

Abstract
It has become clear that different genome regions need not evolve uniformly. This variation is particularly evident in bacterial genomes with multiple chromosomes, in which smaller, secondary chromosomes evolve more rapidly. We previously demonstrated that substitution rates and gene dispensability were greater on secondary chromosomes in many bacterial genomes. In Vibrio, the secondary chromosome is replicated later during the cell cycle, which reduces the effective dosage of these genes and hence their expression. More rapid evolution of secondary chromosomes may therefore reflect weaker purifying selection on less expressed genes. Here, we test this hypothesis by relating substitution rates of orthologs shared by multiple Burkholderia genomes, each with three chromosomes, to a study of gene expression in genomes differing by a major reciprocal translocation. This model predicts that expression should be greatest on chromosome 1 (the largest) and least on chromosome 3 (the smallest) and that expression should tend to decline within chromosomes from replication origin to terminus. Moreover, gene movement to the primary chromosome should associate with increased expression, and movement to secondary chromosomes should result in reduced expression. Our analysis supports each of these predictions, as translocated genes tended to shift expression toward their new chromosome neighbors despite inevitable cis-acting regulation of expression. This study sheds light on the early dynamics of genomes following rearrangement and illustrates how secondary chromosomes in bacteria may become evolutionary test beds.
Keywords: Burkholderia, expression, substitution rates, dispensability, replication timing
Introduction
Why some bacterial genomes have multiple chromosomes remains a mystery (Mackenzie et al. 2004; Egan et al. 2005). Although additional chromosomes minimally offer an additional replication origin, which could speed replication or facilitate a larger genome, a broader question remains: to what extent does selection act on bacterial genome organization? We previously presented a hypothesis that secondary chromosomes may act as evolutionary test beds (Cooper et al. 2010). This concept is supported by higher substitution rates, greater dispensability, and lower codon usage bias for genes on secondary chromosomes. Accordingly, genes on secondary chromosomes may reflect specific or conditional benefits in certain environments, leaving more critical housekeeping genes on the primary chromosome.
The mechanism driving this variation in evolutionary rates is likely replication timing affecting gene dosage during the cell cycle. To maintain synchronous replication among chromosomes of varying size, current evidence suggests that replication initiation is delayed for secondary, smaller chromosomes (Rasmussen et al. 2007). Thus, in addition to the gradient in gene dosage from the single replication origin to the terminus found on any single bacterial chromosome (Sharp et al. 1989), genes on the secondary chromosome will experience reduced copy number than those on the primary chromosome, on average. This gradient becomes exaggerated during periods of rapid growth, which necessitate overlapping replication cycles (Dryselius et al. 2008; Cooper et al. 2010). The result is increased expression for genes replicated earlier—both on the primary chromosome and near the origins of both chromosomes—and reduced expression for genes replicated later (Sharp et al. 1989; Mira and Ochman 2002; Couturier and Rocha 2006; Dryselius et al. 2008).
This gradient in gene dosage and hence expression in bacteria with multiple chromosomes provides a mechanism by which selection could act to influence genome organization. It is well known that highly expressed genes tend to evolve more slowly for multiple reasons, including the cost imposed by translation error (Drummond and Wilke 2008). Less appreciated is that selection for efficient translation may affect synonymous substitution rates (dS) as well as nonsynonymous substitutions (dN), primarily by the need for optimal codon usage. Selection for translational accuracy is thought to be driven by the loss of ribosomal capacity for production of other proteins and the energy required to dispense with, or mitigate the disruption caused by, the misfolded molecule (Rocha and Danchin 2004; Drummond and Wilke 2008). Importantly, this mechanism also explains the pervasive autocorrelation between dN and dS across many organisms and gene categories (Sharp and Li 1987; Drummond et al. 2005; Drummond and Wilke 2008). Thus, we predicted that late replicated chromosome regions will tend to evolve more rapidly because of relatively reduced expression and hence weaker purifying selection for translational efficiency.
One genus of relatively fast-growing bacteria with multiple chromosomes is Burkholderia, and it is well represented among completely sequenced genomes because of the medical, physiological, and agricultural significance of these organisms (Mahenthiralingam et al. 2005). Burkholderia genomes are notable for their plasticity and uneven composition: the primary chromosome harbors conserved genes linked to metabolism and cell growth, whereas the secondary chromosomes carry more niche-specific genes (Holden et al. 2004; Chain et al. 2006). Translocations in such multichromosome genomes have been observed, however (Slater et al. 2009), providing evidence of the mechanism by which selection could shape genome organization. One such reorganization has been reported among Burkholderia cenocepacia strains (Guo et al. 2010), in which the third chromosome of strain AU1054 acquired many essential genes from the primary chromosome. Lastly, our previous study of bacterial genomes with multiple chromosomes included several Burkholderia genomes, which exhibited increased evolutionary rates consistent with reduced expression on secondary chromosomes (Cooper et al. 2010).
Here, we test this model of bacterial genome organization by studying gene-level effects of a balanced translocation between the largest and smallest chromosomes within a Burkholderia genome. If chromosome position governs expression (and ultimately, the evolutionary fate), then genes should adopt the prevailing expression patterns of their new neighbors, even despite local cis-acting regulatory elements. We obtained expression data (Yoder-Himes et al. 2009) for two focal Burkholderia genomes and calculated substitution rates of orthologs among the related clade of species. As expected, expression and substitution rates were inversely correlated, and more rapidly evolving, less expressed genes were enriched on secondary chromosomes. Expression of translocated genes also changed as expected, with significant increases or decreases depending on the new chromosome neighborhood. This study of multichromosome bacteria allowed us to quantify how selection may act on genome organization in the recent aftermath of a genomic rearrangement and provides a glimpse into the evolutionary test bed in action.
Materials and Methods
Genomes
The genomes of two closely related and well-characterized strains of B. cenocepacia (AU1054 and HI2424) were sequenced by the Joint Genome Institute (JGI, http://jgi.doe.gov, last accessed November 30, 2012) and found to possess similar content (Cooper et al. 2010) with a high degree of synteny (Lin et al. 2008). Strain AU1054 was isolated from the blood of a patient with cystic fibrosis, whereas strain HI2424 was isolated from an onion field (LiPuma et al. 2002). However, 517 genes on chromosome 1 and 371 genes on chromosome 3 underwent a balanced translocation. Two other Burkholderia genomes were included, B. multivorans ATCC17616 and B. ambifaria AMMD, to produce a representative set of the B. cepacia complex with sufficient phylogenetic distance but excluding the B. mallei and B. pseudomallei clades (supplementary fig. S1, Supplementary Material online).
The nucleotide and protein gene sequence FASTA files and annotations for the four Burkholderia genomes of interest were obtained from the Integrated Microbial Genome database at the JGI (Markowitz et al. 2010), and a MySQL database was populated with this genomic data.
Ortholog Identification
Beginning with a list of genes in the reference HI2424 genome, a reciprocal BLAST (Altschul et al. 1990) procedure was used to identify orthologs shared by three Burkholderia genomes (B. cenocepacia HI2424, B. ambifaria AMMD, and B. multivorans ATCC17616). Orthologs across three genomes from the genus Bordetella (Bor. bronchiseptica [RB50], Bor. petrii DSM 12804 [petrii], and Bor. avium 197N [197N]) were also identified to assess whether expression and substitution rate gradients were also present in a related taxon with only a single chromosome. This procedure used Perl scripts, Perl DBI, and Bioperl (Stajich et al. 2002), where sequences were retrieved from the MySQL database and compared using the BLAST executables (version 2.2.23) with protein databases created using the formatdb tool.
To be designated as likely orthologs, all members of gene families needed to be reciprocal best hits in all target genomes and occur in the same chromosome. In addition, orthologs were required to share similar chromosome positions (within ±15% of the position, normalized for genome length, and synchronized relative to the origin of replication) among all genomes to properly assess effects of chromosome location; this filter also reinforces the inference of orthology. However, the screen for conservation of gene position did not discriminate among positive or negative DNA strands or leading or lagging strands relative to the origin; only the distance from the origin was considered. This method of ortholog identification is similar yet simpler than previous methods (Lerat et al. 2003; Cooper et al. 2010) because the bacterial genomes are less divergent and are generally syntenic. Larger divergence would likely cause more true orthologs to be discarded by the requirement for synteny because gene order would likely erode with phylogenetic distance.
Substitution Rate Calculation
The amino acid sequences for each ortholog triad were aligned using ClustalW 2.1 (Larkin et al. 2007) via Bioperl modules. The two ClustalW alignments, along with the nucleotide sequences mapped to the protein alignments, were passed to the PAML module codeml (Yang 2007) to calculate the nucleotide substitution rates. The default settings for a pairwise calculation in codeml were used. Any calculated synonymous (dS) or nonsynonymous (dN) rate of nucleotide substitution for a pair of genes exceeding 2.0 was discarded because of the unreliable nature of these values due to saturation. The evolutionary distances of the chosen Burkholderia species caused many inferred values of dS to exceed 2.0; on the other hand, most orthologs exhibited dN > 0. We focus our analysis on the B. cenocepacia HI2424 and B. ambifaria AMMD ortholog pairs, but nearly identical results were found using ortholog pairs from HI2424 and B. multivorans ATCC171616 (supplementary material, Supplementary Material online). Substitution rates for Bordetella orthologs were calculated using the same method.
Gene Expression Data
A prior study of expression in B. cenocepacia using RNA-seq evaluated the transcriptional response under two conditions (Yoder-Himes et al. 2009). The specific strains studied were HI2424 and AU1054, which were grown in both synthetic CF sputum medium and soil medium (SE); the normalized results for SE conditions for both strains were obtained as it likely represents a more natural growth condition. Genes in the HI2424 genome were linked to the AU1054 genes used as a reference in this study using MUMmer 3.0 (Kurtz et al. 2004), which provided data that were read into R (R Development Core Team 2011) to link and cross-reference expression values between genomes.
Expression data from microarrays were obtained for Bor. bronchiseptica (RB50) as part of a study of growth-phase-dependent gene regulation (Nicholson et al. 2009). The bacteria were cultured on Bordet–Gengou agar, and the RNA of interest in our study was isolated during mid-logarithmic phase (15 h). The cDNA was fluorescently labeled and hybridization to a RB50-specific long-oligonucleotide microarray with reporters linked to RB50 gene loci. After background subtraction, the mean values for six replicates were calculated. Subsequent filtering for loci with expression values greater than zero facilitated use of the cube-root transformation.
Statistical Analysis
All regression analyses and comparison of substitution rate distributions, and related plots, were produced using R (R Development Core Team 2011). When the distribution of the data was skewed or deviated from Gaussian, data were cube root transformed to enable parametric statistical analyses.
Results
Orthologs Are Less Common on Secondary Chromosomes
Orthologs shared by three Burkholderia genomes (B. cenocepacia HI2424, B. ambifaria AMMD, and B. multivorans ATCC17616) typically shared similar chromosome positions, but 91 genes, including 17% of genes on chromosome 3, were discarded from future study for a lack of synteny (table 1). As expected, shared orthologs were less common on secondary chromosomes (table 1) but were not statistically enriched or deficient in any location within each chromosome (supplementary fig. S2, Supplementary Material online).
Table 1.
Orthologs Shared by Three Burkholderia Genomes (Burkholderia cenocepacia HI2424, B. ambifaria AMMD, and B. multivorans ATCC17616) and Filtered for Substitution Rates (dN or dS > 2.0) and Synteny (>15% Difference in Position Relative to the Origin)
| Chromosome | Number of Shared Orthologs | Total Genes in HI2424 | Orthologs/Total Genes | Orthologs Excluded by Filters in Pairwise Genome Comparisons |
|
|---|---|---|---|---|---|
| HI2424-AMMD Substitution Rate/Synteny | HI2424-ATCC17616 Substitution Rate/Synteny | ||||
| Chr 1 | 2,604 | 3,253 | 0.80 | 12/4 | 28/5 |
| Chr 2 | 1,411 | 2,709 | 0.52 | 6/10 | 21/35 |
| Chr 3 | 221 | 929 | 0.24 | 2/12 | 4/25 |
Expression Levels Are Lower and Substitution Rates Are Higher Near Replication Termini
Within each of the three Burkholderia chromosomes, expression rates decrease and substitution rates (dN, dS and dN/dS) increase with distance from the origin of replication (supplementary figs. S3–S6, Supplementary Material online). These trends continue and are even exaggerated among chromosomes; for example, mean expression near the terminus of chromosome 1 exceeds mean expression near the origin of chromosome 2, and expression on chromosome 3 tends to be lower still (supplementary fig. S4, Supplementary Material online). Highly significant and pronounced increases in dN and dN/dS were observed from origin to terminus on chromosome 3. Similar, corresponding increases in evolutionary rates (dN, dS, and dN/dS) occur across the chromosomes (supplementary figs. S3, S5, and S6, Supplementary Material online). Although none of these trends explain a large fraction of the variation among genes in expression or evolutionary rates (with r2 values from <0.01 to 0.14, supplementary material, Supplementary Material online), they clearly differentiate among chromosomes and even chromosome regions. To evaluate whether the gradients in expression and evolutionary rate predated the evolution of a genome with multiple chromosomes, we evaluated the relationships between replication timing, expression, and evolutionary rates in Bordetella, a close outgroup with a single chromosome. Indeed, in Bor. bronchiseptica, expression declined and dN increased with distance from the replication origin (supplementary figs. S7 and S8, Supplementary Material online).
These relationships could be influenced by nonindependent measures of expression of genes found in operons, so we examined whether substitution rate patterns persisted among chromosomes for three subsets of our database. These subsets sampled every fourth gene sorted by chromosome location based on the mean operon size of four genes in bacterial genomes (Zheng et al. 2002) and were then divided by chromosomes. This strategy should be considered conservative given that only 58% of genes in these genomes are expected to be in operons of two or more (Dam et al. 2007). All substitution rates (dN, dS, and dN/dS) increased from chromosomes 1 to 3 (supplementary fig. S9 and table S1, Supplementary Material online), and all but one pairwise comparison between chromosomes were statistically significant.
Chromosomes Define Distinct Clusters of Expression and Evolutionary Rates
The inverse relationship between expression and dN has been well defined and is evident in these genomes, independently of chromosome position (fig. 1A). However, overlaying chromosome position upon this regression highlights genes with high expression and low dN overwhelmingly on chromosome 1, and conversely, more genes with low expression and high dN on chromosomes 2 and 3. The same patterns are seen for both dS (fig. 1B) and dN/dS (supplementary fig. S10, Supplementary Material online), and in each case, weakly predictive genome-wide relationships between evolutionary rates and expression become more obvious when grouped by chromosomes. Mean expression for chromosome 1 is 7.8% greater than chromosome 2 and 11.7% greater than chromosome 3, accompanied by 15.2% and 30.6% differences in dN, respectively. These relationships more clearly define the hierarchy of expression rates within these Burkholderia genomes and emphasize the importance of chromosome location.
Fig. 1.—
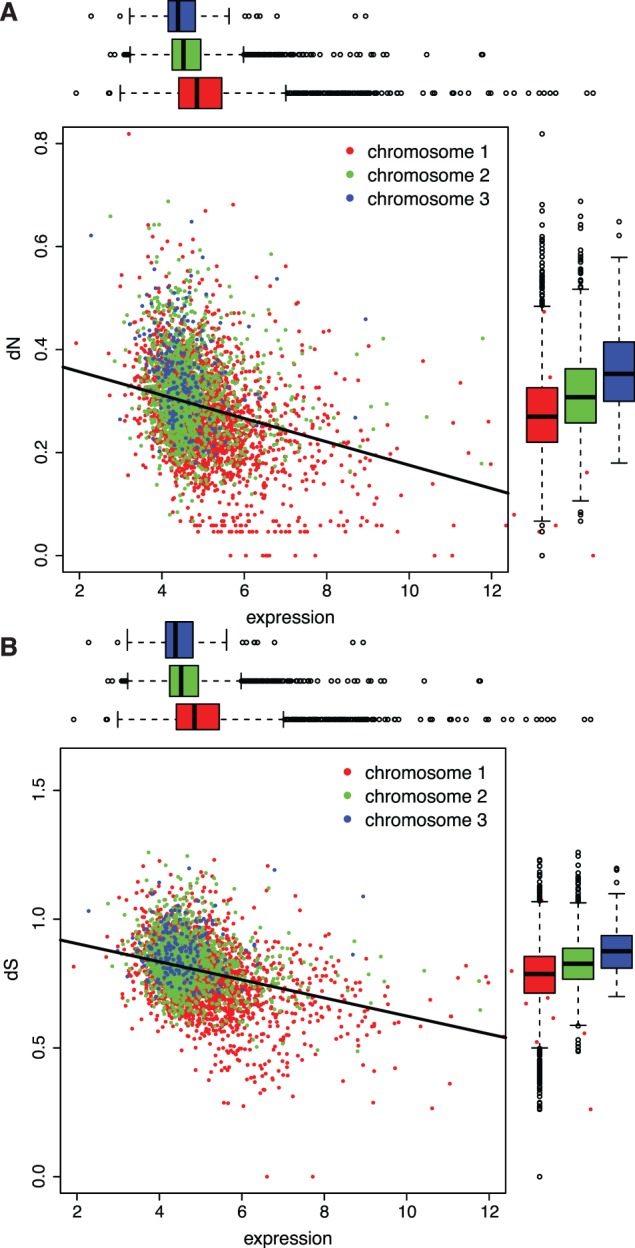
Relationships between substitution rates (A: dN and B: dS) of Burkholderia orthologs and gene expression measured in strain HI2424. Both values were cube root transformed. The overall regressions are significant (dN vs. expression: F = 384, dfs = 1, 4,079, P < 10−16, r2 = 0.086; dS vs. expression F = 553, dfs = 1, 4,079, P < 10−16, r2 = 0.120), and all pairwise comparisons among chromosomes (boxplots) differ significantly (expression: chromosome 1 (c1) to chromosome 2 (c2), t = −14.2, df = 2,885, P < 2.2 × 10−16; c2 to c3, t = −6.44, df = 261, P = 5.75 × 10−10; dN: c1 to c2, t = 11.12, df = 3,556, P < 2.2 × 10−16; c2 to c3, t = 3.51, df = 301, P = 0.000516; dS: c1 to c2, t = −13.72, df = 3,235, P < 2.2 × 10−16; c2 to c3, t = −6.56, df = 276, P = 2.711 × 10−10).
Translocated Genes Exhibit Altered Expression Consistent with the New Chromosome Location
The balanced translocation found in the genome of B. cenocepacia AU1054 relative to related strains involved a portion of chromosome 1 near the replication terminus and approximately half of chromosome 3 (fig. 2A). These genomic regions on average differ significantly in their profiles of substitution rates and expression (fig. 1). Despite highly correlated expression in syntenic regions of both genomes (supplementary figs. S11–S15, Supplementary Material online), genes that moved from chromosome 1 to chromosome 3 exhibited significantly decreased expression relative to genes remaining on chromosome 1 (fig. 2B). In addition, genes that moved from chromosome 3 to chromosome 1 exhibited increased expression relative to genes remaining on chromosome 3 (fig. 2B). Such changes in expression are unlikely the cause of altered strand bias (the fraction on the positive or negative DNA strand), which did not change during the translocation, or differential operon structure of genes within the blocks, which are nearly equivalent (Dam et al. 2007).
Fig. 2.—

Altered expression of translocated genes in Burkholderia cenocepacia matching their new chromosome neighbors. (A) Schematic of the translocation found in strain AU1054 in comparison to its nearest relative HI2424, which represents the consensus genome order of the species complex; dark red and dark blue represent the translocated genomic blocks. (B) Boxplots of expression ratios (AU1054/HI2424) of shared orthologs. The translocation from chromosome 1 (517 genes) was compared with nearby orthologs—the chromosome half nearer the terminus (1,056 genes)—because their expression in HI2424 better represents the translocated fraction. The resident (523 genes) and translocated (371 genes) regions of chromosome 3 in AU1054 exhibit equivalent expression in HI2424. Two sample t-tests were used; for chromosome 1, t = −2.338, df = 1,571, P = 0.0195, for chromosome 3, t = 2.974, df = 892, P = 0.0030.
Discussion
The reciprocal translocation of 888 genes within B. cenocepacia strain AU1054 led to significantly altered expression to match their new chromosome neighbors, which demonstrates the potential for selection to act on bacterial genome organization. Whether this translocation was actually favored by selection is unknown; however, reduced expression of genes caused by the movement from primary to secondary chromosomes (fig. 1 and supplementary fig. S4, Supplementary Material online) is expected to increase their evolutionary rates (Drummond and Wilke 2008). Such reduced expression could be advantageous in a novel environment and be favored by selection, but with the longer-term consequence of reduced purifying selection and greater substitution rates. Similarly, increased expression caused by movement from secondary to primary chromosomes could also enhance phenotypes favored by selection, increase selection for robust translation, and more gradually decrease substitution rates.
As expected, the changes in expression of the translocated genes are relatively subtle: genes moving from chromosome 1 to chromosome 3 declined 2.1% and genes moving from chromosome 3 to 1 increased 3.7%. The changes that would be expected from this translocation can be estimated from expression patterns of its nearest relative, strain HI2424 (supplementary fig. S4, Supplementary Material online). Mean transformed expression is approximately 8% greater near the terminus of chromosome 1 than at the middle of chromosome 3. The observed changes being less than this expectation could be explained by several factors including the inherent, cis-acting regulation of these genes. Given these relatively subtle shifts, it is not surprising that translocated genes are not visible outliers in comparisons of HI2424 and AU1054 expression (supplementary figs. S11–S15, Supplementary Material online).
The strong clustering of gene expression and substitution rate with chromosome location (fig. 1) and position within chromosomes (supplementary figs. S3–S6, Supplementary Material online) are entirely consistent with delayed replication of secondary chromosomes in Burkholderia, as has been shown for Vibrio (Rasmussen et al. 2007). With a single origin of replication per chromosome, smaller replicons are initiated later to maintain synchrony at the conclusion of genome replication, which reduces gene dosage throughout secondary chromosomes, including regions near the origins (Couturier and Rocha 2006; Dryselius et al. 2008; Cooper et al. 2010). This replication dynamic explains why expression is lower on secondary chromosomes even near the origins but especially near the termini (supplementary fig. S4, Supplementary Material online). The strength of gradients in expression and substitution rates from origin to terminus should be generally correlated with the average growth rate of the organism, because fast-growing species require overlapping replication cycles to allow replication to keep up with growth (Helmstetter 1996). However, the genomes of some slow growing bacteria, such as Mycobacterium and Chlamydia, exhibit little or no gradient in substitution rates (dS in particular) (Mira and Ochman 2002), likely because replication is rarely limiting for growth and occupies a smaller fraction of their cell cycles. Consequently, these Burkholderia genomes exhibit obvious evolutionary gradients consistent with a history of periodic rapid growth.
Genomes have likely evolved to become ordered by replication timing on many occasions, some ancient. For example, the primary chromosomes of Burkholderia genomes share similar gradients in expression and substitution rate with their single-chromosome ancestors, reflected by extant Bordetella (supplementary figs. S7 and S8, Supplementary Material online). Such patterns also occur in Vibrio, which have two chromosomes, and their single-chromosome relatives, as well as in many other multipartite bacterial genomes (Cooper et al. 2010). However, the evolutionary advantage of additional chromosomes beyond additional replication origins remains uncertain. These replicons could perhaps become test beds because they may afford a greater capacity for genes with reduced expression that may only be conditionally beneficial, but they may also become test beds de facto owing to reduced purifying selection. Nonetheless, this study demonstrates that the movement of genes between primary (larger) and secondary (smaller) chromosomes alters their expression and almost certainly the function of the organism, which can in turn be shaped directly by selection. Genes translocated from secondary chromosomes to the primary chromosome are predicted to experience increased expression and hence greater purifying selection, whereas genes moving from the primary chromosome to secondary chromosomes should experience reduced expression and potentially faster evolutionary rates.
Supplementary Material
Supplementary table S1 and figures S1–S15 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
This work was supported by CAREER award DEB-0845851 from the National Science Foundation to V.S.C. The genome sequences analyzed herein were produced by the US Department of Energy Joint Genome Institute (http://www.jgi.doe.gov/, accessed November 30, 2012) in collaboration with the user community. The authors are grateful for open access to the RNA-seq analyses conducted by Yoder-Himes et al. (2009) and for constructive feedback from KM Flynn.
Literature Cited
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Chain PS, et al. Burkholderia xenovorans LB400 harbors a multi-replicon, 9.73-Mbp genome shaped for versatility. Proc Natl Acad Sci U S A. 2006;103:15280–15287. doi: 10.1073/pnas.0606924103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper VS, Vohr SH, Wrocklage SC, Hatcher PJ. Why genes evolve faster on secondary chromosomes in bacteria. PLoS Comput Biol. 2010;6:e1000732. doi: 10.1371/journal.pcbi.1000732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couturier E, Rocha EP. Replication-associated gene dosage effects shape the genomes of fast-growing bacteria but only for transcription and translation genes. Mol Microbiol. 2006;59:1506–1518. doi: 10.1111/j.1365-2958.2006.05046.x. [DOI] [PubMed] [Google Scholar]
- Dam P, Olman V, Harris K, Su Z, Xu Y. Operon prediction using both genome-specific and general genomic information. Nucleic Acids Res. 2007;35:288–298. doi: 10.1093/nar/gkl1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Bloom JD, Adami C, Wilke CO, Arnold FH. Why highly expressed proteins evolve slowly. Proc Natl Acad Sci U S A. 2005;102:14338–14343. doi: 10.1073/pnas.0504070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Wilke CO. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell. 2008;134:341–352. doi: 10.1016/j.cell.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryselius R, Izutsu K, Honda T, Iida T. Differential replication dynamics for large and small Vibrio chromosomes affect gene dosage, expression, and location. BMC Genomics. 2008;9:559. doi: 10.1186/1471-2164-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan ES, Fogel MA, Waldor MK. Divided genomes: negotiating the cell cycle in prokaryotes with multiple chromosomes. Mol Microbiol. 2005;56:1129–1138. doi: 10.1111/j.1365-2958.2005.04622.x. [DOI] [PubMed] [Google Scholar]
- Guo FB, Ning LW, Huang J, Lin H, Zhang HX. Chromosome translocation and its consequence in the genome of Burkholderia cenocepacia AU-1054. Biochem Biophys Res Commun. 2010;403:375–379. doi: 10.1016/j.bbrc.2010.11.039. [DOI] [PubMed] [Google Scholar]
- Helmstetter CE. Timing of synthetic activities in the cell cycle. In: Neidhardt FC, et al., editors. Escherichia coli and Salmonella typhimurium: cellular and molecular biology. Washington (DC): ASM Press; 1996. pp. 1627–1649. [Google Scholar]
- Holden MT, et al. Genomic plasticity of the causative agent of melioidosis, Burkholderia pseudomallei. Proc Natl Acad Sci U S A. 2004;101:14240–14245. doi: 10.1073/pnas.0403302101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Lerat E, Daubin V, Moran NA. From gene trees to organismal phylogeny in prokaryotes: the case of the gamma-proteobacteria. PLoS Biol. 2003;1:E19. doi: 10.1371/journal.pbio.0000019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Bourque G, Tan P. A comparative synteny map of Burkholderia species links large-scale genome rearrangements to fine-scale nucleotide variation in prokaryotes. Mol Biol Evol. 2008;25:549–558. doi: 10.1093/molbev/msm282. [DOI] [PubMed] [Google Scholar]
- LiPuma JJ, Spilker T, Coenye T, Gonzalez CF. An epidemic Burkholderia cepacia complex strain identified in soil. Lancet. 2002;359:2002–2003. doi: 10.1016/S0140-6736(02)08836-0. [DOI] [PubMed] [Google Scholar]
- Mackenzie C, Kaplan S, Choudhary M. Multiple chromosomes. In: Miller RV, Day MJ, editors. Microbial evolution: gene establishment, survival, and exchange. Washington (DC): ASM Press; 2004. pp. 82–101. [Google Scholar]
- Mahenthiralingam E, Urban TA, Goldberg JB. The multifarious, multireplicon Burkholderia cepacia complex. Nat Rev Microbiol. 2005;3:144–156. doi: 10.1038/nrmicro1085. [DOI] [PubMed] [Google Scholar]
- Markowitz VM, et al. The integrated microbial genomes system: an expanding comparative analysis resource. Nucleic Acids Res. 2010;38:D382–D390. doi: 10.1093/nar/gkp887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mira A, Ochman H. Gene location and bacterial sequence divergence. Mol Biol Evol. 2002;19:1350–1358. doi: 10.1093/oxfordjournals.molbev.a004196. [DOI] [PubMed] [Google Scholar]
- Nicholson TL, Buboltz AM, Harvill ET, Brockmeier SL. Microarray and functional analysis of growth phase-dependent gene regulation in Bordetella bronchiseptica. Infect Immun. 2009;77:4221–4231. doi: 10.1128/IAI.00136-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. Vienna (Austria): R Foundation for Statistical Computing; 2011. R: a language and environment for statistical computing. [Google Scholar]
- Rasmussen T, Jensen RB, Skovgaard O. The two chromosomes of Vibrio cholerae are initiated at different time points in the cell cycle. EMBO J. 2007;26:3124–3131. doi: 10.1038/sj.emboj.7601747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha EP, Danchin A. An analysis of determinants of amino acids substitution rates in bacterial proteins. Mol Biol Evol. 2004;21:108–116. doi: 10.1093/molbev/msh004. [DOI] [PubMed] [Google Scholar]
- Sharp PM, Li WH. The rate of synonymous substitution in enterobacterial genes is inversely related to codon usage bias. Mol Biol Evol. 1987;4:222–230. doi: 10.1093/oxfordjournals.molbev.a040443. [DOI] [PubMed] [Google Scholar]
- Sharp PM, Shields DC, Wolfe KH, Li WH. Chromosomal location and evolutionary rate variation in enterobacterial genes. Science. 1989;246:808–810. doi: 10.1126/science.2683084. [DOI] [PubMed] [Google Scholar]
- Slater SC, et al. Genome sequences of three agrobacterium biovars help elucidate the evolution of multichromosome genomes in bacteria. J Bacteriol. 2009;191:2501–2511. doi: 10.1128/JB.01779-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stajich JE, et al. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 2002;12:1611–1618. doi: 10.1101/gr.361602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Yoder-Himes DR, et al. Mapping the Burkholderia cenocepacia niche response via high-throughput sequencing. Proc Natl Acad Sci U S A. 2009;106:3976–3981. doi: 10.1073/pnas.0813403106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Szustakowski JD, Fortnow L, Roberts RJ, Kasif S. Computational identification of operons in microbial genomes. Genome Res. 2002;12:1221–1230. doi: 10.1101/gr.200602. [DOI] [PMC free article] [PubMed] [Google Scholar]