

Abstract
A 53-year-old man complained of orthostatic, non-rotating dizziness, and chronic watery diarrhea of several years duration. His nerve-conduction velocity test revealed peripheral sensory-motor polyneuropathy and he showed an autonomic function abnormality. Echocardiographic examination showed ventricular and atrial wall thickening with a granular "sparkling" appearance. Left ventricular systolic function was preserved but pseudonormal diastolic dysfunction was present. Coronary angiography showed normal coronary arteries and an endomyocardial biopsy revealed lesions consistent with cardiac amyloidosis. Colonoscopic biopsy also revealed the deposition of amyloid fibrils. Gene analysis found the transthyretin variant Asp38Ala. His son had same mutation, but three daughters did not. In conclusion, we report a case of familial transthyretin amyloidosis with Asp38Ala.
Keywords: Amyloidosis, Transthyretin, Asp38Ala, Orthostatic hypotension, Polyneuropathy
Introduction
Amyloidosis is a disease condition in which the extracellular deposition of pathological insoluble fibrillar proteins occurs in various organs that can impair tissue structure and function.1) Amyloidosis may be systemic or localized and is currently classified according to the type of precursor protein.2) The three most frequent types of systemic amyloidosis are acquired monoclonal immunoglobulin light-chain amyloidosis, familial transthyretin amyloidosis (ATTR), which can be caused by mutations of transthyretin (TTR), and systemic senile amyloidosis related to wild-type TTR.3) TTR is one of over 20 known proteins that are capable of forming amyloid fibrils in vivo. Although both wild-type and mutated TTR can form amyloid, alterations in the primary structure of the TTR protein due to TTR mutations can result in greatly accelerated amyloid formation and these mutation are the origin of all symptomatic cases of ATTR.3),4) More than 100 amyloidogenic TTR mutations have been reported in association with markedly variable clinical features, disease penetrances, and prognoses.5-7)
Cardiac involvement in amyloidosis is a manifestation of one of several systemic diseases known as amyloidoses.1),8) Regardless of the underlying pathogenesis, cardiac amyloidosis is characterized by extracellular amyloid infiltration throughout the heart. This infiltrative process results in biventricular wall thickening with non-dilated ventricles and may also involve the conduction system.9) However, cardiac involvement may be the predominant feature or be found during investigation in a patient presenting with another major organ involvement.10) Furthermore, the initial suspicion of cardiac amyloidosis is often triggered by the recognition that the heart disease is part of a multiorgan disorder.10)
Here, we report a case of cardiac amyloidosis, suspected based upon two-dimensional echocardiographic findings, which presented with autonomic dysfunction, chronic gastrointestinal symptoms, and uncertain cardiac symptoms.
Case
In July 2010, a 53-year-old man was admitted to our neurology clinic for the evaluation of dizziness and headache of three years duration. He had not been diagnosed with hypertension, diabetes, or pulmonary disease. The patient presented with chronic diarrhea and a residual urine sensation of several years duration, and reported taking an alpha-blocker to treat the latter. In addition, he complained of non-rotating dizziness aggravated by abrupt standing for 1 years. However, despite of the cessation of alpha-blocker treatment, which could have been responsible for the orthostatic hypotension, the non-rotating dizziness did not improve. On physical examination, blood pressure was 108/65 mmHg and his pulse rate was regular at 60 beats/min in the supine position. However, when the patient changed posture from supine to standing, his blood pressure fell below 80/45 mmHg. Nerve-conduction velocity testing revealed peripheral sensory-motor polyneuropathy combined with bilateral carpal tunnel syndrome. He also showed autonomic dysfunction. At admission, he presented atypical chest pain of four months duration, and was referred to the cardiology department. No abnormal findings were found by 12-lead standard electrocardiography, and laboratory studies revealed normal liver and renal functions. In addition, his erythrocyte sedimentation rate (4.0 mm/hr: normal < 10 mm/hr) and C-reactive protein (0.15 mL/dL: normal < 0.5 mL/dL) were also normal. However, two-dimensional transthoracic echocardiography revealed a thickened left ventricle (interventricular septal dimension 1.19 cm, left ventricle posterior wall dimension 1.28 cm), and that the right ventricle and interatrial septum had a granular "sparkling" appearance (Fig. 1). Left ventricular systolic function was preserved (ejection fraction = 54% by the modified Simpson' method) but diastolic dysfunction was present. Pulsed-wave Doppler recording of mitral inflow showed a normal diastolic filling pattern, with an E/A ratio of 1.1 (Fig. 2A), but early diastolic mitral annulus tissue Doppler velocity (Ea) and the E/Ea index were 4 cm/s and 20.3, respectively, indicating a pseudonormal pattern (Fig. 2B). These findings were compatible with infiltrative cardiomyopathy. Coronary angiography showed normal coronary arteries and an endomyocardial biopsy revealed lesions consistent with cardiac amyloidosis. Light microscopic findings (haematoxylin and eosin staining) revealed amyloid appearing as pink-hyaline extracellular deposits between myocytes and in blood vessels (Fig. 3). Electron microscopic findings demonstrated amyloid fibrils at the edge of a myocytes (Fig. 4).
Fig. 1.

Two-dimensional transthoracic echocardiography. Biventricular hypertrophy and the thickened inter-atrial septum are shown in a parasternal long-axis view (A), four-chamber view (B and C).
Fig. 2.

Pulse-waved Doppler echocardiogram (A) and tissue Doppler echocardiogram (B) showing and elevated E/Ea ratio and low mitral annulus velocities, suggestive of diastolic dysfunction with a pseudonormal pattern.
Fig. 3.
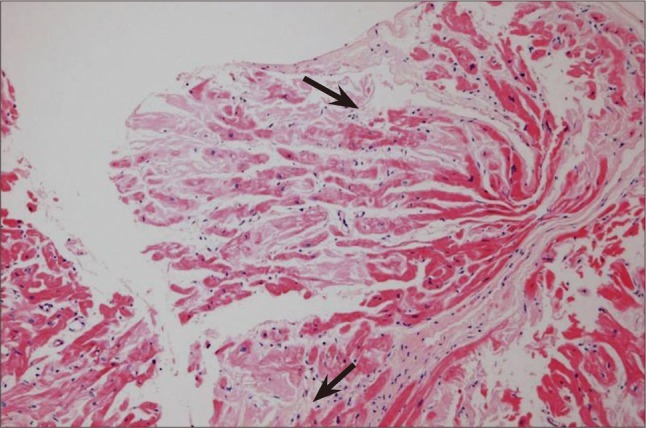
Light microscopy finding of tissue obtained by cardiac biopsy (haematoxylin and eosin stained, original magnification × 40). Amyloid appears as pink-hyaline extracellular deposits (black arrows) between myocytes and in blood vessels.
Fig. 4.

Electron microscopy of cardiac tissue. Electron microscopy demonstrated fibrils typical of amyloid at the edge of a myocytes (A). The edge of a myocyte (lower left) and above it is a mass of amyloid fibrils (B).
In addition, a colonoscopic biopsy was performed to identify the cause of the chronic watery diarrhea, and histopathological findings of a colon mucosal biopsy specimen showed chronic colitis and amyloid fibril depositions.
Gene analysis of the TTR exons found a heterozygous mutation at 173 A > C, and thus, a diagnosis of ATTR with variant Asp38Ala (aspartate to alanine mutation at amino acid 38) (Fig. 5). Family screening for ATTR revealed that his son had the same mutation but that his three daughters did not (Fig. 5); his son had no symptoms and normal echocardiographic findings. The patient is being treated by continued symptomatic care and is being regularly followed.
Fig. 5.

Transthyretin gene analysis in the patient and his offspring (A: patient, B: daughter 1, C: daughter 2, D: daughter 3, E: son). A: The patient was diagnosed as transthyretin amyloidosis (ATTR) variant Asp38Ala. E: The patient's offspring were screening for ATTR, but only his son had the same mutation.
Discussion
This report describes a case of cardiac amyloidosis of ATTR variant Asp38Ala with systemic amyloid deposition in multiple organs and tissues in a Korean family. The distribution of amyloid deposition in the heart and colonic mucosa with a clinical presentation of polyneuropathy may be a characteristic feature of ATTR Asp38Ala. To our knowledge, this is the first case report of ATTR Asp38Ala in a Korean.
In addition, according to the largest series of electrocardiography findings in cardiac amyloidosis, the two main ECG abnormalities were the presence of low voltage and a pseudo-infarct pattern in 46% and 47% of patients, respectively.11) However, the present case had no abnormal electrocardiographic findings.
As mentioned in the Introduction, genetic mutations in the TTR gene lead to heterogeneous clinical manifestations. Of these mutations, ATTR Val30Met (valine to methionine at amino acid 30) is the most common.12) The characteristic presentation of endemic Val30Met comprises sensorimotor polyneuropathy and autonomic neuropathy, but cardiomyopathy is rare. Amyloid deposition in ATTR Val30Met is localized to the subendocardial area including the conduction system, and thus, various types of conduction block frequently appear, which require pacemaker implantation.4),13-18)
On the other hand, ATTR Asp38Ala is clinically characterized by progressive cardiac dysfunction, and peripheral somatic and autonomic neuropathy.19-21) Yazak et al.19) reported postmortem findings for two ATTR Asp38Ala cases, in which amyloid deposition was observed extensively in myocardium, peripheral nerves, sympathetic ganglia, and in the gastrointestinal tract. In addition, pulmonary parenchyma was also diffusely involved in their cases. Tachibana et al.20) presented another case of ATTR Asp38Ala. The patient presented progressive dysesthesia in her legs, and subsequently, leg weakness, severe diarrhea, and shortness of breath gradually appeared with cardiac dysfunctions, which included ventricular wall thickened and reduced fractional shortening on the echocardiogram. However, in the present case, cardiac symptoms were not prominent and pulmonary involvement was absent.
In the previous cases, the symptom onset in ATTR Asp38Ala occurred at an older age than in our patient. Furthermore, peripheral nervous and gastrointestinal symptoms preceded cardiac symptoms, which led to progressive heart failure. These findings suggest that patients diagnosed early in the disease course, and their offspring, may need regular cardiac surveillance.
We report an unusual case of cardiac amyloidosis is a patient with familial transthyretin amyloidosis variant Asp38Ala who presented with autonomic dysfunction, chronic gastrointestinal symptoms, and uncertain cardiac symptoms.
References
- 1.Falk RH, Comenzo RL, Skinner M. The systemic amyloidoses. N Engl J Med. 1997;337:898–909. doi: 10.1056/NEJM199709253371306. [DOI] [PubMed] [Google Scholar]
- 2.Westermark P, Benson MD, Buxbaum JN, Cohen AS, Frangione B, Ikeda S, Masters CL, Merlini G, Saraiva MJ, Sipe JD. Amyloid fibril protein nomenclature -- 2002. Amyloid. 2002;9:197–200. doi: 10.3109/13506120209114823. [DOI] [PubMed] [Google Scholar]
- 3.Westermark P, Benson MD, Buxbaum JN, Cohen AS, Frangione B, Ikeda S, Masters CL, Merlini G, Saraiva MJ, Sipe JD. A primer of amyloid nomenclature. Amyloid. 2007;14:179–183. doi: 10.1080/13506120701460923. [DOI] [PubMed] [Google Scholar]
- 4.Ando Y, Nakamura M, Araki S. Transthyretin-related familial amyloidotic polyneuropathy. Arch Neurol. 2005;62:1057–1062. doi: 10.1001/archneur.62.7.1057. [DOI] [PubMed] [Google Scholar]
- 5.Falk RH, Dubrey SW. Amyloid heart disease. Prog Cardiovasc Dis. 2010;52:347–361. doi: 10.1016/j.pcad.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 6.Shah KB, Inoue Y, Mehra MR. Amyloidosis and the heart: a comprehensive review. Arch Intern Med. 2006;166:1805–1813. doi: 10.1001/archinte.166.17.1805. [DOI] [PubMed] [Google Scholar]
- 7.Rapezzi C, Perugini E, Salvi F, Grigioni F, Riva L, Cooke RM, Ferlini A, Rimessi P, Bacchi-Reggiani L, Ciliberti P, Pastorelli F, Leone O, Bartolomei I, Pinna AD, Arpesella G, Branzi A. Phenotypic and genotypic heterogeneity in transthyretin-related cardiac amyloidosis: towards tailoring of therapeutic strategies? Amyloid. 2006;13:143–153. doi: 10.1080/13506120600877136. [DOI] [PubMed] [Google Scholar]
- 8.Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349:583–596. doi: 10.1056/NEJMra023144. [DOI] [PubMed] [Google Scholar]
- 9.Smith TJ, Kyle RA, Lie JT. Clinical significance of histopathologic patterns of cardiac amyloidosis. Mayo Clin Proc. 1984;59:547–555. doi: 10.1016/s0025-6196(12)61493-1. [DOI] [PubMed] [Google Scholar]
- 10.Falk RH. Diagnosis and management of the cardiac amyloidoses. Circulation. 2005;112:2047–2060. doi: 10.1161/CIRCULATIONAHA.104.489187. [DOI] [PubMed] [Google Scholar]
- 11.Rahman JE, Helou EF, Gelzer-Bell R, Thompson RE, Kuo C, Rodriguez ER, Hare JM, Baughman KL, Kasper EK. Noninvasive diagnosis of biopsy-proven cardiac amyloidosis. J Am Coll Cardiol. 2004;43:410–415. doi: 10.1016/j.jacc.2003.08.043. [DOI] [PubMed] [Google Scholar]
- 12.Benson MD, Uemichi T. Transthyretin amyloidosis. Amyloid Int J Exp Clin Invest. 1996;3:44–56. [Google Scholar]
- 13.Ikeda S, Hanyu N, Hongo M, Yoshioka J, Oguchi H, Yanagisawa N, Kobayashi T, Tsukagoshi H, Ito N, Yokota T. Hereditary generalized amyloidosis with polyneuropathy. Clinicopathological study of 65 Japanese patients. Brain. 1987;110(Pt 2):315–337. doi: 10.1093/brain/110.2.315. [DOI] [PubMed] [Google Scholar]
- 14.Benson MD, Kincaid JC. The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve. 2007;36:411–423. doi: 10.1002/mus.20821. [DOI] [PubMed] [Google Scholar]
- 15.Conceição I, De Carvalho M. Clinical variability in type I familial amyloid polyneuropathy (Val30Met): comparison between late- and early-onset cases in Portugal. Muscle Nerve. 2007;35:116–118. doi: 10.1002/mus.20644. [DOI] [PubMed] [Google Scholar]
- 16.Bittencourt PL, Couto CA, Clemente C, Farias AQ, Palácios SA, Mies S, Goldberg AC. Phenotypic expression of familial amyloid polyneuropathy in Brazil. Eur J Neurol. 2005;12:289–293. doi: 10.1111/j.1468-1331.2004.00941.x. [DOI] [PubMed] [Google Scholar]
- 17.Ikeda S, Nakazato M, Ando Y, Sobue G. Familial transthyretin-type amyloid polyneuropathy in Japan: clinical and genetic heterogeneity. Neurology. 2002;58:1001–1007. doi: 10.1212/wnl.58.7.1001. [DOI] [PubMed] [Google Scholar]
- 18.Suhr OB, Svendsen IH, Andersson R, Danielsson A, Holmgren G, Ranløv PJ. Hereditary transthyretin amyloidosis from a Scandinavian perspective. J Intern Med. 2003;254:225–235. doi: 10.1046/j.1365-2796.2003.01173.x. [DOI] [PubMed] [Google Scholar]
- 19.Yazak M, Take YI, Katoh M, Ikeda SI. Postmortem findings in two familial amyloidosis patients with transthyretin variant Asp38Ala. Amyloid. 2000;7:270–277. doi: 10.3109/13506120009146441. [DOI] [PubMed] [Google Scholar]
- 20.Tachibana N, Tokuda T, Yoshida K, Taketomi T, Nakazato M, Li YF, Masuda Y, Ikeda S. Usefulness of MALDI/TOF mass spectrometry of immunoprecipitated serum variant transthyretin in the diagnosis of familial amyloid polyneuropathy. Amyloid. 1999;6:282–288. doi: 10.3109/13506129909007341. [DOI] [PubMed] [Google Scholar]
- 21.Kishikawa M, Nakanishi T, Miyazaki A, Shimizu A, Kusaka H, Fukui M, Nishiue T. A new amyloidogenic transthyretin variant, [D38A], detected by electrospray ionization/mass spectrometry. Amyloid. 1999;6:278–281. doi: 10.3109/13506129909007340. [DOI] [PubMed] [Google Scholar]