

Abstract
We have analyzed a natural population of the marine bacterium, Alteromonas macleodii, from a single sample of seawater to evaluate the genomic diversity present. We performed full genome sequencing of four isolates and 161 metagenomic fosmid clones, all of which were assigned to A. macleodii by sequence similarity. Out of the four strain genomes, A. macleodii deep ecotype (AltDE1) represented a different genome, whereas AltDE2 and AltDE3 were identical to the previously described AltDE. Although the core genome (∼80%) had an average nucleotide identity of 98.51%, both AltDE and AltDE1 contained flexible genomic islands (fGIs), that is, genomic islands present in both genomes in the same genomic context but having different gene content. Some of the fGIs encode cell surface receptors known to be phage recognition targets, such as the O-chain of the lipopolysaccharide, whereas others have genes involved in physiological traits (e.g., nutrient transport, degradation, and metal resistance) denoting microniche specialization. The presence in metagenomic fosmids of genomic fragments differing from the sequenced strain genomes, together with the presence of new fGIs, indicates that there are at least two more A. macleodii clones present. The availability of three or more sequences overlapping the same genomic region also allowed us to estimate the frequency and distribution of recombination events among these different clones, indicating that these clustered near the genomic islands. The results indicate that this natural A. macleodii population has multiple clones with a potential for different phage susceptibility and exploitation of resources, within a seemingly unstructured habitat.
Keywords: Alteromonas macleodii, metagenome, population genomics, genomic island, constant-diversity, phage
Introduction
Microbiology has been built upon the study of pure cultures, which represent large populations of individual cells of clonal descent that are normally assumed to be genetically homogeneous. This fundamental principle is widely used whether describing the physiological traits of an isolate or sequencing the genome of a particular strain. Nonetheless, the applicability of the pure culture paradigm to the description of natural microbial populations is debateable. To date, the accepted models of bacterial population genetics state that a low phenotypic diversity is expected due to purges involving fitter mutants, known as periodic selection events (Atwood et al. 1951; Cohan 2002; Doolittle 2012). Accordingly, the concept of genomic reconstruction of individual microbes from metagenomic data sets depends on the near homogeneity of abundant environmental strains (Tyson et al. 2004; Iverson et al. 2012). Evidence from metagenomic studies, however, indicate that strain genomes, even if present in large amounts, recruit very unevenly, indicating a large microdiversity in a single sample (Coleman et al. 2006; Legault et al. 2006; Cuadros-Orellana et al. 2007; Albertsen et al. 2012). Besides, large variations due to single-nucleotide polymorphisms (SNPs) and recombination among concurrent lineages have been described abundantly in the literature (see for e.g., Papke et al. 2004; Shapiro et al. 2012). It should be noted that very seldom (if ever) has the genome of a pure culture been compared with a metagenome obtained from the same sample used to isolate the cultured strain. There are a few examples in which the same environment was sampled but at a different time point (Coleman et al. 2006; Cuadros-Orellana et al. 2007). Under these conditions, the population might have changed during the time span that elapsed between strain isolation and metagenomic sampling.
To better understand the natural genomic diversity of a population of marine bacteria, we have studied the Alteromonas macleodii population in a single sample from the water column of the deep South Adriatic (Mediterranean Sea). Alteromonas macleodii is a globally distributed and commonly isolated marine gamma-proteobacterium (Sass et al. 2001; Lopez-Lopez et al. 2005). Mesocosm experiments (Schafer et al. 2000) and metatranscriptomic data (McCarren et al. 2012; Shi et al. 2012) have shown the dominance of r-strategists, such as A. macleodii, in heterotrophic blooms in the relatively nutrient depleted regions of the world’s oceans. Alteromonas macleodii isolates cluster by molecular analysis into two major genotypic groups, or ecotypes, one found in temperate latitudes in the upper water column and another that is for the most part found in the deep water column of the Mediterranean (Lopez-Lopez et al. 2005; Ivars-Martinez et al. 2008a, 2008b; López-Pérez et al. 2012).
The strain A. macleodii deep ecotype (AltDE) has been sequenced and described previously (Ivars-Martinez et al. 2008b). Additional isolates were obtained from the same sample and subsequently classified as A. macleodii. In parallel, a metagenomic fosmid library (∼40,000 fosmids, average size 35–40 kb) was constructed (fig. 1). The fosmid approach is advantageous over direct metagenomic sequencing in that longer, naturally linked genes are available (Ghai et al. 2010). Besides, the representative bias of fosmid cloning is different and likely smaller than that of pure culture. By sequencing metagenomic fosmids and strain genomes, we provide here a snapshot of the diversity present at this single time/space point.
Fig. 1.—

Flow chart of the methods followed for the isolation of Alteromonas macleodii strains, construction of the metagenomic fosmid library, and sequencing of both. Pure culture on marine agar and a metagenomic fosmid library were carried out from the same sample. Some isolates characterized as A. macleodii and metagenomic fosmids assigned to the same species were sequenced.
The results indicate that a limited number of clones are present. However, similar to previous studies (for e.g., Thompson et al. 2005), the strains present are extremely divergent in large genomic regions. Interestingly, a significant portion of the sequence diversity appears to be generated through recombination events, which supports the findings of previous studies (Wilmes et al. 2009; Cadillo-Quiroz et al. 2012; Shapiro et al. 2012). Recently, a model of constant–diversity (C-D) in which phages are instrumental in maintaining clonal diversity by a “kill-the winner” dynamics at the population level was proposed (Rodriguez-Valera et al. 2009). The data presented here indicating diversity in putative phage receptors and microniche specialization support this model.
Materials and Methods
Sample Collection
The A. macleodii genomes and the fosmids analyzed in this work come from the same sample, and therefore, they represent the clonal lineages present within this specific section of the water column. Two hundred liters of seawater was collected from the South Adriatic Sea (41°36’N; 17°22’E) at 1,000 m depth (200 m from the bottom) and sequentially filtered through Millipore 20, 5, and 0.22 µm-pore-size filters (fig. 1). DNA was purified by a classical phenol–chloroform protocol and 10 µg used to construct a genomic library using the CopyControl™ Fosmid Library Production Kit (Epicentre) as described previously (Martin-Cuadrado et al. 2008). A total of 38,704 fosmid clones were obtained. In parallel, from an aliquot of 1 l from the same seawater, marine agar plates were inoculated, and a total of 125 colonies were isolated. These isolates were characterized by 16S rRNA gene PCR amplification and sequencing, and 30 strains were classified as members of the species A. macleodii (more than 97% 16S rRNA similarity). Previously, one of them was selected for sequencing within the Moore Foundation Marine Microbial Genomics initiative and was described as the AltDE (Ivars-Martinez et al. 2008b).
Sequencing and Assembly
A total of three additional A. macleodii strains from the same set of deep Adriatic isolates, named AltDE1, AltDE2, and AltDE3, were sequenced. DNA was extracted by phenol–cloroform as described in Neumann et al. (1992) and checked for quality on a 0.8% agarose gel. The quantity was measured using Quant-iT PicoGreen dsDNA Reagent (Invitrogen). The DNA from each strain was pyrosequenced (Roche 454 GS-FLX system, GATC, Konstanz, Germany) at low coverage (10-fold). Only the sequence of AltDE1 was completed by an additional ¼ run of the Roche 454 GS-FLX system and near 1 Gb using the Solexa GAIIx technology (Macrogen, Korea). Low-quality regions were completely clipped using sff_extract (http://bioinf.comav.upv.es/sff_extract/index.html, last accessed December 12, 2012). In parallel, a total of 15,968 fosmid insert ends from the AD1000 collection were sequenced using Sanger sequencing. A BLASTN screening (cut off: more than 97% identity in 70% of the length) resulted in 447 fosmid ends to belong to A. macleodii species. A total of 245 fosmids were chosen based on their similarity to the deep ecotype strain genomes in one or both ends. Fosmid DNA was individually isolated using the QIAprep spin miniprep kit (Qiagen). The fosmid DNA was sequenced divided into 20 groups of nonoverlapping fosmids. Each group was tagged and sequenced together in two lanes of the Solexa HiSeq2000 technology (Macrogen, Korea). Two different programs were used in the assembly, Geneious Pro 5.0.1 (with default parameters) and MIRA (http://www.chevreux.org/projects_mira.html, last accessed December 12, 2012). Both results were compared for equal assemblies. Finally, 161 fragments larger than 10 kb were assembled and analyzed. A total of 64 were considered complete as they included both sequenced fosmid ends. For the assembled contigs of the genome of AltDE1, oligonucleotides designed from the sequence of the ends of assembled contigs and PCR amplicons were generated and sequenced to obtain one single closed contig. The coverage for each of the genomes, the amount of bases sequenced, and other related details of the sequencing of the fosmids are provided in supplementary table S1, Supplementary Material online.
Pulse Field Gel Electrophoresis
To assess the presence of plasmids, pulse field gel electrophoresis (PFGE) was carried out with AltDE and AltDE1. PFGE of the genomic DNA was performed in a contour-clamped homogeneous electric field (CHEF) system on a CHEF-DR III device (Bio-Rad Laboratories, Hercules, CA) with 1% agarose gels and modified 0.5× TBE buffer (45 mM Tris, 45 mM boric acid, 0.1 mM ethylenediaminetetraacetic acid) at 14°C. Pulse time ramps and run times were varied to identify different plasmid conformations (Hightower et al. 1987; Mathew et al. 1988). A lambda ladder PFGE marker (New England Biolabs) was used as a molecular size marker. After electrophoresis, the gels were stained with ethidium bromide (Sigma Co., St. Louis, MO), rinsed with distilled water, and photographed on an UV transilluminator.
Gene Prediction, Annotation, and Bioinformatics Analysis
Gene prediction of the assembled contigs was done using the ISGA pipeline (http://isga.cgb.indiana.edu/, last accessed December 12, 2012). The predicted protein sequences were compared using BLASTP to the National Center for Biotechnology Information nr protein database (e value: 10−5). ORFs smaller than 100 bp and without significant homology to other proteins were not considered. BioEdit was used to manipulate the sequences (Hall 1999). GC content was calculated using the EMBOSS tool geecee (Rice et al. 2000). For comparative analyses, reciprocal BLASTN and TBLASTXs searches between the genomes and the fosmids were carried out, leading to the identification of regions of similarity, insertions and rearrangements. To allow the interactive visualization of genomic fragment comparisons Artemis v.12 (Rutherford et al. 2000), Artemis Comparison Tool ACTv.9 (Carver et al. 2005) were used to compare the genomes. Average nucleotide identity (ANI) was calculated as defined before (Konstantinidis and Tiedje 2005), using a minimum cut off of 50% identity and 70% of the length of the query gene. Sequences were aligned using MUSCLE version 3.6 (Edgar 2004) and ClustalW (Thompson et al. 1994) and edited manually as necessary. The coverage of the genomes by the metagenomic fosmids was calculated by BLASTN comparison using a cut off of 95% identity in at least 5 kb. "nucmer" and "show-snps" of the MUMmer3+ package were used to identify the indels and the SNPs between the fosmids and the AltDE and AltDE1 genomes.
Analysis of Recombination
For the local alignments among the metagenomic fosmids and the A. macleodii genomes, we used MAUVE (v.2.3.1) (Darling et al. 2004). The Recombination Detection software package RDP4 (beta 16), in which different recombination detection programs (RDP, GENECONV, SiScan, BootScan, MaxChi, Chimaera and 3Seq) are embedded, was used to detect potential recombination strains, parental strains, and possible recombination breakpoints (Martin et al. 2010). These data can be found in supplementary table S4, Supplementary Material online. To be considered as a reliable recombination event, the highest multiple-comparison-corrected P-value cut off was set at 10−5 for at least three different methods.
dN/dS Analysis
Nonsynonymous versus synonymous nucleotide substitution ratio (dN/dS), frequently used as a marker for diversifying selection, was used as an indicator of selective pressure acting on a protein-coding gene. Orthologous protein sequence pairs were aligned using ClustalW, and the protein alignments imposed upon the nucleotide sequences using the program pal2nal (Suyama et al. 2006). Protein-encoding sequences with a high dN/dS ratio (>1) were considered to exhibit diversifying selection (positive selection for variability at some sites).
Biochemical Assays
Urease tests were carried out as described previously (Christian 1946; Ivars-Martinez et al. 2008b), and nitrate respiration was performed as described by Skerman (1967).
Motility Assays
AltDE and AltDE1 were streaked on marine broth (MB) agar plates and grown at 25°C. A single colony of each strain was inoculated into 10 ml MB media and grown in culture overnight at 180 rpm. Cell densities were adjusted to OD600 1.0, and 1 µl of the adjusted culture was stabbed into the center of a 0.35% MB agar plates. The plates were then incubated at 25°C for 24 h before the diameter of the growth zone was measured. Three independent cultures were performed in triplicate (N = 9) for each strain.
Accession Numbers
The sequences have been deposited in GenBank under the following BioProject codes: PRJNA58251 and PRJNA13374 for A. macleodii DE (AltDE), PRJNA65403 for A. macleodii DE1 (AltDE1), and PRJNA175354 for the metagenomic fosmids.
Results
An aliquot from a 200 l sample of South Adriatic seawater was used to inoculate marine agar plates, and 125 colonies were isolated (fig. 1). The isolates were characterized by 16S rRNA gene PCR amplification and sequencing, and 31 (25%) were classified as members of the species A. macleodii (more than 97% 16S rRNA similarity to the type strain). One of them (AltDE) was previously selected for genome sequencing (Ivars-Martinez et al. 2008b). Here, three more A. macleodii strains from the same set of deep Adriatic isolates have been sequenced at low coverage (10-fold) (see supplementary table S1, Supplementary Material online, for sequencing details). Two of them, AltDE2 and AltDE3, appeared to be identical to AltDE (even at the nucleotide level) and were not studied further (data not shown). The fact that three of four genomes sequenced turned out to be identical is already remarkable and points toward a low clonal diversity present in the sample. However, the representational bias of pure culture could be increasing the recovery of the single clone represented by AltDE, particularly when all the isolates were retrieved on a single culture medium.
The third strain sequenced here, AltDE1, showed several synteny discontinuities and was selected to be fully sequenced and assembled (table 1). DNA was isolated from the remaining sample (see Materials and Methods) and was used to generate a large fosmid library, AD1000 (38,704 clones) (Martin-Cuadrado et al. 2008). A large number of fosmids from this library (∼8,000) have been end sequenced (data not shown) and 3.5% could be assigned to A. macleodii by similarity to the available genomes (Ivars-Martinez et al. 2008b; López-Pérez et al. 2012) (supplementary fig. S1, Supplementary Material online). Finally, from 245 fosmids that could be assigned to environmental A. macleodii (eA.macleodii) by a strict similarity threshold (see Materials and Methods), a total of 161 were successfully sequenced, assembled to a length larger than 10 kb, and analyzed. These sequences amount to approximately 8.2 Mb and, together with the two different genomes of the strains from this single sample (AltDE and AltDE1), allow us to analyze the local diversity of cellular lineages of this specific population.
Table 1.
General Features of AltDE and AltDE1 Replicons
| Chr. AltDE | Chr. AltDE1 | Plasmid pAMDE1 | |
|---|---|---|---|
| Size (bp) | 4,480,937 | 4,643,846 | 303,282 |
| G+C (%) | 44.82 | 44.86 | 41.4 |
| Contigs | 1 | 1 | 1 |
| Total ORFs | 4,389 | 4,432 | 316 |
| Function assigned | 4,028 | 3,900 | 113 |
| Hypothetical proteins | 361 | 532 | 203 |
| Shared genes | 3,631 | 3,631 | — |
| Exclusive genes | 758 | 801 | — |
| rRNA operons | 5 | 5 | — |
| tRNA | 63 | 66 | — |
| ANI | — | 98.51 | — |
Note.—Chr., chromosome (Konstantinidis and Tiedje 2005).
Variation along the Core Genome
The ANI over the shared genes (3,631) between AltDE and AltDE1 was 98.51%, illustrating the high level of similarity between the two isolates (table 1 and fig. 2). The average nonsynonymous (dN) to synonymous (dS) substitution rates for this set of genes was 0.1719. This figure is in the usual range found for other couples of strains belonging to the same species (e.g., Escherichia coli 0.072, Helicobacter pylori 0.188, Neisseria meningitidis 0.158, and Salinibacter ruber 0.125) (Jordan et al. 2002b; Pena et al. 2010; Martincorena et al. 2012). One hundred seven genes had dN/dS values >1, which indicates that they are under positive selection (supplementary fig. S2, Supplementary Material online, the complete gene list can be found in supplementary table S2, Supplementary Material online) (Jordan et al. 2002a). No clear pattern was discerned from their annotation. Some housekeeping genes, such as DNA polymerase III delta subunit gene, were paralogous copies and could be undergoing a process of physiological specialization (Sanchez-Perez et al. 2008).
Fig. 2.—

Comparison of Alteromonas macleodii AltDE and AltDE1 genomes and their metagenomic fosmids. Middle panel: Genomic islands (GI) found in only some of the strains are indicated as red rectangles and numbered as in supplementary table S5, Supplementary Material online. Flexible genomic islands (fGIs), which are present in both genomes albeit containing different genes, are shown in black connecting the genome plots. The upper and lower panels indicate the location and similarity of the metagenomic fosmids (eAmacleodii) to each genome (see inset). Location of tRNAs and rRNA genes of each genome is also indicated.
As it is presented in table 2 and figure 3, most fosmids showed near 100% identity along the complete sequence to AltDE or AltDE1 genomes, 22 (14%) and 61 (38%), respectively (see complete fosmid annotation in supplementary table S3, Supplementary Material online). These results suggest that 1) the different clonal lineages are not equally represented in this environment and 2) that the number of abundant clonal lineages is not very high, because only two clones account for more than half of the eAmacleodii metagenomic fosmids retrieved. On the other hand, 76 (47.2%) fosmids could not be assigned to either genome at this high level of similarity although they clearly belong to the deep ecotype clade (95–99% similarity to either or both strains). In general, those fosmids have fewer SNPs compared with AltDE1 than with AltDE (fig. 3). This shows that there are additional clonal lineages besides the ones represented by AltDE and AltDE1. Actually, evidence from the hypervariable genomic islands indicates that there are at least four different clonal lineages (see later). Two additional fully sequenced fosmids (AD1000-357-C11 and AD1000-G14-C7, see supplementary table S3, Supplementary Material online) showed high similarity (98–99%) to Alteromonas isolates from different origins, specifically to 673 (López-Pérez et al. 2012) and SN2, respectively (Math et al. 2012).
Table 2.
eAmacleodii Fosmids Classification
| %id | Number of Fosmids (No. of Synteny Lost by GI, No. of Synteny Lost by Insertion Element) |
|---|---|
| Syntenic | |
| >99% to AltDE1 | 61 |
| >99% to AltDE2 | 22 |
| 98–99% to Alt673 (López-Pérez et al. 2012) | 1 |
| 98–99% to Alteromonas sp. SN2 (Math et al. 2012) | 1 |
| 98–99% both AltDE1/AltDE | 21 |
| Nonsyntenic | |
| >99% to AltDE1 | 29 (6, 5) |
| >99% to AltDE2 | 7 (3, 1) |
| 95–99% both AltDE1/AltDE | 19 (5, 11) |
Fig. 3.—
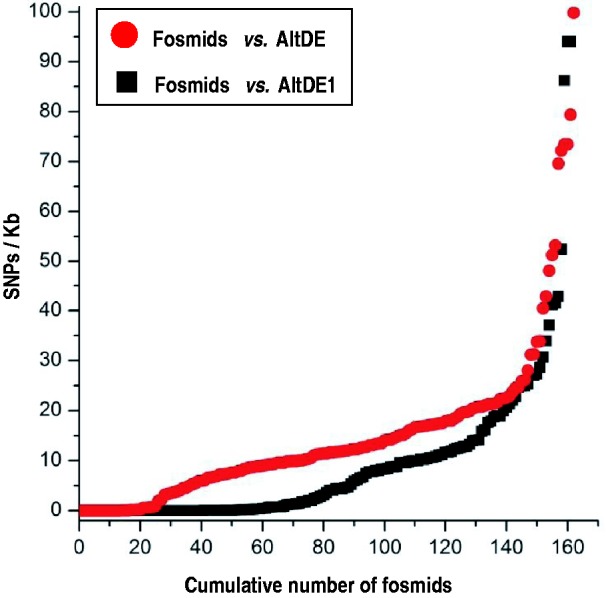
Number of fosmids containing the indicated number of single-nucleotide polymorphisms (SNPs) per kilobase of sequence when compared with the strain genomes.
The metagenomic fosmids covered a significant fraction of the AltDE and AltDE1 core genomes (52.4% and 52.88%, respectively, and also some of the genomic islands (23 fosmids). In these flexible areas of the genomes, we observed fosmids with nearly identical copies of the genomic island versions found in AltDE (two fosmids) and AltDE1 (six fosmids). Considering our data set size, the capture of almost identical versions of these hypervariable regions confirms the existence of a limited number of lineages. Among the 76 fosmids that are not identical to either of the isolated strains, 31 had the synteny lost by the insertion of mobile elements, that is, IS element or integrases genes, especially when the fosmid mapped near a genomic island.
The presence of several overlapping regions allowed us to analyze recombination events among the genomes and the fosmids sequenced (supplementary fig. S3, Supplementary Material online). From a total of 78 local alignments between the genomes and one or more fosmids, 28 indicated recombination events distributed among 56 different fosmids. Half of the recombined segments were shorter than 3 kb and less than 6% were larger than 10 kb (more details about recombination sites can be found in supplementary table S4, Supplementary Material online). It was remarkable that, among the 234 genes in AltDE and 218 in AltDE1 that were associated to recombination events, more than half (59.7% and 57.8%, respectively) localized near or inside the genomic islands (see later) (supplementary fig. S3, Supplementary Material online). Functional classification of the recombinant genes indicated that different categories of transporters were over-represented (supplementary fig. S4, Supplementary Material online). In some cases, multiple recombination events were concentrated within the same gene cluster; for example, an ABC transport gene system for iron/hemin group, a phosphate (pstABSC) and phosphonate transporter gene, an heme exporter gene system (ccmA-F), and also other for the transport of biopolymers (tonB-exbBD). Other transporters that displayed allelic substitutions were three iron outer membrane receptor genes and other for the transport of ammonium. Some of the recombination events with highest probability affected TonB receptor genes (supplementary fig. S5, Supplementary Material online). TonB receptors are involved in the transport of several substrates in marine bacteria (Blanvillain et al. 2007), and they have been described as experiencing recombination-driven phase variation in Mannheimia haemolytica A1 (Graham and Lo 2002). Interestingly, TonB proteins also participate in the adsorption of some phages to the host cells (Hantke and Braun 1978), and also the infection of some phages is TonB dependent, for example, phage H8 of Salmonella enterica (Rabsch et al. 2007). Earlier comparisons of different A. macleodii strains isolated from different geographical points (Lopez-Lopez et al. 2005; Ivars-Martinez et al. 2008a) found recombination among several distinct housekeeping genes. Here, some recombination events with geographically distant strains could be detected as well, for example, fosmid AD1000-52-H12 presented a fragment containing a czcABC system (a cation efflux system of more than 8 kb) only found in the surface 673 strain isolated from the English Channel (López-Pérez et al. 2012), whereas the rest of the sequence had 99% identity with AltDE. Something similar was found in two other fosmids (AD1000-G9-16 and AD1000-249-G9), but in this case, the region exchanged was smaller (3.5 kb and 4.9 kb, respectively).
Flexible Genomic Islands
Most of the previous adaptive islands identified when comparing AltDE with a distant isolate (ATCC 27126, only 84% ANI) (Ivars-Martinez et al. 2008b) represented still nonshared regions. Some of these islands are present in most (or all) of the available strain genomes but containing different genes, that is, they are in the same genomic context and code for the same type of function or structure, but the genes are only distantly related, if at all. For the sake of clarity, we will designate these genomic islands found in many strains but containing different genes "flexible Genomic Islands" (fGIs). fGIs are often linked to phage susceptibility by coding for the synthesis of the exposed structures of the cell (Pasic et al. 2009; Rodriguez-Valera et al. 2009). Their analysis is particularly relevant to understand the biological role of the diversity found among concurrent lineages and how it is generated. Besides, because the gene content of these islands is so markedly different in the different clonal lineages, they can be used to estimate the numbers of different clones present in the sample.
Lipopolysaccharide O-chain (fGI6)
The cluster of genes coding for the synthesis of the polysaccharide O-chain, classically known as the O-antigen, is one of the most diverse gene clusters found in Gram-negative bacteria. The O-chain is also the recognition target of many phages and provides infection specificity (Hooton et al. 2011). As expected, these areas of the genomes of AltDE and AltDE1 are not shared and constitute fGI6 (fig. 2). From the comparison between the metagenomic fosmids and the two genomes, we have found evidence that within this sample, very close relatives to AltDE (within the 98% similarity over the conserved genes) contained at least four total or partially different O-chain gene clusters (fig. 4). Thus, although the conserved genes at one end of fosmids AD1000-35-C02 and AD1000-G18-C11 were close to be 100% identical to AltDE1, they presented a totally or partially different O-chain cluster (fig. 4). Therefore, there are at least two additional clonal lineages at the level of this fGI. However, we also found fosmids that contained almost identical versions to the ones in the sequenced genomes. Specifically, two fosmids, AD1000-19-C11 and AD1000-G12-C2 were 99% identical to the O-chain cluster found in AltDE (except for an IS element). Another fosmid, AD1000-44-F09, was 100% identical to the cluster found in AltDE1. Therefore, as seen for other fGIs (see later), their conservation is similar to that of the core genome within the clonal lineages in which they reside. None of the new versions of the O-chain gene cluster contained in the fosmids had homologs in any of the other A. macleodii genomes or in any other known bacterial genome.
Fig. 4.—

Representation of the flexible genomic island fGI6, involved in the biosynthesis of the O-chain lipopolysaccharide (LPS), in genomes and metagenomic fosmids. (A) fGI6 in the genomes AltDE and AltDE1. (B) Metagenomic fosmids AD1000-35-C02 and AD1000-G18-C11 with different versions for the O-chain gene cluster. Syntenic regions between each fosmid and AltDE1 are highlighted in blue or yellow to identify the overlaps of each fosmid with AltDE1 genome. Numbers under the genes indicate synonymous (green) and nonsynonymous (red) substitutions.
The main differences found among the versions of this fGI are the presence of a different set of glycosyl-transferases (GT) genes involved in the transfer of sugars onto an UndP lipid carrier to form the O-unit (Samuel and Reeves 2003). The similarity among the GTs, mainly of I or II family types, is very low (or undetectable), and they are dispersed along the O-chain gene cluster (fig. 4). The high number of GTs genes found in AltDE (18) is indeed remarkable. In contrast, AltDE1 has only three GTs genes, suggesting that sugar composition may be much simpler in this strain. Among the fosmids, AD1000-35-C02 presents a new set of GTs genes (five near the 3′-terminus). Other genes that were strain specific coded modifiers of the O-chain such as O-acetyl-transferases (modify the O-chain after polymerization), sulfotransferases, or aminotransferases. We also found different versions for the lipopolysaccharide biosynthesis protein WalW that removes N-linked or O-linked acetyl groups from cell wall polysaccharides (Samuel and Reeves 2003). The O-chain processing enzymes are a flippase (Wzx), (which translocates the UndP-linked O-unit onto the periplasmic face of the inner membrane), the O-chain polymerase (Wzy), and the O-chain length determinant protein (Wzz). As has been described in other species (Samuel and Reeves 2003), the O-chain length determinant proteins (Wzz) found in the genomes share only 63% similarity. If the O-unit varies, the Wzx and the Wzy proteins also differ in specificity. We found no similarity among the Wzx protein versions of both genomes and the one found in AD1000-35-C02. Also, very little similarity was found between the polymerases Wzy (<30%). In summary, there is ample evidence indicating that the O-chains of the lineages represented by the metagenomic fosmid and the two genomes are quite different. This could change the range of phages to which they are sensitive, which supports the C-D model (Rodriguez-Valera et al. 2009).
In AltDE and AltDE1 genomes, there was another nonconserved region, which could be also implicated in the modification of the sugars of the O-chain (fGI9 in fig. 2). In AltDE, this region codes for five new GTs together with sulphatases and sulfotransferases and, in AltDE1, four more nonconserved GTs. One notable difference in AltDE1 was the presence of four genes coding for proteins with aspartic acid-rich motifs annotated as thrombospondin type 3 (TSP-3). TSPs are multimeric multidomain cell surface glycoproteins (Kvansakul et al. 2004). Comparing this region with other Alteromonas genomes, we found a high similarity of this area of AltDE1 with Alteromonas sp. SN2, although the synteny is only partially conserved. The presence in the same sample of a very close relative to Alteromonas sp. SN2 is also supported by the presence of fosmid AD1000-G14-C7, which presents a 97.66% of similarity along more than 38 kb. The similarity found in this part of the genomes between AltDE1 and SN2 strains suggests that horizontal gene transfer and recombination events (perhaps mediated by the repetitions found in the TSP-3) happen between these distantly related species.
Exopolysaccharide (fGI5)
Exopolysaccharides are long sugar polymers that can be attached to the cell surface or released as extracellular slime in the surroundings of the cell (Knoshaug et al. 2000), protecting the cells against osmotic stress, antibiotics, desiccation, or toxic compounds (Cochran et al. 2000; Bramhachari and Dubey 2006). Their biosynthesis requires the polymerization of nucleoside diphospho-sugar precursors and the transport of the polymer across the cytoplasmic membrane and the cell wall. Both AltDE and AltDE1 have a gene cluster that appears to code for exocellular polysaccharide (EPS) biosynthesis (supplementary fig. S6, Supplementary Material online). The implication of this cluster in the production of an EPS or biofilm is supported by experimental evidence in Pseudoalteromonas atlantica. In this microbe, the interruption of a glycosyltransferase gene by an IS element in a similar cluster of genes changes the cell phenotype, losing the capacity to produce the extracellular polysaccharide (Higgins et al. 2007, 2009). In both AltDE and AltDE1, this cluster is flanked by two tRNA synthetases: a threonyl-tRNA synthetase at the 5′-terminus and a phenylalanyl-tRNA synthetase (alpha and beta subunits) at the 3′-end. These tRNA synthetases are found adjacent to this cluster in many other genomes (including close relatives to Alteromonas such as Glaciecola agarilytica 4H-3-7+YE-5 and P. atlantica). This may suggest that the complete EPS cluster was inserted in an ancestral lineage before diversifying later by sequential recombination events, as the tRNA-synthetases are well-known recombination hot spots.
This cluster of genes could be divided in two parts: The first one (shaded yellow in supplementary fig. S6, Supplementary Material online) is conserved between the A. macleodii strains and other Alteromonadaceae such as Pseudoalteromonas (P. atlantica spp. SM9913 and P. haloplanktis) and G. agarilytica 4H-3-7+YE-5. It is also partially present in many Vibrios. It contains the eps genes (epsBUEFCDC), a very well-conserved O-chain polymerase gene (marked with a black spot in supplementary fig. S6, Supplementary Material online, 94% similarity between both strains) and other gene for the fusion-protein diguanylate cyclase-phosphodiesterase, which may act as the transcriptional regulator (Kirillina et al. 2004). The nonconserved region contains genes for glycosyltransferases, O-chain flippases (Wzx), O-chain polymerases (Wzy), and acyltransferases. We found also three fosmids containing the complete (or part) of the EPS cluster (supplementary fig. S6, Supplementary Material online). Two of them (AD1000-5-F10 and AD1000-9-F08) are 100% identical to AltDE1, and the third one (AD1000-21-D04) is identical to AltDE, with the exception of the two GTs genes that are absent in the fosmid (supplementary fig. S6, Supplementary Material online). In total, three versions of this fGI were found. Again, all these differences found in the EPS suggest diversity in the exposed cell structures that might act as phage targets (Sutherland 1971).
Flagellum (fGI4)
Both strains AltDE and AltDE1 are motile, using a polar flagellum. Accordingly, a large flagellar gene cluster was found with the canonical organization of nonenteric gamma proteobacterial flagellar gene clusters (Liu and Ochman 2007) (fig. 5). The genes found indicate that the flagellum in these bacteria is of the Pseudomonas aeruginosa type with a single major flagellin (FliC) (Logan 2006).
Fig. 5.—

Flexible genomic island fGI4 involved in the biosynthesis and assembly of flagellar components. The Alteromonas macleodii strains AltDE, AltDE1 genomes, and the overlapping fosmid AD1000-9-A03 are shown. Hypervariable regions associated with glycosylation of flagellin in the AltDE1 genome are highlighted in pink. Numbers below the genes indicate synonymous (green) and nonsynonymous substitutions (red) between the two genomes. The orange inset in the figure shows the alignment among the three versions of flaK genes. Key sensory and DNA-binding regions are highlighted in boxes.
The varying degrees of conservation throughout this large cluster illustrate the overall genomic variation found among these concurrent clones. The genomes are perfectly syntenic in this region except for a large divergent segment in the middle of the cluster (fig. 5), divergent both with respect to decreased similarities among functionally identical genes (e.g., flaA) and also with respect to the presence of genes specific in each. For example, a small glycosylation island (a gene cluster for several glycosyltransferases) was found only in AltDE1 and not in AltDE or the metagenomic fosmid AD1000-9-A03. The absence of these genes, and the insertion of several IS elements in the corresponding region, in AltDE indicate that these functions may be somewhat defective in this genome. This glycosylation cluster in the AltDE1 genome is actually very similar to that seen in Pse. aeruginosa PAK that is involved in the glycosylation of FliC and is only found in a subpopulation of Pse. aeruginosa strains (Arora et al. 2001). Most of the exposed flagellar components are located near the glycosylation island, and many showed large sequence variations between the two strains. For example, nucleotide similarity between both genomes was <60% in the fliC genes, which code for the major protein component of the exposed flagellar body. The two subunits present in AltDE1 also have low similarity (52%) between them. As suggested by the genomic variability found in the flagellar cluster, differences in motility were confirmed experimentally with AltDE1 displaying greater motility than AltDE (2.1 cm vs. 0.98 cm) in 0.35% marine agar (supplementary fig. S7, Supplementary Material online).
So far, the variable regions found in the flagellar cluster are consistent with the idea that these variations lead to different phage susceptibilities. Flagellatropic phages are known to reversibly bind to helical grooves on the bacterial flagellum and use the rotation of the flagellum to spiral toward the cell surface (Samuel et al. 1999). Any change in flagellin structure or glycosylation would alter phage susceptibility (Koskella et al. 2011). Interestingly, we found an intermediate level of variation in genes involved in the regulation of flagellum synthesis. Assembly of the flagellum is a highly ordered process, and temporal regulation of flagellar genes is achieved through sophisticated regulatory networks to coordinate gene expression in a hierarchical manner (Smith and Hoover 2009; Anderson et al. 2010). The regulatory genes found in both strains and the metagenomic fosmids are most similar to the system described in Vibrio parahaemolyticus. In this regulatory system, the product of gene flaK is the single class I gene product, like flrA in other Vibrio species or fleQ in Pseudomonas, and activates σ54-dependent transcription of class II genes, which encode components of the MS ring-switch-export apparatus and the two-component system FlaLM (Kim and McCarter 2004). Subsequently, phosphorylated FlaM activates σ 54-dependent transcription of class III genes, which encode the basal body hook and the flagellins (Smith and Hoover 2009; Syed et al. 2009). flaK of both A. macleodii strains is located next to the glycosylation island (fig. 5) and was remarkably variable (81% similarity). Even more remarkable was the clustering of nonsynonymous substitutions at the regulatory and DNA-binding domains of the FlaK gene (fig. 5). The regulatory domain responds to signals such as those produced by heat or osmotic shock, quorum sensing, and solid surface signalling (Smith and Hoover 2009; Anderson et al. 2010). The different sequences found in both strains indicate a different response to these or other stimuli. The DNA-binding domain recognizes upstream activation sequences that are found in the secondary regulators and many other flagellar genes enhancing their transcription. Because we do not know the recognition sequences of σ 54-dependent activation in Alteromonas, we cannot predict what will be the effects but a significant difference in flagellar regulation between the two strains is a plausible prediction. Similarly, differences were detected in FlaL gene (data not shown), the first of the two component system involved in secondary regulation, also in the region involved in signal recognition (in this specific case intracellular) (Kim and McCarter 2004). The FlaL/FlaM sensor system is involved in regulation at a more refined level and potentially important for environmental sensing (Kim and McCarter 2004). Overall, the sequence variability observed here provides evidence for differences in flagellar regulation between the closely related lineages of A. macleodii (inset of fig. 5) and indicate that they could exhibit different responses to environmental signals under varying conditions. We identified two metagenomic fosmids that overlapped this region. One of them, AD1000-39-B06, is 100% identical to AltDE1. Also AD1000-9-A03 (shown in fig. 5) is nearly identical to AltDE1 in the conserved region, but beyond the flaK subunit, the few genes remaining including the fliC located upstream were more similar to AltDE.
CRISPRs (fGI7)
CRISPRs consist of multiple copies of a short repeat sequence (typically 25–40 nucleotides) separated by similarly sized variable sequences that are derived from invaders such as viruses and conjugative plasmids (Pourcel et al. 2005; Sorek et al. 2008; Tyson and Banfield 2008). These components constitute a putative prokaryotic RNA-interference-based immune system (Makarova et al. 2006; Sorek et al. 2008) protecting against bacteriophages or plasmids (Mojica et al. 2005). In fGI7, AltDE contains a complete set of CRISPR and CRISPR-associated sequence (Cas) proteins and a complete urease gene cluster including the transporter and the urea degradation genes (Ivars-Martinez et al. 2008b). Urea reduction was also demonstrated phenotypically. In our fosmid collection, we found one fosmid (AD1000-11-D07) that was 100% identical to this region of the AltDE strain. All these genes are absent in AltDE1, instead there are several components of the mer operon, that confer mercury resistance to bacteria (merA, which codes for the mercuric reductase, merR and merT) (supplementary fig. S8, Supplementary Material online). Actually, we could not find any CRISPR system, neither complete nor remains, at any location within the AltDE1 genome. However, its numbers are higher, detracting in this case, from the importance of the CRISPR system as phage protection mechanism.
On the other hand, the CRISPR system found in AltDE indicates that, as could be expected, both strains share phages. Three of the 55 CRISPR spacers found in AltDE match the lambda-like prophage found in AltDE1 (see supplementary information, Supplementary Material online). The 32 nucleotides of the spacer 40 match the terminase small subunit at 100% identity, and spacers 2 and 43 match (although with 4 mismatches) two hypothetical proteins in the AltDE1 prophage (supplementary fig. S9, Supplementary Material online). A match between a single CRISPR spacer and a foreign DNA sequence or protospacer provides immunity (Deveau et al. 2010). Interestingly, fosmid AD1000-11-D07 contains the complete CRISPR locus of AltDE. Furthermore, the two strains sequenced at low coverage, AltDE2 and AltDE3, contained all the spacers present in AltDE (data not shown). This evidence indicates very close ancestry (Weinberger et al. 2012).
Metal Resistance (fGI2)
In a previous work (López-Pérez et al. 2012), the genome comparison of different geographical isolates of A. macleodii, including AltDE, showed a hypervariable region located next to the single Phe-tRNA containing a large cluster of genes that code for metal resistance and a hydrogenase cluster of genes. Because genomic islands are frequently located adjacent to tRNA genes, it is thought that the conserved sequence of tRNA genes facilitates their integration and excision (reviewed in Hacker and Kaper 2000). In AltDE and AltDE1, this region corresponds to fGI2, where successive insertions of a cluster of czcABC genes (encoding a heavy metal efflux pump important for resistance to cobalt, zinc and cadmium) were found. AltDE itself has three different clusters (highlighted with different geometric shapes in fig. 6), whereas AltDE1 has only two. In AltDE, there are remains of this tRNA gene that produces the insertion of the third set of czc genes not present in AltAD1. Both genomes contain a set of hydrogenase genes in this region (green genes in fig. 6). The hydrogenase activity of AltDE has been proven by heterologous expression and has been shown to be the most oxygen-resistant hydrogenase described presently (Weyman et al. 2011). We found several metagenomic fosmids identical to the genomes in this region (AD1000-49-F05 and AD1000-50-C08 are identical to AltDE1 and fosmid AD1000-G10-19 to AltDE) and also three other fosmids that presented a great variability. AD1000-42-A03, AD1000-G19-10, and AD1000-G15-18 contain one of the sets of the genes czc identical to both AltDE and AltDE1, but the adjacent genes are different. Fosmid AD1000-42-A03 appears to represent a simpler version of this region as its 5′-end is identical to AltDE but lacks two of the czc clusters. Furthermore, in fosmid AD1000-G15-18, we detected part of the Phe-tRNA gene (arrow in fig. 6) before a totally different region not found in the genomes, suggesting another integration event where the tRNA acted again as the insertion target. Finally, the 5′-end of fosmid AD1000-G19-10 contains 14 genes with no similarity to any Alteromonas including the final transposase. All these genes may have been acquired via horizontal gene transfer using the tRNA as an insertion target.
Fig. 6.—

Alignment of the metal–resistance-related region in Alteromonas macleodii strains AltDE, AltDE1, and overlapping metagenomic fosmids. Regions highlighted by geometric figures (square, diamond, or ellipse) show clusters of czc genes. The clusters indicated by the same geometric figure are identical in sequence. Bold arrows on AltDE and the fosmid AD1000-G15-18 indicate the location of the 3′-end of the tRNA gene that is duplicated and represents a hallmark of an integration event.
Discussion
The analysis of the fosmid ends of the deep Adriatic fosmid library indicates that A. macleodii cells belonging to the deep ecotype clade were quite abundant in this specific sample (3.5%). Although this is a relatively high cell density for this microbial species in a deep Mediterranean habitat (Martin-Cuadrado et al. 2007), recent evidence indicates that A. macleodii blooms under some circumstances (Quaiser et al. 2011; Smedile et al. 2012). To get our results in perspective, we have estimated the total number of A. macleodii cells present in the population studied here. Assuming a total bacterioplankton cell density of 105 cells per ml, 3.5% represents a total number for the 200 l studied here of only 109 A. macleodii cells. This is roughly equivalent to the number of cells present in a typical laboratory culture of about 100 ml at the end of the exponential phase. Within this restricted number, we have found evidence for at least two very different clones represented by isolates AltDE and AltDE1. The sequences of the fosmids show that both are very abundant adding up to 38% and 14% of the population, respectively. The remaining A. macleodii fosmids likely belong to different lineages that are as different as AltDE is from AltDE1. In support of this, we found evidence for the presence of at least two other clones of the deep ecotype from the existence of different O-chain biosynthetic clusters different from those found in the two genomes sequenced. Therefore, we could assume that there were at least four distinct clonal lineages at this level of divergence, that is, core genomes at over 98% similarity and very different flexible gene pools. For the sake of clarity, we will refer to these different lineages as "clonal frames" (Milkman and Bridges 1990). This terminology was coined in pregenomic times to describe bacterial lineages of common ancestry and clonal (asexual) descent but in which replacement of genome fragments by recombination, and drift by neutral genetic mutations, have occurred. Although we cannot estimate precisely the number of clonal frames present in this sample, the evidence indicates that there are at least four and, probably, not a much larger number, being significantly abundant. Supporting this estimation we found 1) a high numbers of fosmids with identical sequence to either of the two available strain genomes (plus two other complete genomes of strains that were identical to AltDE), 2) fosmids with identical sequences to the fGIs of the strain genomes, and 3) more than one identical overlapping sequences even though the maximum coverage found at any region of the genome was 4-fold.
The recovery of identical copies to most of the fGIs in the metagenomic fosmids indicates that they are probably maintained for significantly long periods within these clonal frames. The mechanism of variation that generates fGIs is clearly different from that typical of the core, and the presence in some islands of tRNAs, IS elements, and other conserved sequences indicates the possible facilitation of recombination with distant microbes (i.e., horizontal gene transfer [HGT]). However, this mechanism is not essential to justify the genomic diversity found. Our results suggest that there might be an ample pool of these genomic clusters within a single species (or in this case within the “deep ecotype” of A. macleodii) to provide the observed variation. In support of this premise, we noted that some fGIs do not show typical HGT genomic parameters, such as different GC content (supplementary table S5, Supplementary Material online). Variation in the core was also based on mutation and recombination, albeit of smaller fragments (hundreds of nucleotides rather than kilobases), so there might not be major differences in this respect. The high frequency of recombination found in the TonB receptor genes highlights how selection concentrates successful recombination events in the regions where they are more needed (in the case of TonB receptors to provide extra transport capabilities). Thus, the main difference between the fGIs and the core might reside in the selection pressures that act on them. The flagellum regions described here illustrates this point very nicely. The glycosylation region is rich in IS elements and could be a hot spot for recombination, receiving different sets of glycosylation machineries found in the different clonal frames. On the other hand, the presence within the same gene cluster of regulatory regions that are only different at the key sensory and DNA-binding regions indicates subtle selection pressures that act at the sequence level by mutation or highly targeted recombination.
The differences between the two clonal frames represented by the complete genomes illustrate very well the kind of complementary features that concurrent clones may possess. There is ample evidence, as with any other similar set of highly related microbes, that they differ in surface structures and consequently in phage susceptibility. We proposed recently that this derives from the need to share phage predatory pressure (Rodriguez-Valera et al. 2009), and several recent reports support this proposal (Rodriguez-Brito et al. 2010; Avrani et al. 2011; Marston et al. 2012). Even with four equally abundant clones, the population density calculated for each in this habitat would be in the order of 1,000 cells per ml (see earlier). However, if these microbes live attached to particles as has been proposed (Ivars-Martinez et al. 2008b), the overall population density is largely meaningless, and particles would be hotspots of high cell density, very susceptible to massive phage lysis. Further, as predicted in Rodriguez-Valera et al. (2009), both strains show remarkable evidence of differential niche adaptation, with potential to use different substrates, behavior (motility), and even production of secondary metabolites (see supplementary information, Supplementary Material online), that might be involved in antagonistic relationships and competition with other members of the natural assemblage. The fact that these two significantly divergent clonal frames coexist in this relatively small population and in similar amounts supports the constant diversity model (Rodriguez-Valera et al. 2009). This is important to highlight in view of the common trend to interpret the differences found in genomes of environmentally relevant microbes as always reflecting the adaptation of the strain represented by the sequenced genome to the place or habitat of isolation. It is often the case that many clones are found simultaneously at the same place and time and that they are actually equipped to complement each other rather than just compete for the resources.
Our results are also relevant to genome reconstructions from metagenomic data sets. These methodologies require a very good understanding of the clonal diversity that can be found in a single sample. Our results indicate that large segments of the core genome of A. macleodii in the Mediterranean sample analyzed here could be assembled from metagenomic reads but to get a reliable assembly of the many hypervariable regions and to avoid chimera formation over the recombination hotspots would require an extremely high coverage and would still be largely unreliable.
Supplementary Material
Supplementary information, tables S1–S5, and figures S1–S10 are available at Genome Biology and Evolution online (http://gbe.oxfordjournals.org/).
Acknowledgments
This work was supported by projects MAGYK (BIO2008-02444), MICROGEN (Programa CONSOLIDER-INGENIO 2010 CDS2009-00006), CGL2009-12651-C02-01 from the Spanish Ministerio de Ciencia e Innovación, DIMEGEN (PROMETEO/2010/089), ACOMP/2009/155 from the Generalitat Valenciana, MaCuMBA from European Community (EC) (Ref. FP7-KBBE-2012-6-311975), and French ANR (ANR-08-GENM-024-001). FEDER funds supported this project. I.G-H. was supported by MAGYK from Ministerio de Ciencia e Innovación. A-B.M-C. was supported by CONSOLIDER-INGENIO 2010 CDS2009-00006. R.G. was supported by a Juan de la Cierva scholarship from the Spanish Ministerio de Ciencia e Innovación.
Literature Cited
- Albertsen M, Hansen LB, Saunders AM, Nielsen PH, Nielsen KL. A metagenome of a full-scale microbial community carrying out enhanced biological phosphorus removal. ISME J. 2012;6(6):1094–1106. doi: 10.1038/ismej.2011.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JK, Smith TG, Hoover TR. Sense and sensibility: flagellum-mediated gene regulation. Trends Microbiol. 2010;18(1):30–37. doi: 10.1016/j.tim.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora SK, Bangera M, Lory S, Ramphal R. A genomic island in Pseudomonas aeruginosa carries the determinants of flagellin glycosylation. Proc Natl Acad Sci U S A. 2001;98(16):9342–9347. doi: 10.1073/pnas.161249198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood KC, Schneider LK, Ryan FJ. Periodic selection in Escherichia coli. Proc Natl Acad Sci U S A. 1951;37(3):146–155. doi: 10.1073/pnas.37.3.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avrani S, Wurtzel O, Sharon I, Sorek R, Lindell D. Genomic island variability facilitates Prochlorococcus-virus coexistence. Nature. 2011;474(7353):604–608. doi: 10.1038/nature10172. [DOI] [PubMed] [Google Scholar]
- Blanvillain S, et al. Plant carbohydrate scavenging through tonB-dependent receptors: a feature shared by phytopathogenic and aquatic bacteria. PLoS One. 2007;2(2):e224. doi: 10.1371/journal.pone.0000224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramhachari PV, Dubey SK. Isolation and characterization of exopolysaccharide produced by Vibrio harveyi strain VB23. Lett Appl Microbiol. 2006;43(5):571–577. doi: 10.1111/j.1472-765X.2006.01967.x. [DOI] [PubMed] [Google Scholar]
- Cadillo-Quiroz H, et al. Patterns of gene flow define species of thermophilic Archaea. PLoS Biol. 2012;10(2):e1001265. doi: 10.1371/journal.pbio.1001265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver TJ, et al. ACT: the artemis comparison tool. Bioinformatics. 2005;21(16):3422–3423. doi: 10.1093/bioinformatics/bti553. [DOI] [PubMed] [Google Scholar]
- Cochran WL, McFeters GA, Stewart PS. Reduced susceptibility of thin Pseudomonas aeruginosa biofilms to hydrogen peroxide and monochloramine. J Appl Microbiol. 2000;88(1):22–30. doi: 10.1046/j.1365-2672.2000.00825.x. [DOI] [PubMed] [Google Scholar]
- Cohan FM. What are bacterial species? Annu Rev Microbiol. 2002;56:457–487. doi: 10.1146/annurev.micro.56.012302.160634. [DOI] [PubMed] [Google Scholar]
- Coleman ML, et al. Genomic islands and the ecology and evolution of Prochlorococcus. Science. 2006;311(5768):1768–1770. doi: 10.1126/science.1122050. [DOI] [PubMed] [Google Scholar]
- Cuadros-Orellana S, et al. Genomic plasticity in prokaryotes: the case of the square haloarchaeon. ISME J. 2007;1(3):235–245. doi: 10.1038/ismej.2007.35. [DOI] [PubMed] [Google Scholar]
- Christian W. Urea descomposition as a means of diferentiating Proteus and paracolon cultures from each other and from Salmonella and Shigella types. J Bacteriol. 1946;52:461–466. doi: 10.1128/jb.52.4.461-466.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AC, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14(7):1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveau H, Garneau JE, Moineau S. CRISPR/Cas system and its role in phage-bacteria interactions. Annu Rev Microbiol. 2010;64:475–493. doi: 10.1146/annurev.micro.112408.134123. [DOI] [PubMed] [Google Scholar]
- Doolittle WF. Population genomics: how bacterial species form and why they don't exist. Curr Biol. 2012;22(11):R451–R453. doi: 10.1016/j.cub.2012.04.034. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghai R, et al. Metagenome of the Mediterranenan deep chlorophyl maximum stuidied by direct and fomid library 454 pyrosequencing. ISME J. 2010;4:1154–1166. doi: 10.1038/ismej.2010.44. [DOI] [PubMed] [Google Scholar]
- Graham MR, Lo RY. A putative iron-regulated TonB-dependent receptor of Mannheimia (Pasteurella) haemolytica A1: possible mechanism for phase variation. Vet Microbiol. 2002;84(1–2):53–67. doi: 10.1016/s0378-1135(01)00415-1. [DOI] [PubMed] [Google Scholar]
- Hacker J, Kaper JB. Pathogenicity islands and the evolution of microbes. Annu Rev Microbiol. 2000;54:641–679. doi: 10.1146/annurev.micro.54.1.641. [DOI] [PubMed] [Google Scholar]
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- Hantke K, Braun V. Functional interaction of the tonA/tonB receptor system in Escherichia coli. J Bacteriol. 1978;135(1):190–197. doi: 10.1128/jb.135.1.190-197.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins BP, Carpenter CD, Karls AC. Chromosomal context directs high-frequency precise excision of IS492 in Pseudoalteromonas atlantica. Proc Natl Acad Sci U S A. 2007;104(6):1901–1906. doi: 10.1073/pnas.0608633104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins BP, Popkowski AC, Caruana PR, Karls AC. Site-specific insertion of IS492 in Pseudoalteromonas atlantica. J Bacteriol. 2009;191(20):6408–6414. doi: 10.1128/JB.00771-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hightower RC, Metge DW, Santi DV. Plasmid migration using orthogonal-field-alternation gel electrophoresis. Nucleic Acids Res. 1987;15(20):8387–8398. doi: 10.1093/nar/15.20.8387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooton SP, Timms AR, Rowsell J, Wilson R, Connerton IF. Salmonella typhimurium-specific bacteriophage PhiSH19 and the origins of species specificity in the Vi01-like phage family. Virol J. 2011;8:498. doi: 10.1186/1743-422X-8-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivars-Martinez E, et al. Biogeography of the ubiquitous marine bacterium Alteromonas macleodii determined by multi-locus sequence analysis (MLSA) Mol Ecol. 2008a;17:4092–4106. doi: 10.1111/j.1365-294x.2008.03883.x. [DOI] [PubMed] [Google Scholar]
- Ivars-Martinez E, et al. Comparative genomics of two ecotypes of the marine planktonic copiotroph Alteromonas macleodii suggests alternative lifestyles associated with different kinds of particulate organic matter. ISME J. 2008b;2(12):1194–1212. doi: 10.1038/ismej.2008.74. [DOI] [PubMed] [Google Scholar]
- Iverson V, et al. Untangling genomes from metagenomes: revealing an uncultured class of marine Euryarchaeota. Science. 2012;335(6068):587–590. doi: 10.1126/science.1212665. [DOI] [PubMed] [Google Scholar]
- Jordan IK, Rogozin IB, Wolf YI, Koonin EV. Essential genes are more evolutionarily conserved than are nonessential genes in bacteria. Genome Res. 2002a;12(6):962–968. doi: 10.1101/gr.87702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan IK, Rogozin IB, Wolf YI, Koonin EV. Microevolutionary genomics of bacteria. Theor Popul Biol. 2002b;61(4):435–447. doi: 10.1006/tpbi.2002.1588. [DOI] [PubMed] [Google Scholar]
- Kim YK, McCarter LL. Cross-regulation in Vibrio parahaemolyticus: compensatory activation of polar flagellar genes by the lateral flagellar regulator LafK. J Bacteriol. 2004;186(12):4014–4018. doi: 10.1128/JB.186.12.4014-4018.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirillina O, Fetherston JD, Bobrov AG, Abney J, Perry RD. HmsP, a putative phosphodiesterase, and HmsT, a putative diguanylate cyclase, control Hms-dependent biofilm formation in Yersinia pestis. Mol Microbiol. 2004;54(1):75–88. doi: 10.1111/j.1365-2958.2004.04253.x. [DOI] [PubMed] [Google Scholar]
- Knoshaug EP, Ahlgren JA, Trempy JE. Growth associated exopolysaccharide expression in Lactococcus lactis subspecies cremoris Ropy352. J Dairy Sci. 2000;83(4):633–640. doi: 10.3168/jds.S0022-0302(00)74923-X. [DOI] [PubMed] [Google Scholar]
- Konstantinidis KT, Tiedje JM. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci U S A. 2005;102(7):2567–2572. doi: 10.1073/pnas.0409727102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koskella B, Taylor TB, Bates J, Buckling A. Using experimental evolution to explore natural patterns between bacterial motility and resistance to bacteriophages. ISME J. 2011;5(11):1809–1817. doi: 10.1038/ismej.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvansakul M, Adams JC, Hohenester E. Structure of a thrombospondin C-terminal fragment reveals a novel calcium core in the type 3 repeats. EMBO J. 2004;23(6):1223–1233. doi: 10.1038/sj.emboj.7600166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legault BA, et al. Environmental genomics of "Haloquadratum walsbyi" in a saltern crystallizer indicates a large pool of accessory genes in an otherwise coherent species. BMC Genomics. 2006;7(1):171. doi: 10.1186/1471-2164-7-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Ochman H. Stepwise formation of the bacterial flagellar system. Proc Natl Acad Sci U S A. 2007;104(17):7116–7121. doi: 10.1073/pnas.0700266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan SM. Flagellar glycosylation—a new component of the motility repertoire? Microbiology. 2006;152(Pt 5):1249–1262. doi: 10.1099/mic.0.28735-0. [DOI] [PubMed] [Google Scholar]
- Lopez-Lopez A, Bartual SG, Stal L, Onyshchenko O, Rodriguez-Valera F. Genetic analysis of housekeeping genes reveals a deep-sea ecotype of Alteromonas macleodii in the Mediterranean Sea. Environ Microbiol. 2005;7(5):649–659. doi: 10.1111/j.1462-2920.2005.00733.x. [DOI] [PubMed] [Google Scholar]
- López-Pérez M, et al. Genomes of surface isolates of Alteromonas macleodii: the life of a widespread marine opportunistic copiotroph. Sci Rep. 2012;2:696. doi: 10.1038/srep00696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct. 2006;1:7. doi: 10.1186/1745-6150-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marston MF, et al. Rapid diversification of coevolving marine Synechococcus and a virus. Proc Natl Acad Sci U S A. 2012;109(12):4544–4549. doi: 10.1073/pnas.1120310109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Cuadrado AB, et al. Metagenomics of the deep Mediterranean, a warm bathypelagic habitat. PLoS One. 2007;2(9):e914. doi: 10.1371/journal.pone.0000914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Cuadrado AB, et al. Hindsight in the relative abundance, metabolic potential and genome dynamics of uncultivated marine archaea from comparative metagenomic analyses of bathypelagic plankton of different oceanic regions. ISME J. 2008;2(8):865–886. doi: 10.1038/ismej.2008.40. [DOI] [PubMed] [Google Scholar]
- Martin DP, et al. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics. 2010;26(19):2462–2463. doi: 10.1093/bioinformatics/btq467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena I, Seshasayee AS, Luscombe NM. Evidence of non-random mutation rates suggests an evolutionary risk management strategy. Nature. 2012;485(7396):95–98. doi: 10.1038/nature10995. [DOI] [PubMed] [Google Scholar]
- Math RK, et al. Comparative genomics reveals adaptation by Alteromonas sp. SN2 to marine tidal-flat conditions: cold tolerance and aromatic hydrocarbon metabolism. PLoS One. 2012;7(4):e35784. doi: 10.1371/journal.pone.0035784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew MK, Hui CF, Smith CL, Cantor CR. High-resolution separation and accurate size determination in pulsed-field gel electrophoresis of DNA. 4. Influence of DNA topology. Biochemistry. 1988;27(26):9222–9226. doi: 10.1021/bi00426a022. [DOI] [PubMed] [Google Scholar]
- McCarren P, Hall ML, Whitehead L. The chemical tuning of a weak zinc binding motif for histone deacetylase using electronic effects. Chem Biol Drug Des. 2012;80:203–214. doi: 10.1111/j.1747-0285.2012.01382.x. [DOI] [PubMed] [Google Scholar]
- Milkman R, Bridges MM. Molecular evolution of the Escherichia coli chromosome. III. Clonal frames. Genetics. 1990;126(3):505–517. doi: 10.1093/genetics/126.3.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Soria E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol. 2005;60(2):174–182. doi: 10.1007/s00239-004-0046-3. [DOI] [PubMed] [Google Scholar]
- Neumann B, Pospiech A, Schairer HU. Rapid isolation of genomic DNA from gram-negative bacteria. Trends Genet. 1992;8(10):332–333. doi: 10.1016/0168-9525(92)90269-a. [DOI] [PubMed] [Google Scholar]
- Papke RT, Koenig JE, Rodriguez-Valera F, Doolittle WF. Frequent recombination in a saltern population of Halorubrum. Science. 2004;306(5703):1928–1929. doi: 10.1126/science.1103289. [DOI] [PubMed] [Google Scholar]
- Pasic L, et al. Metagenomic islands of hyperhalophiles: the case of Salinibacter ruber. BMC Genomics. 2009;10:570. doi: 10.1186/1471-2164-10-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena A, et al. Fine-scale evolution: genomic, phenotypic and ecological differentiation in two coexisting Salinibacter ruber strains. ISME J. 2010;4(7):882–895. doi: 10.1038/ismej.2010.6. [DOI] [PubMed] [Google Scholar]
- Pourcel C, Salvignol G, Vergnaud G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology . 2005;151(Pt 3):653–663. doi: 10.1099/mic.0.27437-0. [DOI] [PubMed] [Google Scholar]
- Quaiser A, Zivanovic Y, Moreira D, Lopez-Garcia P. Comparative metagenomics of bathypelagic plankton and bottom sediment from the Sea of Marmara. ISME J. 2011;5(2):285–304. doi: 10.1038/ismej.2010.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabsch W, et al. FepA- and TonB-dependent bacteriophage H8: receptor binding and genomic sequence. J Bacteriol. 2007;189(15):5658–5674. doi: 10.1128/JB.00437-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. EMBOSS: the European molecular biology open software suite. Trends Genet. 2000;16(6):276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Brito B, et al. Viral and microbial community dynamics in four aquatic environments. ISME J. 2010;4(6):739–751. doi: 10.1038/ismej.2010.1. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Valera F, et al. Explaining microbial population genomics through phage predation. Nat Rev Microbiol. 2009;7(11):828–836. doi: 10.1038/nrmicro2235. [DOI] [PubMed] [Google Scholar]
- Rutherford K, et al. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16(10):944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- Samuel AD, et al. Flagellar determinants of bacterial sensitivity to chi-phage. Proc Natl Acad Sci U S A. 1999;96(17):9863–9866. doi: 10.1073/pnas.96.17.9863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel G, Reeves P. Biosynthesis of O-antigens: genes and pathways involved in nucleotide sugar precursor synthesis and O-antigen assembly. Carbohydr Res. 2003;338(23):2503–2519. doi: 10.1016/j.carres.2003.07.009. [DOI] [PubMed] [Google Scholar]
- Sanchez-Perez G, Mira A, Nyiro G, Pasic L, Rodriguez-Valera F. Adapting to environmental changes using specialized paralogs. Trends Genet. 2008;24(4):154–158. doi: 10.1016/j.tig.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Sass AM, Sass H, Coolen MJ, Cypionka H, Overmann J. Microbial communities in the chemocline of hypersaline deep-sea basin (Uranian Basin, Mediterranean Sea) Appl Environ Microbiol. 2001;67:3083–3091. doi: 10.1128/AEM.67.12.5392-5402.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer H, Servais P, Muyzer G. Successional changes in the genetic diversity of a marine bacterial assemblage during confinement. Arch Microbiol. 2000;173(2):138–145. doi: 10.1007/s002039900121. [DOI] [PubMed] [Google Scholar]
- Shapiro BJ, et al. Population genomics of early events in the ecological differentiation of bacteria. Science. 2012;336(6077):48–51. doi: 10.1126/science.1218198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, McCarren J, DeLong EF. Transcriptional responses of surface water marine microbial assemblages to deep-sea water amendment. Environ Microbiol. 2012;14(1):191–206. doi: 10.1111/j.1462-2920.2011.02598.x. [DOI] [PubMed] [Google Scholar]
- Skerman V. A guide to the identification of the genera bacteria. 2nd ed. Baltimore (MD): Williams and Willkins; 1967. [Google Scholar]
- Smedile F, et al. Metagenomic analysis of hadopelagic microbial assemblages thriving at the deepest part of Mediterranean Sea, Matapan-Vavilov Deep. Environ Microbiol. 2012;3(10):1462–2920. doi: 10.1111/j.1462-2920.2012.02827.x. [DOI] [PubMed] [Google Scholar]
- Smith TG, Hoover TR. Deciphering bacterial flagellar gene regulatory networks in the genomic era. Adv Appl Microbiol. 2009;67:257–295. doi: 10.1016/S0065-2164(08)01008-3. [DOI] [PubMed] [Google Scholar]
- Sorek R, Kunin V, Hugenholtz P. CRISPR—a widespread system that provides acquired resistance against phages in bacteria and archaea. Nat Rev Microbiol. 2008;6(3):181–186. doi: 10.1038/nrmicro1793. [DOI] [PubMed] [Google Scholar]
- Sutherland I. EPS polymers are also targets of recognition of phages and or phage hydrolases that burrow homes in the EPS to allow infection of the cell. Enzymic hydrolysis of colanic acid. Eur J Biochem. 1971;23:582–587. doi: 10.1111/j.1432-1033.1971.tb01657.x. [DOI] [PubMed] [Google Scholar]
- Suyama M, Torrents D, Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34(Web Server issue):W609–W612. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed KA, et al. The Vibrio cholerae flagellar regulatory hierarchy controls expression of virulence factors. J Bacteriol. 2009;191(21):6555–6570. doi: 10.1128/JB.00949-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JR, et al. Genotypic diversity within a natural coastal bacterioplankton population. Science. 2005;307(5713):1311–1313. doi: 10.1126/science.1106028. [DOI] [PubMed] [Google Scholar]
- Tyson GW, Banfield JF. Rapidly evolving CRISPRs implicated in acquired resistance of microorganisms to viruses. Environ Microbiol. 2008;10(1):200–207. doi: 10.1111/j.1462-2920.2007.01444.x. [DOI] [PubMed] [Google Scholar]
- Tyson GW, et al. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature. 2004;428(6978):37–43. doi: 10.1038/nature02340. [DOI] [PubMed] [Google Scholar]
- Weinberger AD, et al. Persisting viral sequences shape microbial CRISPR-based immunity. PLoS Comput Biol. 2012;8(4):e1002475. doi: 10.1371/journal.pcbi.1002475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyman PD, et al. Heterologous expression of Alteromonas macleodii and Thiocapsa roseopersicina [NiFe] hydrogenases in Escherichia coli. Microbiology. 2011;157(Pt 5):1363–1374. doi: 10.1099/mic.0.044834-0. [DOI] [PubMed] [Google Scholar]
- Wilmes P, et al. Natural acidophilic biofilm communities reflect distinct organismal and functional organization. ISME J. 2009;3:266–270. doi: 10.1038/ismej.2008.90. [DOI] [PubMed] [Google Scholar]