific-expression (ASE) data to assesses the enrichment of both rare and common causal effects for individuals harboring both protective and risk haplotypes (Figure 1).
Figure 1.
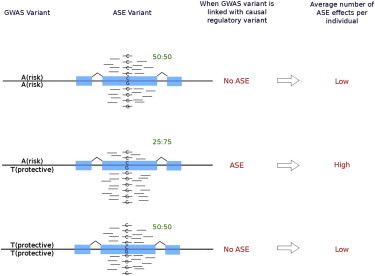
ASE Test for Disease-Associated Variants
ASE that is shared among multiple individuals can indicate the presence of an eQTL (i.e., individuals with ASE are heterozygous for the common causal regulatory variant), multiple rare regulatory variants (i.e., each individual has a private variant impacting expression), or epigenetic effects where one haplotype is silenced relative to the other. We have developed a method that assesses enrichment of ASE effects for individuals harboring both the risk and the protective alleles (i.e., individuals who are heterozygous for the GWAS variant) as compared to homozygous individuals. Here, the expectation is that functional differences will be more manifest for individuals who possess both the risk and protective alleles for genes involved in the etiology of the trait. This enrichment is represented in the figure in that more ASE events are present in individuals heterozygous for the GWAS variant; green ratios describe the relative transcript abundance. This test complements eQTL approaches in that it can add support to the presence of an eQTL, as well as indicate an enrichment of other potential causal effects (independent of frequency) (i.e., rare or private variants) underlying the difference in risk and protective haplotypes.
As a result of the high linkage disequilibrium (LD) in the region and the possibility that the three FL-associated variants (rs10484561, rs2647012, and rs6457327) could be linked to potential eQTLs, we expanded the eQTL analysis to include variants in LD. Using HapMap CEU (Utah residents with ancestry from northern and western Europe from the CEPH collection) genotype data (release 28), we identified 290 SNPs in LD (r2 > 0.5) with the three FL-associated variants. To confirm the role of these linked variants in FL risk, we tested them for association by using genotype data from a previous GWAS of FL.1 SNPs not genotyped or not passing quality-control criteria in the FL GWAS were imputed with BEAGLE 3.310 with the use of phased genotype data for 85 CEU samples from phase I of the 1000 Genomes Project. Those variants that did not show statistical evidence of association with FL (trend p value < 1.67 × 10−2 based on a Bonferroni correction for the three loci tested with α = 0.05) or for which no association data were available were further discarded, leading to the inclusion of an additional 158 variants in LD (55, 45, and 61 variants linked to rs10484561, rs2647012, and rs6457327, respectively) in the eQTL analysis.
To test the correlation between genetic variation and expression levels in the FL-associated loci, we used publicly available gene-expression and genotype data from HapMap CEU individuals. Whole-genome expression data in transformed lymphoblastoid cell lines (LCLs) obtained by RNA sequencing (RNA-seq) were downloaded from two data sets. The first data set (GSE16921)11 contained processed gene-expression RPKM (reads per kilobase per million mapped reads) values from 41 CEU samples and was downloaded from the Gene Expression Omnibus. eQTL transcript association data from 60 CEU samples, 29 of which were shared with GSE16921, were obtained from a second RNA-seq data set (E-MTAB-197) that was generated by Montgomery et al.12 and downloaded from their website. Whole-genome genotyping data for the same HapMap CEU individuals were directly downloaded from HapMap. Although eQTLs can have a distant (trans) effect, we focused here on proximal (cis) eQTLs, and we therefore tested each of the 161 investigated SNPs as an eQTL, and all expression probes were located within 1 Mb of the SNP. For the E-MTAB-197 data set, eQTL association data, measured by Spearman’s rank correlation, were directly obtained from the website of Montgomery et al. For GSE16921, the correlation between expression and genotype for each SNP-probe pair was tested with the Spearman’s rank correlation test with t-distribution approximation. All the correlations between genotype and expression were estimated with respect to the minor allele. p values were adjusted for multiple comparisons with the Benjamini-Hochberg false-discovery-rate (FDR) correction (p values and thresholds adjusted with this correction are henceforth referred to as BH p values and thresholds, respectively). eQTLs were considered significant at a BH p value threshold = 0.01.
Using the GSE16921 RNA-seq expression data set, we found a total of 192 SNP-probe pairs, corresponding to 66 individual SNPs, that reached statistical significance in the eQTL analysis (Table S1, available online). Interestingly, all 45 SNPs linked to rs2647012 showed significant expression changes in nearby genes. In particular, these FL protective variants were consistently associated with higher expression levels of HLA-DQA1 (MIM 146880), HLA-DQB1 (MIM 604305), HLA-DRB1 (MIM 142857), and HLA-DRB5 (MIM 604776) and lower expression levels of HLA-DQA2 (MIM 613503), HLA-DQB2, and HLA-DRB6 (Table S1). Variants linked to the FL-protective SNP rs6457327 significantly correlated with higher HCG22 (MIM 613918) expression (Table S1). However, none of the rs10484561-linked SNPs exhibited significant association with expression changes in any surrounding genes in this RNA-seq data set. To corroborate these findings, we used eQTL data obtained from the E-MTAB-197 data set. In this independent RNA-seq data set, 50 eQTLs reached statistical significance at the BH threshold of p < 0.01 (Table S1). Considerable reproducibility was found between both RNA-seq data sets for the shared SNP-gene combinations at rs2647012-linked SNPs. Thus, we again observed higher HLA-DQA1, HLA-DQB1, HLA-DRB1, and HLA-DRB5 expression levels and lower HLA-DQA2 expression levels for rs2647012-linked variants (Table S1). Associations between rs2647012 SNPs and HLA-DQB2 and HLA-DRB6, as well as associations between rs6457327 variants and HCG22, could not be confirmed because no information for these genes was included in E-MTAB-197. However, additional correlations were observed between rs2647012 SNPs and HLA-DRA (MIM 142860) expression and between rs6457327 SNPs and C6orf26 and HLA-B (MIM 142830) expression in E-MTAB-197 (Table S1). As with the previous data set, no eQTLs were found for the rs10484561-linked SNPs at a BH threshold of p ≤ 0.01.
Overall, the most significant eQTL associations in both RNA-seq data sets were seen for rs2647012-linked variants and HLA-DQB1 (Table S2 and Figure S1). Of the 45 SNPs investigated in this locus, 32 were associated with differential HLA-DQB1 levels and 30 consistently correlated with increased HLA-DQB1 expression. Of the HLA-DQB1 eQTLs, the strongest FL-associated SNP was rs4947344, which showed differential expression for multiple HLA genes (Table S2 and Figure S2). To explore the potential trans-effect of these eQTLs, we also investigated the correlation between rs2647012-linked SNPs and genome-wide expression levels in the GSE16921 data set. We observed that the strongest correlations were found with genes nearby (Figure 2 and Figure S3), further suggesting that rs2647012 variants are cis-regulatory. To further corroborate the potential regulatory effect of FL-associated SNPs on HLA-DQB1, we developed a complementary ASE-based approach (Figure 1). Using this strategy, we first assessed ASE in HapMap CEU individuals heterozygous at HLA-DQB1 exonic SNPs. ASE, calculated as previously reported,13 was assessed exclusively for sites identified as heterozygous in matching individuals sequenced as part of the 1000 Genomes Project Low-Coverage Pilot data set. Only heterozygous variants with a read depth ≥ 8 and observed imbalance level from 15% to 85% were included in the analysis. In total, 162,114 pairs of heterozygous SNP-sample pairs were tested for allelic imbalance, and 3,441 (2.1%) pairs showed evidence of allelic imbalance at a p value level < 2.12 × 10−4, corresponding to a FDR of 1%. Among the 317 HLA-DQB1 SNP-sample pairs for which ASE data were available, 132 (41.6%) were imbalanced at p < 2.12 × 10−4 (FDR = 1%). The presence of ASE can reflect the presence of nearby cis-regulatory polymorphisms.13 If ASE is marking a cis-eQTL, individuals with ASE should also be heterozygous for that eQTL. However, the enrichment of allelic effects does not explicitly require the presence of a common eQTL and could be tagging multiple rare effects (Figure 1). Therefore, we tested whether the ASE-significant signals for linked alleles in HLA-DQB1 were more enriched in samples heterozygous for the FL-associated variants than in homozygous samples. We found convincing evidence of enrichment of allelic imbalance for samples heterozygous for the FL-associated variants linked to rs2647012 (Table S3 and Figure S4), and rs4947344 was the FL-associated variant most significantly enriched with ASE-significant signals (p value = 1.28 × 10−4, Table S3). A second rs2647012-linked SNP, rs9275292, which approached statistical significance in the previous eQTL analysis (BH p value = 1.08 × 10−2), also showed significant enrichment of allelic imbalance (p value = 9.58 × 10−5, Table S3). No ASE data were available for HLA-DRB1, HLA-DRB6, or HLA-DQA2, yet we found 312 pairs of heterozygous HLA-DQA1 SNP-sample pairs, 33 (10.6%) of which had significant ASE. However, there was no enrichment of allelic effect on HLA-DQA1 for samples heterozygous for rs2647012-linked SNPs.
Figure 2.
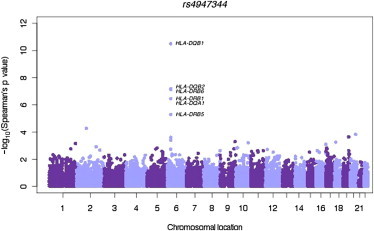
Genome-wide SNP-Probe Associations for rs4947344 in the GSE16921 Data Set
SNP-probe association plot for rs4947344 and probes located across the genome. The strongest associations, represented as −log10 of the Spearman’s observed p value, were observed for HLA class II genes located in cis with respect to rs4947344. A genome-wide association plot for all the rs2647012-linked variants is shown in Figure S3.
rs10484561 and rs2467012 lie in close proximity within an intergenic region and are 29 and 28 kb, respectively, upstream of HLA-DQB1 (Figure S5). Although located in the same region as rs2647012, none of the SNPs linked to rs10484561 showed eQTLs for HLA-DQB1 (Table S4), suggesting that different mechanisms are most likely involved for these two FL-associated loci. However, rs10484561 did show moderate enrichment of ASE effects (Figure S6). To elucidate the potential mechanisms on expression regulation underlying the HLA-DQB1 eQTLs linked to rs2647012, we compared the regulatory context of the rs2647012 and rs10484561 regions by using ENCODE (Encyclopedia of DNA Elements) experimental data. LCL analyses using chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq) revealed several histone marks and transcription-factor binding sites associated with enhancer and silencer activities in the region, overlapped by both rs2647012- and rs10484561-linked variants (Tables S3 and S4). However, there was no significant enrichment of specific transcription factors or epigenetic marks to explain the eQTL- and ASE-effect differences observed between rs2647012- and rs10484561-linked SNPs. A search using the HaploReg resource14 provided similar results without pinpointing particular regulatory motifs that could account for the differences observed between both loci.
However, because the FL-protective rs2647012-linked variants are correlated with increased HLA-DQB1 expression, our results suggest that reduced HLA-DQB1 expression might play a key role in the pathogenesis of lymphoma. In contrast, the colocalizing variant rs10484561 does not seem to modulate HLA-DQB1 through a common eQTL effect or through expression of other HLA class II genes. The functional relevance of this variant might relate to its high LD (D′ > 0.9) with the HLA-DQB1∗0501 and HLA-DRB1∗0101 coding alleles.4 The structural changes of these HLA alleles at the amino acid level might affect antigen binding and overall immune response, providing a feasible mechanism by which rs10484561-linked variants might act. Furthermore, the moderate enrichment of ASE effects indicates the potential for rare regulatory variants interacting with protein-coding variants that contribute to this effect. In the HLA class I region, association between the FL-protective rs6457327 SNPs and HLA-expression changes were more inconsistent. The E-MTAB-197 data set indicated a correlation between the SNPs and C6orf26 and HLA-B; however, these associations were not observed in the GSE16921 data set, which instead showed association with HCG22. SNP rs6457327 has been associated with the transformation of FL to diffuse large-B cell lymphoma,15,16 a more aggressive form of NHL, as well as with survival.16 However, the strength of the disease-risk association between rs6457327 and FL has been less robust than that between HLA class II variants and FL.1 It is possible that variants in this region exert more subtle effects, and therefore, larger data sets are needed for unequivocally determining the potential effect of those variants on the expression of nearby genes. Additionally, there is increasing evidence that the effect of eQTLs differs by cell type. Therefore, it is possible that the lack of eQTL evidence for rs10484561 and rs6457327 might be due to different mechanisms involving transcription factors not expressed in LCLs. Although it has been observed that LCLs are good surrogates for primary tissue and that genetic variation in LCLs might be generalized to primary B cells,17 Epstein-Barr-virus transformation might have an impact on gene regulation in B lymphocytes. Therefore, further analysis of primary B cells and other antigen-presenting cells will provide a further understanding of the potential regulatory effects of these FL-associated variants in primary cell types. Finally, it is likely that other factors not examined in this study exert additional functional effects. In particular, gene-gene and gene-environment interactions probably play a role in FL pathogenesis, and follow-up studies that take into account these additional factors are likely to provide further insight.
GWASs have proven very successful for the discovery of SNPs associated with complex diseases, and now there is an increasing need to explore their potential functional relevance. In this study, we focused on three FL susceptibility loci to determine whether expression changes can be linked to FL pathogenesis. We used an ASE-based approach designed to capture enrichment of common and rare regulatory variants and epigenetic effects for individuals carrying both protective and risk alleles. Ultimately, methods that incorporate diverse genomic data will enhance our understanding of mechanisms involved in the etiology of complex diseases.
Acknowledgments
This work was supported by National Institutes of Health grants CA122663, CA154643-01A1, and CA104682 (to C.F.S.) and CA087014 (to P.M.B).
Contributor Information
Stephen B. Montgomery, Email: smontgom@stanford.edu.
Christine F. Skibola, Email: chrisfs@berkeley.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org
E-MTAB-197 data set, http://jungle.unige.ch/rnaseq_CEU60
Encyclopedia of DNA Elements (ENCODE), http://genome.ucsc.edu/ENCODE/
Gene Expression Omnibus, http://www.ncbi.nlm.nih.gov/geo
International HapMap Project, www.hapmap.org
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Conde L., Halperin E., Akers N.K., Brown K.M., Smedby K.E., Rothman N., Nieters A., Slager S.L., Brooks-Wilson A., Agana L. Genome-wide association study of follicular lymphoma identifies a risk locus at 6p21.32. Nat. Genet. 2010;42:661–664. doi: 10.1038/ng.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smedby K.E., Foo J.N., Skibola C.F., Darabi H., Conde L., Hjalgrim H., Kumar V., Chang E.T., Rothman N., Cerhan J.R. GWAS of follicular lymphoma reveals allelic heterogeneity at 6p21.32 and suggests shared genetic susceptibility with diffuse large B-cell lymphoma. PLoS Genet. 2011;7:e1001378. doi: 10.1371/journal.pgen.1001378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Skibola C.F., Bracci P.M., Halperin E., Conde L., Craig D.W., Agana L., Iyadurai K., Becker N., Brooks-Wilson A., Curry J.D. Genetic variants at 6p21.33 are associated with susceptibility to follicular lymphoma. Nat. Genet. 2009;41:873–875. doi: 10.1038/ng.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Skibola C.F., Akers N.K., Conde L., Ladner M., Hawbecker S.K., Cohen F., Ribas F., Erlich H.A., Goodridge D., Trachtenberg E.A. Multi-locus HLA class I and II allele and haplotype associations with follicular lymphoma. Tissue Antigens. 2012;79:279–286. doi: 10.1111/j.1399-0039.2012.01845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Montgomery S.B., Dermitzakis E.T. From expression QTLs to personalized transcriptomics. Nat. Rev. Genet. 2011;12:277–282. doi: 10.1038/nrg2969. [DOI] [PubMed] [Google Scholar]
- 6.Nica A.C., Dermitzakis E.T. Using gene expression to investigate the genetic basis of complex disorders. Hum. Mol. Genet. 2008;17(R2):R129–R134. doi: 10.1093/hmg/ddn285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schadt E.E., Molony C., Chudin E., Hao K., Yang X., Lum P.Y., Kasarskis A., Zhang B., Wang S., Suver C. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 2008;6:e107. doi: 10.1371/journal.pbio.0060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moffatt M.F., Kabesch M., Liang L., Dixon A.L., Strachan D., Heath S., Depner M., von Berg A., Bufe A., Rietschel E. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature. 2007;448:470–473. doi: 10.1038/nature06014. [DOI] [PubMed] [Google Scholar]
- 9.Myers A.J., Gibbs J.R., Webster J.A., Rohrer K., Zhao A., Marlowe L., Kaleem M., Leung D., Bryden L., Nath P. A survey of genetic human cortical gene expression. Nat. Genet. 2007;39:1494–1499. doi: 10.1038/ng.2007.16. [DOI] [PubMed] [Google Scholar]
- 10.Browning B.L., Browning S.R. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am. J. Hum. Genet. 2009;84:210–223. doi: 10.1016/j.ajhg.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheung V.G., Nayak R.R., Wang I.X., Elwyn S., Cousins S.M., Morley M., Spielman R.S. Polymorphic cis- and trans-regulation of human gene expression. PLoS Biol. 2010;8:8. doi: 10.1371/journal.pbio.1000480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Montgomery S.B., Sammeth M., Gutierrez-Arcelus M., Lach R.P., Ingle C., Nisbett J., Guigo R., Dermitzakis E.T. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature. 2010;464:773–777. doi: 10.1038/nature08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Montgomery S.B., Lappalainen T., Gutierrez-Arcelus M., Dermitzakis E.T. Rare and common regulatory variation in population-scale sequenced human genomes. PLoS Genet. 2011;7:e1002144. doi: 10.1371/journal.pgen.1002144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ward L.D., Kellis M. HaploReg: A resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40(Database issue):D930–D934. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wrench D., Leighton P., Skibola C.F., Conde L., Cazier J.B., Matthews J., Iqbal S., Carlotti E., Bödör C., Montoto S. SNP rs6457327 in the HLA region on chromosome 6p is predictive of the transformation of follicular lymphoma. Blood. 2011;117:3147–3150. doi: 10.1182/blood-2010-10-315382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berglund M., Enblad G., Thunberg U. SNP rs6457327 is a predictor for overall survival in follicular lymphoma as well as survival after transformation. Blood. 2011;118:4489. doi: 10.1182/blood-2011-06-362921. [DOI] [PubMed] [Google Scholar]
- 17.Caliskan M., Cusanovich D.A., Ober C., Gilad Y. The effects of EBV transformation on gene expression levels and methylation profiles. Hum. Mol. Genet. 2011;20:1643–1652. doi: 10.1093/hmg/ddr041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.