

Abstract
A member of the amidohydrolase superfamily, BmulJ_04915 from Burkholderia multivorans, of unknown function was determined to hydrolyze a series of sugar lactones: L-fucono-1,4-lactone, D-arabino-1,4-lactone, L-xylono-1,4-lactone, D-lyxono-1,4-lactone and L-galactono-1,4-lactone. The highest activity was shown for L-fucono-1,4-lactone with a kcat value of 140 s−1 and a kcat/Km value of 1.0 × 105 M−1 s−1 at pH 8.3. The enzymatic product of an adjacent L-fucose dehydrogenase, BmulJ_04919, was shown to be L-fucono-1,5-lactone, via NMR spectroscopy. L-fucono-1,5-lactone is unstable and rapidly converts non-enzymatically to L-fucono-1,4-lactone. Due to the chemical instability of L-fucono-1,5-lactone, 4-deoxy-L-fucono-1,5-lactone was enzymatically synthesized from 4-deoxy-L-fucose using L-fucose dehydrogenase. BmulJ_04915 hydrolyzed 4-deoxy-L-fucono-1,5-lactone with a kcat value of 990 s−1 and a kcat/Km value of 8.0 × 106 M−1 s−1 at pH 7.1. The protein does not require divalent cations in the active site for catalytic activity. BmulJ_04915 is the second enzyme from cog3618 of the amidohydrolase superfamily that does not require a divalent metal for catalytic activity. BmulJ_04915 is the first enzyme that has been shown to catalyze the hydrolysis of either L-fucono-1,4-lactone or L-fucono-1,5-lactone. The structures of the fuconolactonase and the fucose dehydrogenase were determined by X-ray diffraction methods.
L-fucose is a widely distributed monosaccharide throughout the biosphere having functions in both plant and mammalian glycoconjugates (1, 2). These glycoconjugates include the ABO blood group antigens in mammals, L-fucose binding lectins, glycolipids and L-fucopyranosyl moieties in cell walls (1). L-Fucose can function as the sole carbon source for many microbes and has been shown to be metabolized via two separate pathways as illustrated in Figure 1 (2–4). In the first pathway, L-fucose is isomerized to L-fuculose and then phosphorylated to L-fuculose-1-phosphate. It is subsequently cleaved to L-lactaldehyde and dihydroxyacetone phosphate (3). In an alternative pathway, L-fucose is oxidized to L-fuconolactone and then hydrolyzed to L-fuconate. This product is subsequently dehydrated to 2-keto-3-deoxy-L-fuconate and then oxidized to 2,4-diketo-3-deoxy-L-fuconate. This compound is then hydrolyzed to pyruvate and L-lactate (2). In the latter pathway, the enzymes that hydrolyze L-fuconolactone and 2,4-diketo-3-deoxy-L-fuconate have not been characterized.
Figure 1.

Metabolism of L-fucose. Pathway I produces dihydroxyacetone phosphate and L-lactaldehyde. Pathway II produces pyruvate and L-lactate through the oxidation of L-fucose to L-fucono-1,5-lactone.
We have determined the three-dimensional structure and functionally annotated BmulJ_04915 from Burkholderia multivorans ATCC 17616. Two homologues of this enzyme, Bamb_1224 from Burkholderia ambifaria AMMD and Patl_0798 from Pseudoalteromonas atlantica T6c, were also functionally annotated. These enzymes are members of the amidohydrolase superfamily (AHS) of enzymes from cog3618. Enzymes from this superfamily catalyze a diverse set of reactions including the hydrolysis of amide or ester bonds, deamination, decarboxylation, isomerization and hydration reactions (5). The structural hallmark for superfamily members is a distorted (β/α)8-barrel, which binds 0–3 divalent metal ions within the active site (5). The AHS has been separated into 24 clusters of orthologous groups (COG) by NCBI (6).
The functionally characterized enzymes from cog3618 include 2-pyrone-4,6-dicarboxylate lactonase (LigI), 4-sulfomuconolactonase (4-SML) and L-rhamnono-1,4-lactonase (7–9). A sequence similarity network for cog3618 at an E-value cutoff of 10−30 is presented in Figure 2A (10). In this figure each node in the network represents a protein and is connected to other proteins by edges when the BLAST E-values are smaller than 10−30. At this stringency the proteins within cog3618 split into two large groups that are arbitrarily designated here as Class I and Class II. When a more stringent E-value of 10−70 is applied to the sequence similarity network, the proteins organize into smaller groups (Figure 2B), which, due to their higher sequence similarity may be isofunctional. In this figure the groups are assigned an arbitrary numerical identifier (i.e. 1, 2, 3 etc.). L-Rhamnono-1,4-lactonase and BmulJ_04915 map to Class I, while LigI and 4-SML map to Class II. Genomic context analysis of the enzymes within Class I identifies many proteins of unknown function that are physically located near other genes that have been previously annotated as ABC-type carbohydrate transport systems, dehydrogenases, dehydratases and hydrolases. Therefore, many of the proteins of unknown function within Class I are likely involved in carbohydrate metabolism and are thus predicted to hydrolyze sugar lactones.
Figure 2.

(A) Cog3618 sequence similarity networks at an E value of 10−30 where each node represents a protein and an edge represents an E value between two proteins of 10−30 or smaller. (B) Cog3618 sequence similarity networks at an E value of 10−70 where each node represents a protein and an edge represents an E value between two proteins of 10−70 or smaller. The nodes are color coded as follows: predicted L-fucono-1,5-lactonase (yellow nodes), BmulJ_04915 (blue square), Bamb_1224 (blue circle), Patl_0798 (blue triangle), L-rhamnono-1,4-lactonase (orange node), LigI (red node) and 4SML (green node).
BmulJ_04915 is predicted to hydrolyze sugar lactones based on genomic context (Figure 3). The protein, BmulJ_04922, from cog1028 is 64% similar in amino acid sequence to 2-keto-3-deoxy-L-fuconate dehydrogenase (XCC4067) from Xanthomonas campestris. BmulJ_04920 is 60% similar in protein sequence to the known L-fuconate dehydratase from cog4948 (XCC4069) (11 and 33). However, BmulJ_04919 from cog1028 is not related to XCC4067; an enzyme that is known to catalyze the oxidation of L-fucose. The closest experimentally annotated homologue to BmulJ_04919 is L-rhamnose dehydrogenase (SKA58_03570) from Sphingomonas sp. SKA58 (9). In this paper we have determined the three-dimensional structures of BmulJ_04915 and BmulJ_04919 and have shown that BmulJ_04919 catalyzes the oxidation of L-fucose to L-fucono-1,5-lactone and that BmulJ_04915 and two homologues (Bamb_1224 and Patl_0789) catalyze the hydrolysis of this product to L-fuconate.
Figure 3.

Genomic neighborhood of BmulJ_04915. Genes are color coded as follows: cloned and purified for this study (red); predicted from strong sequence similarity to genes encoding proteins of known function (blue); predicted genes based on genomic context (yellow); predicted to function in carbohydrate transport (grey).
Materials and Methods
Materials
All chemicals and buffers were purchased from Sigma Aldrich unless otherwise specified. Sugar lactones which were not commercially available were synthesized according to published procedures with the exception of 4-deoxy-L-fucono-1,5-lactone which was enzymatically synthesized (12). The noncommercial lactones included the following: L-fucono-1,4-lactone (1), D-altrono-1,4-lactone (3), D-arabinono-1,4-lactone (6), L-xylono-1,4-lactone (7), L-mannono-1,4-lactone (11), D-talono-1,4-lactone (12), D-allono-1,4-lactone (13), L-rhamnono-1,4-lactone (14), D-lyxono-1,4-lactone (15), L-lyxono-1,4-lactone (16), L-arabinono-1,4-lactone (17), D-xylono-1,4-lactone (19), L-mannono-1,5-lactone (22), L-rhamnono-1,5-lactone (23) and 4-deoxy-L-fucono-1,5-lactone (24). Those sugar lactones that were available commercially included the following: L-galactono-1,4-lactone (2) L-glucono-1,4-lactone (4), D-idono-1,4-lactone (5), D-mannono-1,4-lactone (8), D-glucono-1,4-lactone (9), D-galactono-1,4-lactone (10), D-ribono-1,4-lactone (18), D-glucurono-6,3-lactone (20) and D-erythronolactone (21). These compounds were obtained from CarboSynth or ChromaDex. The structures of these lactones are presented in Scheme 1. The aldose sugars were obtained from either from Sigma Aldrich or Carbosynth and the structures are presented in Scheme 2. These sugars included L-fucose (25), L-galactose (26), L-glucose (27), D-altrose (28), D-arabinose (29), L-xylose (30), L-rhamnose (31),D-mannose (32), L-allose (33), D-talose (34), L-talose (35), D-allose (36), D-galactose (37), L-mannose (38), D-gulose (39), D-glucose (40), L-arabinose (41), L-ribose (42), D-lyxose (43), L-lyxose (44), D-xylose (45) D-ribose (46) and 4-deoxy-L-fucose (47).
Scheme 1.

Scheme 2.

Cloning, Expression and Purification of BmulJ_04915 from B. multivorans ATCC 17616 and Patl_0798 from P. atlantica T6c
The gene for BmulJ_04915 (gi|161520151) was amplified from Burkholderia multivorans ATCC 17616 genomic DNA using 5′-TTAAGAAGGAGATATACCATGGGTGCCCTGCGTATTGACTCAC-3′ as the forward primer and 5′-GATTGGAAGTAGAGGTTCTCTGCCAGACGAGCATCAGCCGGTTC-3′ as the reverse primer. The gene Patl_0798 (gi|109897125) was amplified from Pseudoalteromonas atlantica Strain T6c genomic DNA using 5′-TTAAGAAGGAGATATACCATGGTGATGAGAATTGATGCACACCAACATT-3′ as the forward primer and 5′-GATTGGAAGTAGAGGTTCTCTGCCAATCGG TAGAATCGTTCGGC-3′ as the reverse primer. PCR was performed using KOD Hot Start DNA Polymerase (Novagen). The conditions were: 2 minutes at 95 °C, followed by 40 cycles of 30 seconds at 95 °C, 30 seconds at 66°C, and 30 seconds at 72 °C. The amplified fragments were cloned into the C-terminal TEV cleavable StrepII-6x-His-tag containing vector, CHS30, by ligation-independent cloning (13).
All purification steps were performed at 4 °C and all growth media contained 100 μg/mL kanamycin and 35 μg/mL chloramphenicol. Plasmid was transformed into Rosetta2(DE3)pLysS (Novagen) and plated on LB agar plates. Five to ten colonies were used to inoculate 100 mL of LB media and allowed to grow overnight at 37 °C (250 rpm agitation). The overnight cultures were used to inoculate 4 L of autoinduction media in ten 2 L flasks and incubated (250 rpm agitation) at 25 °C for 24 hours reaching an OD600 of 20–25 (14 and 15). Cells were harvested by centrifugation and stored at -80 °C. Cells were re-suspended (4:1 v/w) in buffer A (50 mM Hepes pH 7.8, 150 mM NaCl, 10% glycerol, 20 mM imidazole) with 0.1% Tween 20 then disrupted by sonication. The cellular extract was clarified by centrifugation at 4 °C and applied to a 10 mL Ni-Sepharose HP (GE Healthcare) column. The column was washed with 10 column volumes of buffer A and proteins were step eluted with 300 mM imidazole in buffer A. TEV protease (16) was added at a ratio of 1/50 (w/w) to the pooled fractions which were subsequently dialyzed overnight against buffer A with 300 mM imidazole. The pooled eluate was then concentrated to 15–30 mg/mL and applied to a 16/60 Superdex 200 column (GE Healthcare) equilibrated against buffer B (20 mM Hepes pH 7.8, 150 mM NaCl, 5% glycerol). Fractions with greater than 95% purity by SDS-PAGE were passed through a 5 mL Ni-Sepharose HP (GE Healthcare) column to remove uncut protein and TEV protease. Protein not retained by the column was concentrated to 15–30 mg/mL by centrifugal ultrafiltration. Aliquots were snap frozen in liquid nitrogen and stored at −80° C.
Cloning, Expression, and Purification of Bamb_1224 from Burkholderia ambifaria AMMD
The gene for Bamb_1224 was cloned from B. ambifaria AMMD (gi|115351277) with the primer pair 5′-CAGGACCATATGACGTTCCGCATCGACGCTCATCAA-3′ and 5′-GCAGGACAAGCTTTTGAGCGTGTTGATCGCCGGACGGCCGA-3′. Restriction sites for Nde1 and HindIII were inserted into the forward and reverse primers, respectively. The PCR product was purified with a PCR clean-up system (Promega), doubly digested with Nde1 and HindIII, and then ligated into a pET-30 a (+) vector (Novagen) previously digested with Nde1 and HindIII. The recombinant plasmid harboring this gene was transformed into BL21 (DE3) cells (Novagen) via electroporation.
A single colony was used to inoculate 5 mL cultures containing LB and 50 μg/mL kanamycin, and allowed to grow overnight at 37 °C. The overnight cultures were used to inoculate 1 L of LB medium supplemented with 50 μg/mL kanamycin. These cultures were incubated until an OD600 of 0.4–0.6 was obtained and then 1.0 mM isopropyl-β-thiogalactoside (IPTG) was added to induce gene expression at ambient temperature (25 °C) for an additional 18 hours. Cells were harvested by centrifugation at 4 °C and then resuspended in 20 mM HEPES, pH 7.5. Approximately 0.1 mg/mL phenylmethylsulfonyl fluoride (PMSF) was added to the cell suspension and then the cells lysed via multiple rounds of sonication in 50 mM HEPES (pH 7.4), 0.5 M NaCl and 40 mM imidazole. Nucleic acids were removed from the supernatant solution by the addition of 2% w/v protamine sulfate solution over the course of 30 minutes at 4 °C. The protamine sulfate bound DNA was removed by centrifugation at 4 °C and the supernatant was collected and applied to a 5 mL HisTrap HP(GE Healthcare) nickel affinity column. Protein was eluted from the nickel affinity column with 50 mM HEPES (pH 7.4), 0.5 M NaCl and 500 mM imidazole over a gradient of 25 column volumes.
Cloning, Expression, and Purification of BmulJ_04919 from Burkholderia multivorans ATCC 17616
The gene for BmulJ_04919 was cloned from B. multivorans ATCC 17616 (gi| 189353674) with the primer pair 5′-GCAGGAGCCATATGGATCTGAATCTGCAGGACAAGGTCGT-3′ and 5′-GCAGGAGCAAGCTTTCAGACGAGCGCACGATCGAGATGCGTAT-3′. Restriction sites for Nde1 and HindIII were inserted into the forward and reverse primers, respectively. The PCR product was purified with a PCR clean-up system (Promega), doubly digested with Nde1 and HindIII, and then ligated into a pET-30 a (+) vector (Novagen) previously digested with Nde1 and HindIII. The recombinant plasmid harboring this gene was transformed into BL21 (DE3) cells (Novagen) via electroporation.
A single colony was used to inoculate 5 mL cultures containing LB and 50 μg/mL kanamycin, and allowed to grow overnight at 37 °C. The overnight cultures were used to inoculate 1 L of LB medium supplemented with 50 μg/mL kanamycin. These cultures were incubated until an OD600 of 0.4–0.6 was obtained and then 1.0 mM IPTG was added to induce gene expression at ambient temperature (25 °C) for an additional 18 hours. Cells were harvested by centrifugation at 4 °C and then resuspended in 20 mM HEPES, pH 7.5. Approximately 0.1 mg/mL PMSF was added to the cell suspension and then the cells lysed via multiple rounds of sonication. Nucleic acids were removed from the supernatant solution by the addition of 2% w/v protamine sulfate solution over the course of 30 minutes at 0 °C. The protamine sulfate bound DNA was removed by centrifugation at 4 °C. Solid ammonium sulfate was added to 50% of saturation and the precipitated protein isolated by centrifugation. The protein was resuspended in a minimum amount of 20 mM HEPES (pH 7.5) and then applied to a High Load 26/60 Superdex 200 gel filitration column (GE Healthcare). Fractions containing the protein of expected size were pooled and purified further by ion exchange chromatography with a ResourceQ column (6 mL) at pH 7.5.
Crystallization and Data collection of BmulJ_04915 and BmulJ_04919
All crystallization experiments were carried out by the sitting drop vapor diffusion method at 18 °C in 96 well Intelliplates (Art Robbins Instruments). Equal volumes of crystallization reagent and protein (0.5 + 0.5 μL) were mixed and equilibrated against 70 μL of the crystallization agent in the reservoir. Crystals of BmulJ_04915 (30 mg/mL in 20 mM HEPES pH 7.8, 150 mM NaCl, 5% glycerol, 0.5 mM ZnCl2, 9 mM TCEP) were obtained with 20% Peg3350 (w/v), 100 mM HEPES, pH 7.5. Hexagonal rods (0.2 × 0.05 × 0.05 mm) grew sporadically over 1–2 months. Crystals were soaked in mother liquor supplemented with 20% glycerol (v/v) for 10 seconds prior to flash-cooling in liquid nitrogen. Crystals of unliganded BmulJ_04919 (14.5 mg/mL in 50 mM TRIS pH 7.5) were obtained with 0.2 M calcium acetate, 10% (w/v) PEG 8000, 0.1 M HEPES pH 7.5. Triganol bipyramids appeared in 1–2 days and grew to maximal size in a week (0.2 × 0.1 × 0.1 mm). Crystals were soaked in mother liquor supplemented with 20% ethylene glycol for 10 seconds prior to flash-cooling in liquid nitrogen. Crystals of the BmulJ_04919 (14.5 mg/mL in 50 mM TRIS pH 7.5, 5 mM NADP+, 25 mM L-fucose) ternary complex were obtained in 0.2 M MgCl2, 20% Peg8000 (w/v), 0.1 M TRIS pH 8.5. Crystals of the ternary complex formed as small (0.05 × 0.025 × 0.025 mm) misshapen plates that grew at the periphery of the drop over 1–2 weeks. Crystals were soaked in mother liquor supplemented with 5 mM NADP+, 25 mM L-fucose and 20% (v/v) ethylene glycol for two minutes prior to flash-cooling in liquid nitrogen. All X-ray diffraction data were collected under at 100 K using synchrotron radiation (λ=0.98) and intensities were integrated with MOSFLM and scaled with SCALA (4DNM,4GKB) or HKL3000 (4GVX) (17–19). Data for BmulJ_04915 were collected on a Quantum 315 CCD detector (Area Detector Systems Corp) at NSLS beamline X29A (National Synchrotron Light Source, Brookhaven National Laboratory). Data for BmulJ_04919 were collected on a Rayonix 225-HE detector (Rayonix) at APS beamline 31-ID (Advanced Photon Source, Argonne National Laboratory).
Structure Determination of BmulJ_04915 and BmulJ_04919
Initial phases for BmulJ_04915 were determined by molecular replacement (MR) using PHENIX/PHASER and a monomeric ensemble model generated by an overlay of the structures represented by PDB IDs 2QAH, 2FFI and 3CJP; all homologs with less than 20% sequence identity (20 and 21). Starting with the MR phases, the autobuild wizard of PHENIX was able to build a majority of the structure with an R/Rfree of 26.9 and 30.8%, respectively. Initial phases for unliganded BmulJ_04919 were determined by MR using MOLREP and a tetrameric search model (3FTP) (22). A BmulJ_04919 homology model, constructed utilizing the SWISSPDB homology modeling server, was subsequently placed according to the MOLREP MR solution (23). This model was subjected to simulated annealing using torsion angle dynamics within PHENIX, generating an initial model with an R/Rfree of 39.2 and 42.5%, respectively. The structure of the NADP/L-Fucose BmulJ_04919 ternary complex was determined using a single subunit of the unliganded complex. Iterative cycles of refinement in PHENIX and model building in COOT were utilized to obtain the final crystallographic structures (24). Ligand geometry restraints were built using ELBOW within PHENIX.
Measurement of Enzyme Activity
The enzymatic hydrolysis of lactones was monitored with a SpectraMax340 UV—visible spectrophotometer using a colorimetric pH indicator assay at 30 °C. Protons released from the carboxylate product during lactone hydrolysis were measured using the pH indicators cresol purple or bromothymol blue (25). Reactions measured with cresol purple contained 2.5 mM BICINE buffer (pH 8.3), 0.2 M NaCl, 0–1.0 mM lactone, 0.1 mM cresol purple and the lactonase. Reactions measured with bromothymol blue contained 2.5 mM MOPS buffer (pH 7.1), 0.2 M NaCl, 0–0.5 mM lactone, and 0.1 mM bromothymol blue. The final concentration of DMSO was 1%. Changes in absorbance at 577 nm (ε= 1764 M−1 cm−1) and 616 nm (ε = 1135 M−1 cm−1) were monitored in 96-well plates for cresol purple and bromothymol blue, respectively. Background rates arising from acidification by atmospheric CO2 were observed and subtracted from the initial rates. The dehydrogenase activity catalyzed was monitored with a SpectraMax340 UV—visible spectrophotometer by measuring the reduction of NADP+ or NAD+ at 340 nm (ε = 6220 M−1 cm−1) at 30 °C. The assays were performed in 50 mM BICINE buffer (pH 8.0), varying concentrations of sugar substrates, and 0.5 mM NADP+. Kinetic constants for NADP+ and NAD+ were determined in the same manner as described above except that the assay conditions were as follows: 50 mM BICINE buffer (pH 8.0), varying concentrations of nucleotide, and 200 μM L-fucose.
Data Analysis
The kinetic constants were determined from a fit of the initial velocity data to equation 1 using the non-linear least squares fitting program in SigmaPlot 9.0, where v is the initial velocity, Et is the total enzyme concentration, kcat is the turnover number, [A] is the substrate concentration, and Km is the Michaelis constant.
| (1) |
Metal Analysis
The metal content of BmulJ_04915 was determined with an Elan DRC II ICP-MS as previously described (26). Protein samples for ICP-MS analysis were digested with HNO3 and then refluxed for 30 minutes (27). All buffers were passed through a column of Chelex 100 (Bio-Rad) to remove trace metal contamination. EDTA and 1,10-phenanthroline (1.0 mM) were incubated with 1.0 μM BmulJ_04915 in 50 mM buffer at pH values ranging from 6 to 10 to remove divalent metal ions. The buffers for these experiments included CHES (pH 6.0), HEPES (pH 7.0), BICINE (pH 8.0) or CHES (pH 9.0 and 10.0). The effect of added divalent cations on the catalytic activity of BmulJ_04915 was determined by adding Mn2+, Zn2+, Co2+, Cu2+ or Ni2+ (0 – 500 μM) directly to the assay mixtures. The purified enzyme was also incubated with 50 – 500 molar equivalents of these divalent cations for 24 hours at 4 °C in 50 mM HEPES (pH 7.5) and subsequently assayed for catalytic activity.
Reaction Product of BmulJ_04919 with L-Fucose
The product of the reaction catalyzed by BmulJ_04919 was determined through 1H, 13C, Hetero Nuclear Multiple Bond Correlation (HMBC) and Correlation NMR spectroscopy using a Bruker Avance III 500 MHz with an H-C-N cryoprobe. WATERGATE solvent suppression was performed on reaction samples in 500–1000 μL D2O with 100 mM acetate buffer (pH 5.5), 25 mM NAD+, 25 mM L-fucose and 100 μM BmulJ_04919. After incubating for 10 minutes, the enzyme was removed from the reaction mixture using a 3 KDa cutoff VWR Centrifugal Filter. NAD+ and NADH were removed by the addition of Dowex-1 X2 anion exchange resin that was previously equilibrated with 50 mM acetate buffer (pH 5.5). The pH of the sample was then adjusted to pH 4.2 with 1 M acetate buffer (pH 4.2).
L-Fucono-1,5-lactone was identified as a substrate for BmulJ_04915 using 1H NMR spectroscopy. The reactions were conducted with 50 mM phosphate buffer (pH 6.5), 15 mM L-fucose, 15 mM NAD+, 10 μM BmulJ_04919 and 10 μM BmulJ_04915 in a final reaction volume of 200 μL. 1H-NMR spectra were collected over 1 minute intervals with WATERGATE solvent suppression.
Enzymatic Synthesis of 4-Deoxy-L-fucono-1,5-lactone
4-Deoxy-L-fucono-1,5-lactone (24) was synthesized enzymatically from 4-deoxy-L-fucose (47) using BmulJ_04919 as the catalyst. The reaction was conducted in 50 mM phosphate buffer (pH 7.0), 15 mM 4-deoxy-L-fucose and 15 mM NAD+ in a final volume of 1 mL for 20 minutes. The enzyme was removed from the reaction mixture using a 3 KDa cutoff VWR Centrifugal Filter. NAD+ and NADH were removed by the addition of Dowex-1 X2 anion exchange resin. The structure of the lactone was determined using Hetero Nuclear Multiple Bond Correlation (HMBC) and Correlation NMR spectroscopy with a Bruker Avance III 500 MHz with an H-C-N cryoprobe using WATERGATE solvent suppression
Sequence Similarity Network for cog3618
Proteins belonging to cog3618 were identified from the NCBI protein database using the query, “cog3618”. The proteins within cog3618 were subjected to an all-by-all BLAST at a specified E-value (10−50, 10−60 etc.) using the NCBI standalone BLAST program. The BLAST files were opened and visualized in the similarity network program Cytoscape (28).
Results
Purification of BmulJ_04915
The gene for BmulJ_04915 was cloned, expressed in E. coli and the protein purified to homogeneity. The purified enzyme contained ~0.7 molar equivalents of Zn2+. The addition of chelating agents or divalent metal ions (Mn2+, Zn2+, Co2+, Cu2+ or Ni2 ) directly to the assay mixture did not affect the rate of the enzyme-catalyzed hydrolysis of L-fucono-1,4-lactone (1). The purified enzyme was incubated with EDTA or 1,10-phenanthroline at pH values ranging from 6 – 10 to chelate the metal ions bound to the protein. The addition of chelators did not diminish the catalytic activity of this enzyme and ICP-MS analysis demonstrated that the addition of 1,10-phenanthroline removed >95% of the Zn2+ that was initially bound to the protein. The apo-enzyme had the same catalytic activity as the as-purified enzyme. Therefore, BmulJ_04915 does not require a divalent cation for substrate turnover.
Substrate Specificity
BmulJ_04915 is a member of cog3618 from the amidohydrolase superfamily. Other enzymes from cog3618 have been shown to catalyze the hydrolysis of lactones (7–9). The genomic context is indicative of an enzyme that participates in the metabolism of carbohydrates and thus this enzyme was predicted to catalyze the hydrolysis of sugar lactones. The enzyme was screened against a small and focused library of 23 lactones as potential substrates for this enzyme (see Scheme 1). BmulJ_04915 exhibited catalytic activity for the hydrolysis of L-fucono-1,4-lactone (1), D-arabino-1,4-lactone (6), L-xylono-1,4-lactone (7) and L-galactono-1,4-lactone (2). The best substrate from this initial set of compounds is L-fucono-1,4-lactone. The kinetic constants are provided in Table 1.
Table 1.
Catalytic constants for BmulJ_04915, Bamb_1224 and Patl_0798
| Enzyme | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| Compound | pH | BmulJ_04915 | Bamb_1224 | Patl_0798 | ||||||
| 8.3 | kcat (s−1) | Km (mM) | kcat/Km (M−1 s−1) | kcat (s−1) | Km (mM) | kcat/Km (M−1 s−1) | kcat (s−1) | Km (mM) | kcat/Km (M−1 s−1) | |
|
| ||||||||||
| L-fucono-1,4-lactone (1) | 140 ± 8 | 1.4 ± 0.2 | 1.0 (0.1) × 105 | 89 ± 4 | 0.9 ± 0.1 | 1.0 (0.1) × 105 | 139 ± 6 | 1.2 ± 0.1 | 1.2 (0.2) × 105 | |
| L-galactono-1,4-lactone (2) | 32 ± 2 | 1.1 ± 0.2 | 3.0 (0.3) × 104 | 55 ± 2 | 1.3 ± 0.1 | 4.0 (0.3) × 104 | 119 ± 7 | 1.5 ± 0.2 | 8.0 (0.5) × 104 | |
| D-arabinono-1,4-lactone (6) | 59 ± 2 | 0.7 ± 0.05 | 8.4 (0.5) × 104 | 19 ± 1 | 0.8 ± 0.1 | 2.4 (0.2) × 104 | 25 ± 3 | 0.75 ± 0.02 | 3.3 (0.2) × 104 | |
| L-xylono-1,4-lactone (7) | 14 ± 1 | 1.5 ± 0.3 | 1.0 (0.1) × 104 | 6.0 ± 0.5 | 1.3 ± 0.3 | 5.0 (0.5) × 103 | 10 ± 0.3 | 1.5 ± 0.4 | 6.6 (0.2) × 104 | |
| 4-deoxy-L-fucono-1,5-lactone (24) | NDa | ND | ND | ND | ND | ND | ND | ND | ND | |
|
| ||||||||||
| 7.1 | kcat (s−1) | Km (mM) | kcat/Km (M−1 s−1) | kcat (s−1) | Km (mM) | kcat/Km (M−1 s−1) | kcat (s−1) | Km (mM) | kcat/Km (M−1 s−1) | |
|
| ||||||||||
| L-fucono-1,4-lactone (1) | 4.0 ± 0.1 | 0.6 ± 0.1 | 7 (0.1) × 104 | 4.1 ± 0.3 | 0.9 ± 0.1 | 5.0 (0.1) × 103 | 3.0 ± 0.2 | 0.6 ± 0.1 | 4.5 (0.5) × 103 | |
| L-galactono-1,4-lactone (2) | 6.0 ± 0.5 | 1.3 ± 0.2 | 5.0 (0.5) × 103 | 6.2 ± 2.0 | 0.5 ± 0.1 | 4.0 (0.5) × 103 | 5.7 ± 0.3 | 2.6 ± 0.3 | 2.2 (0.1) × 103 | |
| D-arabinono-1,4-lactone (6) | 2.0 ± 0.2 | 1.0 ± 0.3 | 1.6 (0.3) × 103 | 2.2 ± 0.2 | 0.7 ± 0.1 | 3.2 (0.2) × 103 | 1.8 ± 0.2 | 1.6 ± 0.3 | 1.0 (0.1) × 103 | |
| L-xylono-1,4-lactone (7) | ND | ND | 1.0 (0.1) × 103 | ND | ND | 7.0 (0.3) × 102 | ND | ND | 1.0 (0.2) × 103 | |
| 4-deoxy-L-fucono-1,5-lactone (24) | 994 ± 64 | 0.13 ± 0.03 | 8.0 (1.0) × 106 | 1530 ± 47 | 0.21 ± 0.02 | 7.2 (0.6) × 106 | 1320 ± 80 | 0.45 ± 0.02 | 2.6 (0.2) × 106 | |
Kinetic constants were not determined for this substrate
Structure of BmulJ_04915
The structure of BmulJ_04915 was determined by molecular replacement to 2.15 A resolution (Table 2). There is a monomer in the asymmetric unit that is consistent with the protein being a monomer in solution. The final model contains residues 3–289, all of which are well defined in electron density. The crystal from which BmulJ_04915 was determined requires TCEP and Zn2+ (added prior to knowledge that BmulJ_04915 was not a metalloenzyme) to form and was only sporadically reproducible. A well-defined Zn2+ ion, coordinated by Asp-176 and His-222 of one molecule and Cys-71 and Asp-72 of a crystallographically related molecule, specifies how Zn2+ was critical to the crystallization. BmulJ_04915 forms a distorted (β/α)8-TIM barrel with an active site at the C-terminal end of the β-barrel consistent with other members of the amidohydrolase family (Figure 4A). There are few inserted secondary structural elements, with most of the defining structural features being extended loops connecting the (β/α′)8-TIM barrel elements. Residual density within the active site was modeled as a bound HEPES molecule derived from the crystallization buffer. The piperazine ring lies against the face of Trp-22 (loop between β-strand 1/α-helix 1) while the sulfonate is interacting with Arg-106 and the three active site histidine residues (His-9, His-11, His-166) (Figure 4B). A search using the SSM server returns a number of amidohydrolases at > 2.0 A RMSD and less than 20% sequence identity demonstrating that the structure of BmulJ_04915 is significantly different from those previously determined (29). The closest structure is that of the 2-pyrone-4,6-dicarboxylic acid hydrolase (LigI) from Sphingomonas paucimobilis (RMSD 2.2A over 229 Cα, 20% sequence identity) which also catalyzes the hydrolysis of a lactone (7).
Table 2.
Data Collection and Refinement Statistics for the BmulJ_04915 and BmulJ_04919 Crystal Structures
| DATASET STATISTICSa | BmulJ_04915 | apo-BmulJ_04919 | BmulJ_04919 Ternary complex |
| Space Group | P3221 | P61 | C2 |
| Unit Cell (Å, °) | a=75.1 c=142.3 | a=100.0 c=205.8 |
a=68.4 b= 118.5 c=118.2 β=92.9 |
| Resolution (Å) | 40-2.15 (2.27-2.15) | 40-1.5 (1.52-1.50) | 40-1.5 (1.52-1.5) |
| Completeness (%) | 99.9 (99.8) | 100.0 (100.0) | 96.7 (98.7) |
| Redundancy | 11.4 (11.7) | 8.9 (9.3) | 2.8 (2.7) |
| Mean(I)/sd(I) | 17.3 (3.5) | 10.8 (2.8) | 10.0 (1.8) |
| Rsym | 0.094 (0.777) | 0.110 (0.836) | 0.103 (0.494) |
| STRUCTURE STATISTICS | |||
| Resolution (Å) | 40-2.15 (2.23-2.15) | 40-1.5 (1.52-1.50) | 40-1.5 (1.52-1.50) |
| Unique reflections | 25899 (2790) | 185136 (6256) | 145306 (4808) |
| Rcryst (%) | 19.0 (25.3) | 15.1 (23.4) | 15.2 (20.3) |
| Rfree (%, 5% of data) | 22.8 (28.9) | 17.2 (24.9) | 17.9 (22.7) |
| Contents of model (Native Range)b | 1-296 | 1-258 | 1-258 |
| Observed residues | 3-289 | A1-191, A203-258, B1-258, C1-258, D1-194, D203-D258 | A1-258, B1-258, C1-258, D1-258 |
| Waters | 194 | 1098 | 1045 |
| Atoms total | 2526 | 8876 | 9232 |
| Average B-factor (Å2) | |||
| Wilson B Factor (Å2) | 25.6 | 13.8 | 8.9 |
| TLS groups (#) | 11.0 | 14.0 | 25.0 |
| Protein/Waters/NADP/Fucose | 30.6/39.8/-/- | 14.1/29.1/-/- | 9.4/23.9/7.7/6.8 |
| RMSD | |||
| Bond lengths (Å)/Angles (°) | 0.01/1.11 | 0.008/1.12 | 0.006/1.17 |
| MOLPROBITY STATISTICS | |||
| Ramachandran Favored/Outliers (%) | 98.6 (0.35) | 96.9 (0.0) | 96.9 (0.0) |
| Rotamer Outliers (%) | 1.4 | 1.4 | 1.4 |
| Clashscored | 8.38 (91st pctl.) | 5.37 (91st pctl.) | 3.81 (95th pctl.) |
| Overall scored | 1.56 (97th pctl.) | 1.59 (78th pctl.) | 1.47 (87th pctl.) |
| PDB ID | 4DNM | 4GKB | 4GVX |
Statistics in parenthesis are for the highest resolution bin
numbering outside of listed native range are polyhistidine tags or cloning artifacts
Scores are ranked according to structures of similar resolution as formulated in MOLPROBITY
Figure 4.

Structure of BmulJ_04915. (A) Ribbon diagram of BmulJ_04915. HEPES molecule shown as spheres. (B) Residues directly adjacent to bound HEPES in the active site of BmulJ_04915. Numbers in parenthesis designate the approximate location in the secondary structure from which the residue originates.
Purification and Properties of BmulJ_04919
A gene for a protein currently annotated as a short chain dehydrogenase of unknown specificity (BmulJ_04919) is positioned in the same putative operon as BmulJ_04915. This gene was cloned, overexpressed and the protein purified to homogeneity. BmulJ_04919 is a member of cog1028, which includes other enzymes with the following experimentally verified functions: 2-dehydro-3-deoxy-L-fuconate dehydrogenase (11), 2-dehydro-3-deoxy-L-rhamnonate dehydrogenase (9), L-rhamnose dehydrogenase (9), 2,3-dihydro-2,3-dihydroxybenzoate dehydrogenase (30), D-gluconate dehydrogenase (31) and sorbitol-6-phosphate dehydrogenase (32). The closest homolog to BmulJ_04919 with a verified catalytic activity in cog1028 appears to be L-rhamnose dehydrogenase from Sphingomonas sp. SKA58. BmulJ_04919 is unrelated to the putative L-fucose dehydrogenase (XCC4065 from Xanthomonas campestris) from cog0667 (11 and 33). The oxidized product of the reaction catalyzed by BmulJ_04919 is likely to be the physiological substrate for BmulJ_04915.
Substrate Specificity of BmulJ_04919
It was anticipated that BmulJ_04919 would catalyze the formation of L-fucose-1,5-lactone from L-fucose and NADP+ since the pyranose form of L-fucose is predominant in solution (Figure 6A). To determine the substrate specificity of this enzyme, a small library of pentose and hexose sugars was tested as potential substrates by monitoring the reduction of NADP+ at 340 nm (Scheme 2). L-fucose (25), L-galactose (26) and D-arabinose (29) were the only sugars oxidized by this enzyme. The dehydrogenase has the highest activity with L-fucose and the kinetic constants are presented in Table 3.
Figure 6.

1H-NMR time course of the enzymatic conversion of L-fucose to L-fucono-1,5-lactone and L-fucono-1,5-lactone to L-fuconate. The resonances corresponding to the C-6 methyl groups of both α-(1.125 ppm) and β-L-fucose (1.15 ppm), L-fucono-1,5-lactone (1.295 ppm), L-fucono-1,4-lactone (1.225 ppm) and L-fuconate (1.182 ppm) are provided. (A) L-fucose, the substrate for BmulJ_04919, prior to addition of enzyme at pH 6.5. (B) Five minutes after the addition of BmulJ_04919 to L-fucose, the enzymatic product, L-fucono-1,5-lactone, and the non-enzymatic product L-fucono-1,4-lactone are at equal concentrations. (C) Five minutes after the addition of BmulJ_04915 to the reaction mixture. The L-fucono-1,5-lactone is no longer present in the reaction mixture and the L-fucono-1,4-lactone appears to be unchanged. L-fuconate (1.182 ppm) appears to be the major product. The α-anomer of L-fucose appears to be unchanged, however, the β-anomer has been reduced to at least half of the original concentration.
Table 3.
Catalytic constants for BmulJ_04919 at pH 8.0a
| Substrate | kcat (s−1) | Km (μM) | kcat/Km (M−1 s−1) |
|---|---|---|---|
| L-fucose (25) | 10 ± 0.3 | 6.2 ± 0.8 | 1.5 (0.2) × 106 |
| D-arabinose (29) | 21.0 ± 0.1 | 94 ± 3 | 2.3 (0.1) × 105 |
| L-galactose (26) | 11 ± 0.5 | 285 ± 36 | 3.7 (0.3) × 104 |
| 4-deoxy-L-fucose (47) | 26 ± 1 | 786 ± 92 | 4.0 (0.1) × 104 |
| NADP+ | 11 ± 0.2 | 4.0 ± 0.5 | 3.0 (0.4) × 106 |
| NAD+ | 5 ± 1 | 28 ± 4 | 5.3 (0.1) × 105 |
When NADP+ or NAD+ was varied the L-fucose concentration was fixed at 200 μM. When the sugar substrates were varied the NADP+ concentration was fixed at 500 μM.
Identification of the Initial Reaction Product of BmulJ_04919
The only product of an L-fucose dehydrogenase that has ever been structurally characterized is L-fucono-1,4-lactone (1) (35). However, it has been proposed that L-fucono-1,5-lactone is the true product of the reaction, but this compound has never been observed due to the rapid non-enzymatic conversion to L-fucono-1,4-lactone (1) or hydrolysis to L-fuconate (34 and 35). To help stabilize product formation, the reaction was conducted at pH 5.5 and then quenched at pH 4.2. The initial product was structurally characterized by NMR spectroscopy and shown to be L-fucono-1,5-lactone with the resonance for the methyl group at 1.32 ppm, which quickly rearranges to the 1,4-lactone (1.24 ppm) at pH values above 5.0. For further characterization, the reaction was quenched at pH 4.5, which greatly slowed the intermolecular rearrangement.
The enzymatically produced L-fucono-1,5-lactone is rapidly converted to L-fucono-1,4-lactone (1). Enzyme, NAD+, and L-fucose were added together at pH 5.5 and the reaction monitored by 1H-NMR spectroscopy until L-fucono-1,5-lactone was the predominate product. The enzyme was removed from the reaction mixture and the pH adjusted to 6.5. The time course for the subsequent change in the 1H-NMR spectrum for the C-6 methyl carbons of L-fucose (25) and the reaction products is presented in Figure 5. At 1.1 and 1.2 ppm there is a pair of doublets for the α-and β-anomers of L-fucose (25), respectively. At 1.24 and 1.32 ppm are the two doublets for L-fucono-1,4-lactone (1) and L-fucono-1,5-lactone, respectively (Figure 5A). At the earliest time points, L-fucono-1,5-lactone is clearly dominant relative to L-fucono-1,4-lactone (1). However, after five minutes approximately half of the L-fucono-1,5-lactone has been converted to the L-fucono-1,4-lactone (Figure 5B). After one hour nearly all of the L-fucono-1,5-lactone was converted to L-fucono-1,4-lactone (1) (Figure 5C). The nonenzymatic transformation of L-fucono-1,5-lactone to L-fucono-1,4-lactone (1) does not utilize the hydrolyzed product, L-fuconate, as a reactive intermediate. The rate constant is approximately 0.12 min−1 at pH 6.5.
Figure 5.
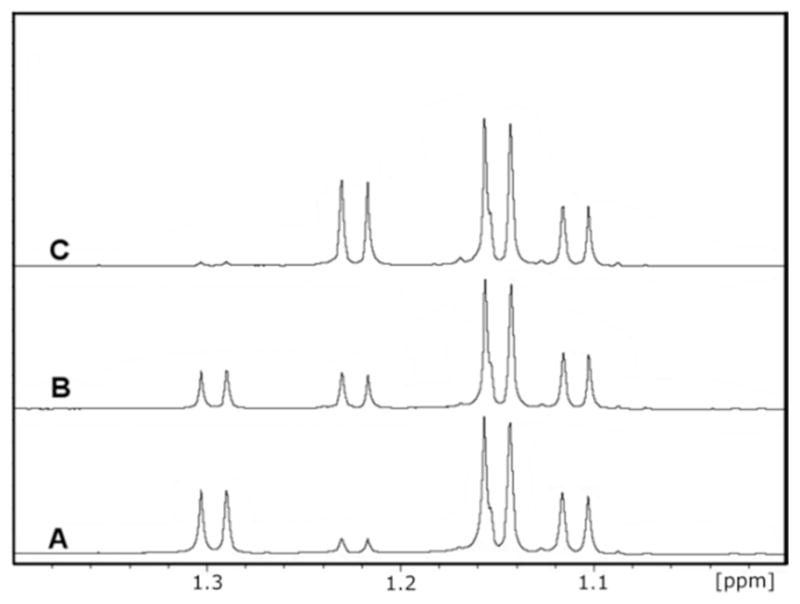
1H-NMR time course for the non-enzymatic conversion of L-fucono-1,5-lactone to L-fucono-1,4-lactone. The resonances corresponding to the C-6 methyl groups of both α-(1.12 ppm) and β-L-fucose (1.15 ppm), L-fucono-1,5-lactone (1.32 ppm) and L-fucono-1,4-lactone (1.24 ppm) are presented. (A) The major reaction product of BmulJ_04915 at pH 4.2 is shown to be L-fucono-1,5-lactone. (B) Five minutes after the reaction was adjusted to pH 6.5 the L-fucono-1,4-lactone is of equal concentration to that of the original enzymatic product. (C) 60 minutes after the pH of the reaction was adjusted to pH 6.5, the major peak is L-fucono-1,4-lactone.
Identification of Physiological Substrate for BmulJ_04915
To determine whether BmulJ_04915 preferentially hydrolyzes the 1,4-or 1,5-lactone of L-fuconate, this enzyme was incubated with L-fucose (25) and NAD+. Shown in Figure 6A is the 1H-NMR spectrum of the methyl carbon for the α-and β-anomers of L-fucose (25) before the addition of BmulJ_04919. After about five minutes, the concentration of the two lactones was approximately equal to one another as shown in Figure 6B. The lactonase (BmulJ_04915) was added and the spectrum of the reaction product was monitored as a function of time. The resonances for the L-fucono-1,5-lactone disappeared at a significantly higher rate than the ones for L-fucono-1,4-lactone (1) (Figure 6C). The formation of L-fuconate was confirmed by the new resonance at approximately 1.18 ppm.
To further demonstrate that BmulJ_04915 catalyzes the hydrolysis of the 1,5-lactone faster than the 1,4-lactone, 4-deoxy-L-fucono-1,5-lactone (24) was synthesized enzymatically using 4-deoxy-L-fucose (47) as a substrate for BmulJ_04919. This compound is missing the hydroxyl group at C-4 and thus cannot form a 1,4-lactone. The product of this reaction was shown to be 4-deoxy-L-fucono-1,5-lactone (24) by NMR spectroscopy (Figure 7A). As no 4-hydroxyl group is available, the 6-member lactone is relatively stable and ndergoes slow non-enzymatic hydrolysis. A new methyl doublet appears at 1.25 ppm when 4-deoxy-L-fucose is oxidized to 4-deoxy-L-fucono-1,5-lactone by BmulJ_04919 (Figure 7B). After hydrolysis with BmulJ_04915, this resonance disappears and the methyl resonance for 4-deoxy-L-fuconate appears at 1.07 ppm (Figure 7C). The 4-deoxy-L-fucono-1,5-lactone (24) was isolated and the kinetic constants for the hydrolysis of this compound by BmulJ_04915, Bamb_1224 and Patl_0798 were determined at pH 7.1. The values of kcat and kcat/Km for the hydrolysis of 4-deoxy-L-fucono-1,5-lactone are approximately 250-and 115-fold greater than for L-fucono-1,4-latone assayed at pH 7.1 (Table 1).
Figure 7.

1H-NMR time course of the enzymatic conversion of 4-deoxy-L-fucose to 4-deoxy-L-fucono-1,5-lactone and 4-deoxy-L-fuconate. The resonances corresponding to the C-6 methyl groups of both α-(1.02 ppm) and β-4-deoxy-L-fucose (1.06 ppm), 4-deoxy-L-fucono-1,5-lactone (1.25 ppm), and 4-deoxy-L-fuconate (1.07 ppm) are provided. (A) 4-deoxy-L-fucose, prior to addition of enzymes at pH 6.5. (B) One minute after the addition of BmulJ_04919 to 4-deoxy-L-fucose, the enzymatic product is 4-deoxy-L-fucono-1,5-lactone. (C) One minute after the addition of BmulJ_04915 to the reaction mixture. 4-deoxy-L-fucono-1,5-lactone is no longer present in the reaction mixture and 4-deoxy-L-fuconate (1.07 ppm) appears to be the major product.
Structure of BmulJ_04919
Unliganded BmulJ_04919 crystallized in space group P61 with a tetramer per asymmetric unit and was phased by molecular replacement using a tetrameric model (PDB code: 3FTP, 3-ketoacyl-(acyl-carrier-protein) reductase) and refined to a resolution of 1.5 A (Table 2). The NADP/L-fucose BmulJ_04919 ternary complex crystallized in space group C2 with two dimers per asymmetric unit and was phased by molecular replacement using a single subunit of the unliganded structure and refined to a resolution of 1.5 A. Density was sufficient to build the entire structure, excluding a portion of a helix-turn-helix motif (residues ~192–202) in two subunits of the unliganded structure. The subunit structure of BmulJ_04919 consists mainly of a Rossmann-fold dinucleotide cofactor binding motif where a central, twisted β-sheet consisting of 7 parallel β-strands (strand topology 3-2-1-4-5-6-7) is flanked by 4 helices (α1,2,3,7,9) on one side and 5 helices (α3,4,5,6) on the other (Figures 8). A structure similarity search utilizing the SSM server indicates a similarity to a number of short-chain dehydrogenase/reductase (SDR) family enzymes with an RMSD of <1.5 (29). The highest scores were for human retinal short-chain dehydrogenase/reductase (PDB code:1YDE, RMSD 1.15 A, sequence identity 35%, Z score 16.2) and Thermoplasma acidophilum D-aldohexose dehydrogenase, AldT (PDB code:2DTE, RMSD 1.31 A, sequence identity 32%, Z-score 15.3). Structurally, the largest differences between these enzymes are modest deviations in the positions of the β4-α4 loop, α4, the α7/α8 helix turn helix, and α2. For example, in AldT α2 is converted to a loop and 10 residues are deleted compared to the same region in BmulJ_04919 (36). Examination of the intermolecular interactions of BmulJ_04919 within the two crystal forms is consistent with BmulJ_04919 existing as a tetramer in solution (Figure S1). In addition, the top structural homologs as indicated by the SSM search also exist as similar tetramers in their crystal structures. The tetramer is composed of a dimer of dimers with 222 point group symmetry where the majority of interactions are between the A–B subunit and the A–D subunit. There are only minor interactions between the A and C subunit, where the C-terminus of the A subunit interacts with C-terminus of the C subunit and α5 forming one wall of the L-fucose binding pocket (Figure S1). Interestingly the C-terminus of AldT was found to entirely cover the sugar binding pocket and was postulated to stabilize the active-site conformation and promote the catalytic reaction, with possibly enhanced dissociation at thermophilic temperatures (36). In contrast, the C-terminus of BmulJ_04919 remains 6–7 A from the sugar binding pocket and appears well anchored, suggesting it does not move during the reaction.
Figure 8.

Ribbon diagram of BmulJ_04919 with bound NADP (magenta sticks) and L-fucose (green sticks).
NADP+ binding Site of BmulJ_04919
Clear and continuous electron density for NADP+ was visualized in the BmulJ_04919 ternary complex (Figure 9A). As is common in SDR enzymes, the NAD(P)+ is bound at the C-terminal edge of the seven stranded β-sheet in an extended conformation (Figure 9B) (37). In addition to the general topology, the Rossman-fold also includes a highly variable Gly-rich sequence essential for coordination of the cofactor pyrophosphate. In BmulJ_04919 this sequence lies between the end of β-strand 1 and start of α-helix 1 with the sequence G14GASGIGG21. Clear binding determinants can be identified which support the preference of BmulJ_04919 for NADP+ over NAD+. The 2′-phosphate of NADP+ forms a salt bridge with the guanidinium group of Arg-39 and a hydrogen bond with the side chain of His-40, both originating from the loop between β2 and α2 (Figure 9C). Utilization of this loop for cofactor discrimination is typical for SDR enzymes (37). Arg-39 is positioned by a hydrogen bond to Glu-63, originating from the loop between β-strand 3 and α-helix 3. The side chain of Arg-39 additionally stacks against the face of the adenosine moiety, which in combination with van der Walls contacts with Leu-64 and a hydrogen bond from Glu-63 to the adenosine amino group complete the binding pocket for the adenosine moiety.
Figure 9.

Interactions of BmulJ-04919 with ligands. (A) 2.5σ Fo-Fc kick map for NADP+ bound to the NADP+ L-fucose BmulJ_04919 ternary complex. (B) Interactions of NADP+ with the secondary structure elements of BmulJ_04919. (C) 2′-Adenosine phosphate binding site of BmulJ-04919. NADP+ is shown as stick with orange carbons, and residues of BmulJ_04919 adjacent to the 2′-adenosine phosphate are shown as sticks with green carbons. (D) 2.5σ Fo-Fc kick map for L-fucose bound to the NADP+ L-fucose BmulJ_04919 ternary complex. L-fucose is shown as sticks with yellow, numbered carbons. (E) Stereo view of the interactions of L-fucose with BmulJ_04919. Proteins atoms, L-fucose and NADP+ are shown as sticks with green, yellow, and white carbons respectively.
L-Fucose binding site of BmulJ_04919
Clear and continuous electron density was observed for L-fucose (Figure 9D). The unliganded structure superimposes well with the ternary complex with an root-mean-square-deviation of 0.22 A for 247 common Cα excluding residues 192–202 which are either missing or more distant from the active site in the unliganded structure (see below). The binding site for L-fucose utilizes structural elements from the C-terminal region. L-fucose binds in a C-3-endo conformation and is highly coordinated with each hydroxyl forming at least two hydrogen bonds to protein atoms (Figure 9E). L-fucose is bound with C-3 positioned directly against the nicotinamide (3.3 A) for the 4-pro-S hydride transfer typical of ‘classical’ SDR enzymes (37). In addition, the protein relay connecting the acid/base hydroxyl-tyrosinate (Tyr-163) to a lysine (Lys-157) to a conserved water molecule stabilized by Asn-113, again typical of ‘classical’ SDR enzymes, is observed (37). Determinants that are unique to L-fucose binding include: Asn-94 from the β4-α4 loop coordinating the hydroxyl at C-4, Gln-147 from the α5-α6 loop coordinating the hydroxyl groups at positions C-3 and C-4, the backbone carbonyl of Ala-184 and the side chain of Glu-185 (β6-α7 loop) coordinating the hydroxyl groups at positions C-2 and C-3 respectively, and Lys-141 from the N-terminal end of α5 coordinating both hydroxyl groups at C-2 and C-3. In addition, the side chains of Asn-94, Leu-190, Tyr-191, and Trp-194 form a hydrophobic pocket which interacts with the methylene group of L-fucose. Indeed, the largest structural change from the apo-structure to the ternary complex is the movement of α7-α8 to optimize these interactions. In the apo-structure this region is either disordered or is some 3–4 A more distant from the L-fucose binding site. As α7 also interacts with the NADP+ and most SDR reaction mechanisms proceed through an ordered ‘bi-bi’ mechanism (NAD(P)+ binding first and leaving last), one can envision NADP+ binding initiating the stabilization of α7-α8 in a conformation competent for binding L-fucose, followed by active site closure upon forming the numerous interactions with the substrate. Based on the number and specificity of the interactions, the structure of the ternary complex suggests that BmulJ_04919 would be fairly specific for L-fucose and that these binding determinates could be utilized to model the substrate specificity of homologs of BmulJ_04919.
Discussion
Metal Content
LigI from Sphingomonas paucimobilis was the first enzyme from cog3618 to be mechanistically and structurally characterized and this enzyme does not require the binding of divalent metal ion in the active site for catalytic activity (7). BmulJ_04915, also a member of cog3618, shares ~20% sequence identity with LigI and it is the second example of an enzyme within the AHS that does not require metal for catalytic function. We were unable to detect the binding of metals via ICP-MS in the samples of BmulJ_04915 purified in the absence of added metal and thus this protein does not require the metal for catalytic activity.
The Active Site of BmulJ_04915
Nearly all members of the AHS are recognized by the conservation of five amino acid residues that form the active site and are utilized for metal binding and/or proton transfers. These residues include two histidine residues from the C-terminal end of β-strand 1, two additional histidine residues from the C-terminal ends of β-strands 5 and 6, and a conserved aspartate residue from the C-terminal end of β-strand 8 (5). In BmulJ_04915, three of the four histidine residues are conserved (His-9, His-11 and His-166) from the C-terminal ends of β-strands 1 and 6, in addition to the aspartate (Asp-244) from the end of β-strand 8. The histidine from the end of β-strand 5 is absent and replaced with Leu-141 (Figures 4B and 10A). With the absence of a metal ion bound in the active site, these four residues must now acquire new functional roles.
Figure 10.

(A) Active site of BmulJ_04915 structurally aligned with that of 2-pyrone-4,6-dicarboxylic acid lactonase (LigI). The active site is color-coded as follows: green for LigI (PDB entry 4d8l) and white for BmulJ_04915. Numbers in parenthesis designate the approximate location in the secondary structure from which the residue originates. (B) L-Fucono-1,5-lactone modeled into the active site of BmulJ_04915.
A structural alignment of the LigI and BmulJ_04915 active sites reveals relatively little conservation between the two binding pockets, aside from the residues predicted to confer catalytic activity (Figure 10A). Low conservation of the active site can be explained by analysis of the two significantly different substrates utilized by these enzymes. LigI catalyzes the hydrolysis of a planar dicarboxylated 6-membered lactone, while BmulJ_04915 catalyzes the hydrolysis of a non-planar 6-membered lactone. The structure of BmulJ_04915 could not be obtained with bound substrate or product, but it is possible to manually dock the L-fucono-1,5-lactone substrate in the active site to obtain a view of the structural determinants for substrate binding (Figure 10B). Modeling L-fucono-1,5-lactone into the active site of BmulJ_04915 was based on the structural alignment with LigI (with bound product) and the general path of the bound HEPES molecule (Figures 10A and 4B). The sulfonate of the HEPES molecule bound to BmulJ_04915 is positioned analogous to the newly formed carboxylate of the product of the reaction catalyzed by LigI (4-carboxy-2-hydroxymuconate) interacting with three histidine residues (His-9, His-11 and His-166), Arg-106 and Asp-244 (Figure 4B). In this model Gln-110 and Lys-169 are positioned to interact with hydroxyl groups at C-3 and C-4 of L-fucono-1,5-lactone. On either face of the lactone, Leu-141 and Leu-247 constrict the active site. Finally, Trp-22 lies against the aliphatic face of the lactone (C3-C4-C5) near the entrance to the active site.
Proposed Mechanism of Action
The four AHS conserved residues (His-9, His-11, His-166, and Asp244) align with their counterparts in LigI and assume similar roles in catalysis. The three conserved histidine residues polarize the carbonyl group of the sugar lactone substrate via electrostatic interactions (7). The aspartate residue from β-strand 8 (Asp-244) is positioned to deprotonate an active site water molecule for nucleophilic attack on the C-1 carbonyl group of the lactone substrate. A fifth active site residue conserved within many members of cog3618 is an arginine from β-strand 4 (Arg-106). This arginine is proposed to assist in the cleavage of the lactone functional group through electrostatic interactions with the hydroxyl leaving group of the product. The proposed mechanism is presented in Scheme 3.
Scheme 3.

Natural Substrate of BmulJ_04915
BmulJ_04915 exhibits significant catalytic activity for a series of 5-member sugar lactones (Table 1) that share the same stereochemistry at C-2 and C-3 and has the highest activity for the L-fucono-1,4-lactone. The best substrate for BmulJ_04919 is β-L-fucose, which undergoes oxidation to L-fucono-1,5-lactone and subsequent non-enzymatic transformation to L-fucono-1,4-lactone. It has been postulated that the instability of D-galactono-1,5-lactone is due to an intermolecular rearrangement that occurs in a distorted chair conformation. The rearrangement is thought to arise from the axial hydroxyl group in C-4, which is positioned for nucleophilic attack on C-1 (38). A similar rearrangement is assumed for L-fucono-1,5-lactone and is fully consistent with the NMR data reported in this paper. The 1H-NMR spectra show a direct conversion of L-fucono-1,5-lactone to L-fucono-1,4-lactone without transformation to L-fuconate as an intermediate. The NMR spectra clearly show that BmulJ_04915 hydrolyzes L-fucono-1,5-lactone at a rate faster than with L-fucono-1,4-lactone (Figure 6).
It was not possible to determine the kinetic constants for the hydrolysis of L-fucono-1,5-lactone due to chemical instability and thus a stable mimic was utilized. 4-Deoxy-L-fucono-1,5-lactone was synthesized enzymatically and provided a pyranosyl analog that had greater stability due to the absence of the axial hydroxyl group on C-4, which allowed for the kinetic constants to be determined at pH 7.1. 4-Deoxy-L-fucono-1,5-lactone was shown to be hydrolyzed by BmulJ_04915 approximately two orders of magnitude faster than the corresponding L-fucono-1,4-lactone. Thus, even though L-fucono-1,5-lactone is kinetically unstable, it appears to be the natural substrate of BmulJ_04915.
The closest homologue (~27% sequence identity) of known function to BmulJ_04915 is SKA58_03595 from Sphingomonas sp. SKA58, which has been experimentally annotated as an L-rhamnono-1,4-lactonase (9). Kinetic constants for L-rhamnono-1,4-lactonase have not apparently been reported nor has a complete substrate profile been established. L-rhamnono-1,4-lactonase is adjacent to an L-rhamnose dehydrogenase from cog1028 that shares 30% sequence identity with BmulJ_04919. Given the sequence similarities, it is predicted that these homologues of BmulJ_04915 and BmulJ_04919 will act in a similar manner to produce a 1,5-lactone, which is subsequently hydrolyzed to the acid sugar.
Additional L-Fucono-1,5-Lactonases
L-fucono-1,5-lactonase activity was demonstrated in groups 1, 9 and 14 of cog3618 with enzymes Bamb_1224, Patl_0798 and BmulJ_04915 respectively (Figure 3, Table 1). Groups 1, 9 and 14 are grouped together as part of Class I enzymes at a low stringency E-value of 10−30 and only begin to separate into more distinct groups at E-values greater than 10−70 (Figure 2B). A list of candidate L-fucono-1,5-lactonase sequences has been established. These proteins shared at least 30% sequence identity and map to the Class I enzymes in the sequence similarity network. All candidate sequences were aligned with the experimentally verified L-fucono-1,5-lactonase, BmulJ_04915. Sequences were considered L-fucono-1,5-lactonase if the following active site residues were conserved: His-9, His-11, Trp-22, Arg-106, Gln-110, His-166, Thr-207, Glu-208 and Asp-244. The 174 sequences that conform to the specific active site residues are now postulated to be L-fucono-1,5-lactonase proteins and are represented as yellow nodes in Figure 2 and are listed in Table S1 of the Supporting Information.
Supplementary Material
Acknowledgments
Funding
This work was supported in part by the Robert A. Welch Foundation (A-840) and the National Institutes of Health (GM 71790 and GM 93342).
X-ray diffraction data for this study were measured at beamlines X29 of the National Synchrotron Light Source (NSLS), Brookhaven National Laboratory, and Lilly Research Laboratory Collaborative Access Team (LRL-CAT) at the Advanced Photon Source, Argonne National Laboratory. Use of the NSLS beamline is supported by the Center for Synchrotron Biosciences grant, P30-EB-009998, from the National Institute of Biomedical Imaging and Bioengineering (NIBIB) and by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-98CH10886. Use of the Advanced Photon Source at Argonne National Laboratory was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly Company, which operates the facility.
Footnotes
Accession Codes
The X-ray coordinates L-fucono-1,5-lactonase (BmulJ_04915) have been deposited in the Protein Data Bank as 4DLF, 4DLM, 4DO7 and 4DNM. X-ray coordinates for L-fucose dehydrogenase (BmulJ_04919) have been deposited as 4GKB, 4GLO and 4GVX.
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Table S1 and Figure S1. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Becker DJ, Lowe JB. Fucose: biosynthesis and biological function in mammals. Glycobiology. 2003;13:41R–53R. doi: 10.1093/glycob/cwg054. [DOI] [PubMed] [Google Scholar]
- 2.Chan JY, Nwokoro NA, Schachter H. L-Fucose metabolism in mammals. The conversion of L-fucose to two moles of L-lactate, of L-galactose to L-lactate and glycerate, and of D-arabinose to L-lactate and glycollate. J Biol Chem. 1979;254:7060–7068. [PubMed] [Google Scholar]
- 3.Ghalambor MA, Heath EC. The metabolism of L-Fucose. II. The enzymatic cleavage of L-fuculose 1-phosphate. J Biol Chem. 1962;237:2427–2433. [PubMed] [Google Scholar]
- 4.Heath EC, Ghalambor MA. The Metabolism of L-Fucose. I. The Purification and Properties of L-Fuculose Kinase. J Biol Chem. 1962;237:2423–2426. [PubMed] [Google Scholar]
- 5.Seibert CM, Raushel FM. Structural and Catalytic Diversity within the Amidohydrolase Superfamily. Biochemistry. 2005;44:6383–6391. doi: 10.1021/bi047326v. [DOI] [PubMed] [Google Scholar]
- 6.Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. The COG Database: New Developments in Phylogenetic Classification of Proteins from Complete Genomes. Nucleic Acids Res. 2001;29:22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hobbs ME, Malashkevich V, Williams HJ, Xu C, Sauder JM, Burley SK, Almo SC, Raushel FM. Structure and Catalytic Mechanism of LigI: Insight into the Amidohydrolase Enzymes of cog3618 and Lignin Degradation. Biochemistry. 2012;51:3497–3507. doi: 10.1021/bi300307b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Halak S, Basta T, Buerger S, Contzen M, Wray V, Pieper DH, Stolz A. 4-Sulfomuconolactone hydrolases from Hydrogenophaga intermedia S1 and Agrobacterium radiobacter S2. J Bacteriol. 2007;189:6998–7006. doi: 10.1128/JB.00611-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watanabe S, Saimura M, Makino K. Eukaryotic and bacterial gene clusters related to an alternative pathway of non-phosphorylated L-rhamnose metabolism. J Biol Chem. 2008;283:20372–20382. doi: 10.1074/jbc.M801065200. [DOI] [PubMed] [Google Scholar]
- 10.Atkinson HJ, Morris JH, Ferrin TE, Babbitt PC. Using sequence similarity networks for visualization of relationships across diverse protein superfamilies. PLoS One. 2009;4:e4345. doi: 10.1371/journal.pone.0004345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yew WS, Fedorov AA, Fedorov EV, Rakus JF, Pierce RW, Almo SC, Gerlt JA. Evolution of enzymatic activities in the enolase superfamily: L-fuconate dehydratase from Xanthomonas campestris. Biochemistry. 2006;45:14582–14597. doi: 10.1021/bi061687o. [DOI] [PubMed] [Google Scholar]
- 12.Xiang DF, Kolb P, Fedorov AA, Xu C, Fedorov EV, Narindoshvili T, Williams HJ, Shoichet BK, Almo SC, Raushel FM. Structure-based function discovery of an enzyme for the hydrolysis of phosphorylated sugar lactones. Biochemistry. 2012;51:1762–1773. doi: 10.1021/bi201838b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aslanidis C, de Jong PJ. Ligation-independent Cloning of PCR Products (LIC-PCR) Nucleic Acids Res. 1990;18:6069–6074. doi: 10.1093/nar/18.20.6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expression Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 15.Fox BG, Blommel PG. Autoinduction of protein expression. Curr Protoc Protein Sci. 2009;Chapter 5(Unit 5):23. doi: 10.1002/0471140864.ps0523s56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tropea JE, Cherry S, Waugh DS. Expression and purification of soluble His(6)-tagged TEV protease. Methods Mol Biol. 2009;498:297–307. doi: 10.1007/978-1-59745-196-3_19. [DOI] [PubMed] [Google Scholar]
- 17.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Cryst. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 18.Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Cryst. 2011;67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evans P. Scaling and assessment of data quality. Acta Cryst. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 20.Zwart PH, Afonine PV, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, McKee E, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Storoni LC, Terwilliger TC, Adams PD. Automated structure solution with the PHENIX suite. Methods Mol Biol. 2008;426:419–435. doi: 10.1007/978-1-60327-058-8_28. [DOI] [PubMed] [Google Scholar]
- 21.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lebedev AA, Vagin AA, Murshudov GN. Model preparation in MOLREP and examples of model improvement using X-ray data. Acta Cryst. 2008;64:33–39. doi: 10.1107/S0907444907049839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 24.Koehn RK, Diehl WJ, Scott TM. The differential contribution by individual enzymes of glycolysis and protein catabolism to the relationship between heterozygosity and growth rate in the coot clam, Mulinia lateralis. Genetics. 1988;118:121–130. doi: 10.1093/genetics/118.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chapman E, Wong CH. A pH sensitive colorimetric assay for the high throughput screening of enzyme inhibitors and substrates: a case study using kinases. Bioorg Med Chem. 2002;10:551–555. doi: 10.1016/s0968-0896(01)00306-6. [DOI] [PubMed] [Google Scholar]
- 26.Hall RS, Xiang DF, Xu C, Raushel FM. N-Acetyl-D-glucosamine-6-phosphate deacetylase: Substrate activation via a single divalent metal ion. Biochemistry. 2007;46:7942–7952. doi: 10.1021/bi700543x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kamat SS, Bagaria A, Kumaran D, Hampton GP, Fan H, Sali A, Sauder JM, Burley SK, Lindahl PA, Swaminathan S, Raushel FM. Catalytic mechanism and three-dimensional structure of adenine deaminase. Biochemistry. 2011;50:1917–1927. doi: 10.1021/bi101788n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Cryst. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 30.Sundlov JA, Garringer JA, Carney JM, Reger AS, Drake EJ, Duax WL, Gulick AM. Determination of the crystal structure of EntA, a 2,3-dihydro-2,3-dihydroxybenzoic acid dehydrogenase from Escherichia coli. Acta Crystallogr Sect D. 2006;D62:734–740. doi: 10.1107/S0907444906015824. [DOI] [PubMed] [Google Scholar]
- 31.Klasen R, Bringer-Meyer S, Sahm H. Biochemical characterization and sequence analysis of the gluconate:NADP 5-oxidoreductase gene from Gluconobacter oxydans. J Bacteriol. 1995;177:2637–2643. doi: 10.1128/jb.177.10.2637-2643.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Novotny MJ, Reizer J, Esch F, Saier MH., Jr Purification and properties of D-mannitol-1-phosphate dehydrogenase and D-glucitol-6-phosphate dehydrogenase from Escherichia coli. J Bacteriol. 1984;159:986–990. doi: 10.1128/jb.159.3.986-990.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yokochi N, Nishimura S, Yoshikane Y, Ohnishi K, Yagi T. Identification of a new tetrameric pyridoxal 4-dehydrogenase as the second enzyme in the degradation pathway for pyridoxine in a nitrogen-fixing symbiotic bacterium, Mesorhizobium loti. Arch Biochem Biophys. 2006;452:1–8. doi: 10.1016/j.abb.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 34.Schachter H, Sarney J, McGuire EJ, Roseman S. Isolation of diphosphopyridine nucleotide-dependent L-fucose dehydrogenase from pork liver. J Biol Chem. 1969;244:4785–4792. [PubMed] [Google Scholar]
- 35.Tsuji Y, Koike A, Yamamoto K, Tochikura T. Purification and some properties of L-fucose dehydrogenase from Agrobacterium radiobacter and its application to the assay of bound-fucose in glycoconjugates. Biochem Biophys Acta. 1992;1117:167–173. doi: 10.1016/0304-4165(92)90075-6. [DOI] [PubMed] [Google Scholar]
- 36.Yasutake Y, Nishiya Y, Tamura N, Tamura T. Structural insights into unique substrate selectivity of Thermoplasma acidophilum D-aldohexose dehydrogenase. J Mol Biol. 2007;367:1034–1046. doi: 10.1016/j.jmb.2007.01.029. [DOI] [PubMed] [Google Scholar]
- 37.Lesk AM. NAD-binding domains of dehydrogenases. Curr Opin Struct Biol. 1995;5:775–783. doi: 10.1016/0959-440x(95)80010-7. [DOI] [PubMed] [Google Scholar]
- 38.Ueberschar KH, Blachnitzky EO, Kurz G. Reaction Mechanism of D-Galactose Dehydrogenases from Pseudomonas saccharophila and Pseudomonas fluorescens. Eur J Biochem. 1974;48:389–405. doi: 10.1111/j.1432-1033.1974.tb03780.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.