

Abstract
In order to deal with the complexity of biological systems at the atomic level, limiting assumptions are often made which do not reflect the reality of the system under study. One example is the assumption that the entropy of binding of the macromolecule is not influenced significantly by the different ligands. Recent experimental data on ligands binding to HIV-1 protease challenge this assumption.
Introduction
What often appears trivial on first evaluation becomes more challenging as tacit assumptions are rejected, and the complexity of the problem is revealed in all its glory. During the past decades, I have progressed toward a more complete understanding of the complexities of molecular recognition by attempting to design ligands for protein binding sites. These “lessons” learned in the school of hard knocks might be of some benefit to those still on the road to harsh reality.
First, science is a game of successive approximations. Our current state of “understanding” is transient, and dogmas almost always require revision an/or refinement as our experiences increase. Reductionism has certainly proven a useful paradigm in science, but the complexity of biological systems requires a different mindset. Certainly, much of the underlying physics and chemistry of molecular recognition have been routinely minimized to force problems of molecular recognition within our paradigm/computer. The sins we have committed to tackle problems beyond our current understanding are many:
Sins of omission Earlier in molecular modeling, solvation was omitted as too computationally complex; later electrostatics were omitted as giving the wrong answers.
Sins of commission Ignoring what has been demonstrated; monopole electrostatics cannot reproduce the electrostatic fields generated by quantum mechanics.
Sins of arrogance Oversimplification as a means of confronting computational complexity—since we cannot generate the partition function for the configurations of water in solvation, we invent empirical, implicit solvent models to obtains the wrong answer quicker!
Sins of ignorance Pretty pictures that support one’s hypothesis are seductive, but rarely scientific. If it doesn’t work on model systems where experimental data is available for validation, simply increase the size of the system; errors cancel, don’t they.
Sins of myopia Evolution optimizes rates of interactions in molecular systems rather than affinities. This fact makes the discipline of medicinal chemistry feasible. The concept of intelligent design is misleading, at least within our limiting ability to understand; evolution solves problems anyway it finds. We routinely use “Occam’s Razor” to choose between alternative hypotheses. My experience suggests that Mother Nature never shaved with Occam’s Razor. Evolution is not logical, but opportunistic!
Virtual screening example
Consider the usual results of virtual screening when a crystal structure of the therapeutic target is available. For those in the pharmaceutical industry, a validated hit rate of 25% for a lead in a therapeutic project is quite reasonable. For an academic, the implications are quite different. A signal-to-noise of 25% implies that something significant is missing from your virtual-screening algorithm. While the approach is valuable in practice, the limitations in both search and scoring have been well documented [1].
Binding entropy
ΔG = ΔH − TΔS for the system, not just the subsystem that is convenient to consider.
| (1) |
A basic conundrum facing structure-based drug design has been the tacit assumption that the ΔG of binding is not influenced by modulating the binding entropy of the receptor (i.e. that TΔSreceptor is a constant). The consistent conformation of HIV-1 protease seen in hundreds of crystal structures of complexes with inhibitors (PIs) would be consistent with this interpretation. Mutants of HIV-1 protease modify their affinity for an inhibitor by directly changing the interactions between the inhibitor and the active-site residues, or alternatively, by changing the dynamic response of the protein to the inhibitor. Evolution can modulate the enzyme’s affinities for PIs by changes either in ΔH, ΔS or a combination of both. In particular, changes in the ΔSreceptor of the protease (Eq. 1), has been ignored as a means of impacting the affinity of an inhibitor. Modulation of the enzyme dynamics by mutation as a mechanism for resistance has been clearly demonstrated in recent EPR studies of HIV-1 protease interactions with a set of inhibitors in the Fanucci [2–4] and Kent [5] labs.
In order to understand the significance of the DEER EPR studies used, a simplistic overview of this methodology follows. By incorporating a pair of spin labels at selected positions in a protein, the instantaneous distribution of distances between the spins can be determined by DEER EPR experiments on frozen samples. While two types of interactions occur in molecular systems with interacting electron spins: dipole–dipole interactions that dominate longer (15–80 Å) distances are the objective of DEER experiments [6].
Assuming that the distances between electrons is sufficient (>12Å) that through-space dipolar interactions dominate, the isotropic-exchange coupling can be modeled approximately as a decaying exponential with inter-spin separation r, whereas the Hamiltonian for the dipolar coupling between point dipoles gives an r−3 dependence; for a free electron g value the following equation gives the dipolar coupling frequency:
where θ is the angle between the inter-spin and applied magnetic field vectors, νdd is given in MHz and r is measured in nm.
By assuming that the spin label is rotationally averaging the relative orientation θ of the two electron spin dipoles, the distance between spin dipoles is easily calculated by the formula above [7]. If, however, the ensemble of rotational conformers is not averaged as has been determined for some flexible SDSL nitroxide labels [6, 8], then the angle θ between the two electron-spin dipoles cannot be ignored, and significant errors in distance estimates can occur. The relative orientations of the dipoles must be considered in data collection, and analysis of the spectra as a function of the orientation of the sample in the applied magnetic field resolves the orientation issue [9].
Time resolution versus NMR
Pulse DEER EPR experiments allow quantitative determination of the distribution of distances between two spin labels due to their time resolution [7, 10]; this is in marked contrast to NMR where only the averaged distance of the conformer ensemble is determined. Characterizing structural dynamics with determination of distance distributions provides a much more stringent validation criterion by enabling direct comparison of experimental results with MD simulations. From measurement of the dipolar interaction between two spins, both distance and orientation between two paramagnetic centers in systems can be obtained [9]. In particular, the double electron–electron resonance (DEER) protocol using a pulse EPR spectrometer (Fig. 1) measures the strength of dipolar coupling between the two electron spins is proportional to 1/r3, where r is the interspin distance. By measuring the strength of the dipolar interaction between two spin probes, distances between 20 and 75 Å have been measured [6, 11]. Recently, this approach was used to measure population percentages for ensembles of flap conformations of HIV-1 protease when binding different inhibitors [3, 5, 12], or the impact of protease mutations [2] using SDSL and DEER spectroscopy. Pulsed EPR measurements techniques have emerged as an essential tool in the study of macromolecular dynamics, changes in the ensemble populations of macromolecular conformers and as spectroscopic rulers [13] to determine the structure of complex assemblies of larger proteins [9]. DEER experiments provide precise distance constraints for model building of a set of dynamic conformers of the protease consistent with the experimental data [4]. One significant advantage of EPR double-spin experiments over similar NMR studies is the enhanced sensitivity due to the increased magnitude of the electron spin versus a nuclear spin resulting in the ability to measure longer distances (2–8 Å vs. 8–75 Å) (Fig. 2).
Fig. 1.
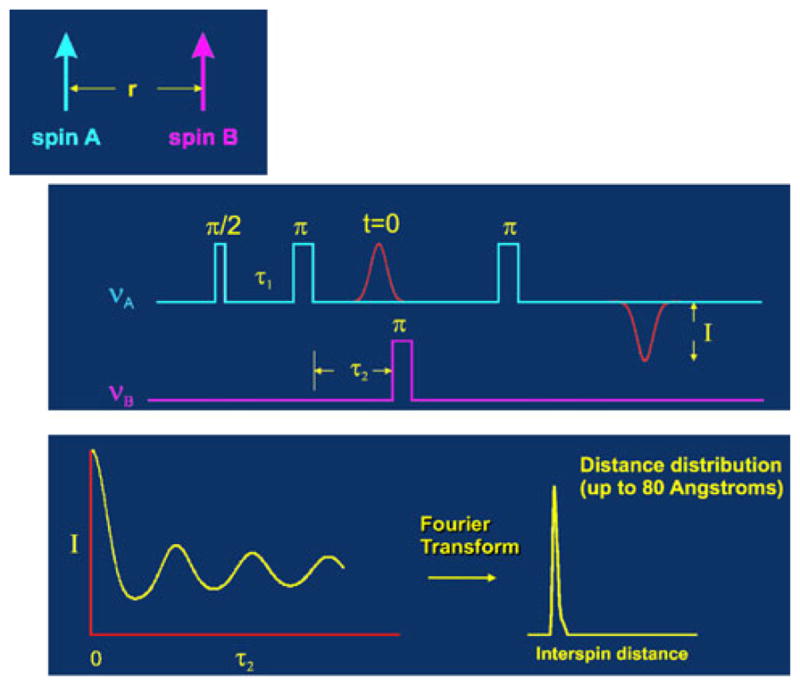
Four-pulse DEER (double electron–electron resonance) [27]
Fig. 2.

SDSL-DEER studies on HIV-1 protease with spin-labeled “flap” residues to measure conformational ensembles populated in solution [3]
DEER distance distributions in HIV-1 protease
Galiano et al. [14] incorporated a SDSL label at K55C and used DEER EPR spectroscopy to determine the distance range between spin labeled 55/55′ of conformations of the flap regions when binding a protease inhibitor. K55 was selected as a site within the flap region tolerant of amino acid substitution while retaining WT activity. Their DEER results characterized the range of flap conformation when uninhibited, showing distances that spanned 26–48 Å, indicative of a broad range of conformational states, i.e. entropically stabilized compared to the protease when inhibitor-bound. A Gaussian distance-distribution with an average distance of 35.5 Å and a width of 10 Å was obtained for the protease itself. Upon addition of ritonavir, the average distance shortened by 3 Å (to 32.6 Å) and the distribution narrowed to 3.0 Å. This distribution breadth was interpreted to reflect spin-label disorder rather than backbone disorder, as it is known that backbone motions are restricted in the inhibitor-bound state. This width is in sharp contrast to the ±0.7 Å precision estimated in the Smythe et al. [15] paper using two conformational constrained TOAC spin labels. Thus, the inherent flexibility of the spin labels used in the SDSL approach clearly limits the precision of distance distributions obtained by DEER (Fig. 3).
Fig. 3.

DEER results as determined with different protease inhibitors by Blackburn et al. [3]
The original Galiano et al. study [16] was extended by Blackburn et al. [3] to measure the impact of 9 different HIV-1 protease inhibitors on the distribution of closed and open conformers of HIV-1 protease. No strong correlation was seen between the closed conformations and the values of Ki for the inhibitors. A strong correlation was seen with the number of hydrogen bonds the inhibitors made in the complex. One caveat, these studies were done with the D25N mutation to inactivate the protease and inhibit autoproteolysis. This issue was subsequently addressed in the study by Torbeev et al. [5] that studied both the wild-type and the D25N enzyme. Regardless, the tacit assumption of structure-based design that the ΔS of binding of the protease is a constant, independent of inhibitor, has clearly been challenged, if not eliminated. The Fanucci group [2] has also characterized the impact of drug-selected mutations on “flap” conformations in the case of the V6 and MDR769 drug-resistance constructs as shown in Fig. 4. Mutations that confer drug resistance obviously utilize allosteric effects on the dynamics of the enzyme to modulate binding affinities of PIs.
Fig. 4.

Location of mutations leading to drug resistance in two protease constructs examined by DEER EPR [2]
Torbeev et al. [5] also incorporated SDSL labels at location Ile 50/50′ of the HIV dimer by chemical synthesis. In complexes with three different inhibitors, MVT-101, JG365 and KVS-1, the distances between the two spin labels indicated an equilibrium between the closed form (flaps down) seen in crystal structures and two other forms with the flaps both open or one open-one closed that depended on the bound inhibitor. This study is fully consistent with the observations of Blackburn et al. [3] that clearly indicated that the dynamic equilibrium of the protease in solution is modulated by complex formation with inhibitors. The precision of the distance distributions, however, was again compromised by the flexibility of the SDSL labels as indicated by the width of the DEER peaks (Fig. 5). Regardless of this technical limitation, the implications of these studies are unavoidable. Even in HIV-1 protease where the crystallographic evidence strongly supports a common structure for the protease when interacting with inhibitors, each inhibitor and each mutant protein has a distinct set of interactions that change the distribution of solution conformers and, thus, impacts the entropy of binding.
Fig. 5.

DEER distance distributions for SDSL spin labels at locations Ile 50/50′ [5]
Implications for other studies
These results help rationalize the study by Tang and Marshall where PLS predictive models of entropy and enthalpy were derived independently from complexes with ITC thermodynamic data and high-resolution structures of the complex [17]. The predictive ability of the two models was quite unreliable for either entropy or enthalpy, but quite predictable for binding energy when the two models were combined. The implication is that the parameters for the models were derived from static crystal structures of the complexes without consideration of multiple conformers of the proteins.
The work of the Martin group that challenged the utility of preorganization [18–24] also bears review in the face of the studies on HIV-1 protease. To quote from a review [18], “Preorganized ligands may bind to proteins with higher affinities than their flexible counterparts. However, we are aware of no convincing experimental evidence that this process must be entropically favored as is widely purported. Indeed, we have shown that the entropies of binding of preorganized ligands may be disfavored relative to their flexible controls even though the constrained ligands may bind with more favorable free energies.” Again, the assumption was made that the similarity in the crystal structures of the preorganized ligands versus the controls indicated no changes in the binding entropy of the protein. This assumption, reasonable at the time, must be questioned in the face of the strong thermodynamic argument that the correct preorganization of the ligand should result in tighter binding to its receptor as well as the data on HIV-1 protease presented above.
While this possibility has not been considered, the Martin group has been diligent is seeking to understand their observations. In a recent comprehensive study combining NMR and MD simulations [24], they concluded, “Small, almost “invisible” changes in the bound state geometry appear to impact the binding energetics… We find no evidence in this study that the compensating contributions in the measured enthalpy-entropy components arise from the same physical basis, and thus enthalpy-entropy compensation is not an intrinsic property of preorganization. That is, the reduced mobility from preorganization of the ligand leads to a more favorable binding entropy, but this reduced ligand mobility does not appear to be the origin of the less favorable binding enthalpy through an effect of the restraint on the conformational dynamics.” Without determining the impact of the different ligands on the conformational ensemble of the protein as was done with HIV protease, this is a reasonable conclusion.
Conclusions
It is difficult to ignore the obvious. When we limit the physical bases of molecular recognition to those we can conveniently incorporate into structure-based design, then we can expect to have limited success only when our limiting assumptions are consistent with the system under study. Unfortunately, biology does not occur at zero degrees Kelvin where we can ignore entropy. With the ever-increasing evidence for disorder in protein structures that modulates functionality [25] and their role in human disease [26], one should anticipate a continuum of examples of changes in protein entropy upon ligand binding. Unfortunately, such changes in the entropy of the protein target on binding of ligands cannot be ignored as we try to predict binding affinities. It is only one of several sins of omission that we commit in structure-based drug design. The remaining problems of monopole electrostatics and inadequate sampling will have to wait another diatribe.
Acknowledgments
Many have contributed to my limited understanding of the limitations of what we have collectively attempted. Certainly, David Barry, Denise Beusen, Heinz Bosshard, Dick Cramer, Richard Dammkoehler, Wayne Hubbell, Martin Karplus, Charlie Molnar, Phil Needleman, Tudor Oprea, Jay Ponder, Jake Schaefer, Harold Scheraga, Andy Vinter, Svante Wold, and many others (too numerous to mention) have generously pointed out critical mistakes in my thinking along my path to humility. My scientific gratitude to the EPR community whose work on HIV-1 protease has so clearly elucidated the problem. My thanks as well to the modeling community for indulging both my passion and my naivety. If one really knew how difficult was the journey, would one have taken that first step?
References
- 1.Feng JA, Marshall GR. SKATE: a docking program that decouples systematic sampling from scoring. J Comput Chem. 2010;31:2540–2554. doi: 10.1002/jcc.21545. [DOI] [PubMed] [Google Scholar]
- 2.Kear JL, Blackburn ME, Veloro AM, Dunn BM, Fanucci GE. Subtype polymorphisms among HIV-1 protease variants confer altered flap conformations and flexibility. J Am Chem Soc. 2009;131:14650–14651. doi: 10.1021/ja907088a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blackburn ME, Veloro AM, Fanucci GE. Monitoring inhibitor-induced conformational population shifts in HIV-1 protease by pulsed EPR spectroscopy. Biochemistry. 2009;48:8765–8767. doi: 10.1021/bi901201q. [DOI] [PubMed] [Google Scholar]
- 4.Galiano L, Ding F, Veloro AM, Blackburn ME, Simmerling C, Fanucci GE. Drug pressure selected mutations in HIV-1 protease alter flap conformations. J Am Chem Soc. 2009;131:430–431. doi: 10.1021/ja807531v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Torbeev VY, Raghuraman H, Mandal K, Senapati S, Perozo E, Kent SB. Dynamics of “flap” structures in three HIV-1 protease/inhibitor complexes probed by total chemical synthesis and pulse-EPR spectroscopy. J Am Chem Soc. 2009;131:884–885. doi: 10.1021/ja806526z. [DOI] [PubMed] [Google Scholar]
- 6.Banham JE, Baker CM, Ceola S, Day IJ, Grant GH, Groenen EJ, Rodgers CT, Jeschke G, Timmel CR. Distance measurements in the borderline region of applicability of CW EPR and DEER: a model study on a homologous series of spin-labelled peptides. J Magn Reson. 2008;191:202–218. doi: 10.1016/j.jmr.2007.11.023. [DOI] [PubMed] [Google Scholar]
- 7.Jeschke G, Koch A, Jonas U, Godt A. Direct conversion of EPR dipolar time evolution data to distance distributions. J Magn Reson. 2002;155:72–82. doi: 10.1006/jmre.2001.2498. [DOI] [PubMed] [Google Scholar]
- 8.Lopez CJ, Fleissner MR, Guo Z, Kusnetzow AK, Hubbell WL. Osmolyte perturbation reveals conformational equilibria in spin-labeled proteins. Protein Sci. 2009;18:1637–1652. doi: 10.1002/pro.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lovett JE, Bowen AM, Timmel CR, Jones MW, Dilworth JR, Caprotti D, Bell SG, Wong LL, Harmer J. Structural information from orientationally selective DEER spectroscopy. Phys Chem Chem Phys. 2009;11:6840–6848. doi: 10.1039/b907010a. [DOI] [PubMed] [Google Scholar]
- 10.Chiang YW, Borbat PP, Freed JH. The determination of pair distance distributions by pulsed ESR using Tikhonov regularization. J Magn Reson. 2005;172:279–295. doi: 10.1016/j.jmr.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 11.Ward R, Bowman A, El-Mkami H, Owen-Hughes T, Norman DG. Long distance PELDOR measurements on the histone core particle. J Am Chem Soc. 2009;131:1348–1349. doi: 10.1021/ja807918f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding F, Layten M, Simmerling C. Solution structure of HIV-1 protease flaps probed by comparison of molecular dynamics simulation ensembles and EPR experiments. J Am Chem Soc. 2008;130:7184–7185. doi: 10.1021/ja800893d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lovett JE, Hoffmann M, Cnossen A, Shutter AT, Hogben HJ, Warren JE, Pascu SI, Kay CW, Timmel CR, Anderson HL. Probing flexibility in porphyrin-based molecular wires using double electron electron resonance. J Am Chem Soc. 2009;131:13852–13859. doi: 10.1021/ja905796z. [DOI] [PubMed] [Google Scholar]
- 14.Galiano L, Bonora M, Fanucci GE. Interflap distances in HIV-1 protease determined by pulsed EPR measurements. J Am Chem Soc. 2007;129:11004–11005. doi: 10.1021/ja073684k. [DOI] [PubMed] [Google Scholar]
- 15.Smythe ML, Nakaie CR, Marshall GR. α-versus 310-helical conformation of alanine-based peptides in aqueous solution: an electron spin resonance investigation. J Am Chem Soc. 1995;117:10555–10562. [Google Scholar]
- 16.Galiano L, Bonora M, Fanucci GE. Interflap distances in HIV-1 protease determined by pulsed EPR measurements. J Am Chem Soc. 2007;129:11004–11005. doi: 10.1021/ja073684k. [DOI] [PubMed] [Google Scholar]
- 17.Tang YT, Marshall GR. PHOENIX: a scoring function for affinity prediction derived using high-resolution crystal structures and calorimetry measurements. J Chem Inf Model. 2011;51:214–228. doi: 10.1021/ci100257s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin SF. Preorganization in biological systems: are conformational constraints worth the energy? Pure Appl Chem. 2007;79:193–200. [Google Scholar]
- 19.Benfield AP, Teresk MG, Plake HR, DeLorbe JE, Millspaugh LE, Martin SF. Ligand preorganization may be accompanied by entropic penalties in protein-ligand interactions. Angew Chem Int Ed Engl. 2006;45:6830–6835. doi: 10.1002/anie.200600844. [DOI] [PubMed] [Google Scholar]
- 20.Benfield AP, Whiddon BB, Clements JH, Martin SF. Structural and energetic aspects of Grb2-SH2 domain-swapping. Arch Biochem Biophys. 2007;462:47–53. doi: 10.1016/j.abb.2007.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clements JH, DeLorbe JE, Benfield AP, Martin SF. Binding of flexible and constrained ligands to the Grb2 SH2 domain: structural effects of ligand preorganization. Acta Crystallogr D Biol Crystallogr. 2010;66:1101–1115. doi: 10.1107/S0907444910035584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeLorbe JE, Clements JH, Teresk MG, Benfield AP, Plake HR, Millspaugh LE, Martin SF. Thermodynamic and structural effects of conformational constraints in protein-ligand interactions. Entropic paradoxy associated with ligand preorganization. J Am Chem Soc. 2009;131:16758–16770. doi: 10.1021/ja904698q. [DOI] [PubMed] [Google Scholar]
- 23.Delorbe JE, Clements JH, Whiddon BB, Martin SF. Thermodynamic and structural effects of macrocyclization as a constraining method in protein-ligand interactions. ACS Med Chem Lett. 2010;1:448–452. doi: 10.1021/ml100142y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ward JM, Gorenstein NM, Tian J, Martin SF, Post CB. Constraining binding hot spots: NMR and molecular dynamics simulations provide a structural explanation for enthalpy-entropy compensation in SH2-ligand binding. J Am Chem Soc. 2010;132:11058–11070. doi: 10.1021/ja910535j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kissinger CR, Dunker AK, Shakhnovich E. Disorder in protein structure and function. Pac Symp Biocomput. 1999;4:517–519. [Google Scholar]
- 26.Uversky VN, Oldfield CJ, Dunker AK. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu Rev Biophys. 2008;37:215–246. doi: 10.1146/annurev.biophys.37.032807.125924. [DOI] [PubMed] [Google Scholar]
- 27.Jeschke G. Distance measurements in the nanometer range by pulse EPR. Chemphyschem. 2002;3:927–932. doi: 10.1002/1439-7641(20021115)3:11<927::AID-CPHC927>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]