

Abstract
The substrate specificities of two incorrectly annotated enzymes belonging to cog3964 from the amidohydrolase superfamily (AHS) were determined. This group of enzymes is currently misannotated as either dihydroorotase or adenine deaminase. Atu3266 from Agrobacterium tumefaciens C58 and Oant2987 from Ochrobactrum anthropi ATCC 49188 were determined to catalyze the hydrolysis of acetyl-R-mandelate and similar esters with values of kcat/Km that exceed 105 M−1 s−1. These enzymes do not catalyze the deamination of adenine or the hydrolysis of dihydroorotate. Atu3266 was crystallized and the structure determined to a resolution of 2.62 Å. The protein folds as a distorted (β/α)8-barrel and binds two zincs in the active site. The substrate profile was determined via a combination of computational docking to the three-dimensional structure of Atu3266 and screening of a highly focused library of potential substrates. The initial weak hit was the hydrolysis of N-acetyl-D-serine (kcat/Km = 4 M−1s−1). This was followed by the progressive identification of acetyl-R-glycerate (4 × 102 M−1s−1), acetyl glycolate (kcat/Km = 1.3 × 104 M−1 s−1) and ultimately acetyl-R-mandelate (kcat/Km =2.8 × 105 M−1 s−1).
The development of a comprehensive strategy for the functional annotation of proteins and enzymes whose sequences have been deposited in public databases has proven to be a difficult and persistent challenge. Utilization of homology-based sequence comparisons for functional annotation of newly sequenced genes can often lead to the annotation of the wrong function when unreasonable threshold values are used (1–3). The end result is often a misrepresentation of the potential metabolic transformations contained within a given organism. In addition to the misannotation of enzyme function there is a significant fraction of the total gene inventory that is simply not annotated (4–6). This observation suggests that a substantial segment of the metabolic landscape remains to be discovered.
In many cases, functional annotation of newly sequenced genes can be strategically approached by the integration of structural genomics (7, 8), computational docking (9, 10), high-throughput screening (11), and genome context analysis (12). We have focused our efforts toward the development of a simple and integrated strategy for functional annotation that is based on an assault on the amidohydrolase superfamily (AHS) of enzymes. This superfamily was first identified and recognized from the three-dimensional structural similarities among urease, adenosine deaminase and phosphotriesterase (13). The amidohydrolase superfamily is characterized structurally by an active site that contains a mono- or binuclear metal center embedded at the C-terminal end of a distorted (β/α)8-barrel protein fold. Typically identified by an HxH motif after the C-terminal end of β-strand 1, the enzymes from the AHS also contain other coordinating ligands to the metal center at the ends of β-strands 4, 5, 6 and 8 (14). The metal center serves in the activation of a hydrolytic water molecule for nucleophilic attack and to stabilize the transition state. Most of the reactions catalyzed by this diverse superfamily involve the hydrolysis of C-O, C-N or P-O bonds. However, some members of this superfamily also catalyze decarboxylation, hydration or isomerization reactions (15–17). Enzymes from the AHS catalyze reactions in the metabolism of carbohydrates, amino acids, nucleic acids and the degradation of organophosphate esters.
The AHS has been organized into 24 Clusters of Orthologous Groups (COG) (18, 19). One of these clusters, cog3964, represents one of the smaller homologous groups of amidohydrolases, with approximately 200 sequences identified to date. A sequence similarity network for this COG is presented in Figure 1 at an E-value cutoff of 10−70. Some members of cog3964 have been annotated as dihydroorotases, which catalyze the interconversion of dihydroorotate and N-carbamoyl aspartate (20, 21), while other members have been annotated as catalyzing the deamination of adenine (22). Experimentally annotated dihydroorotases are found in cog0044 and cog0418. Structurally and experimentally characterized adenine deaminases are found in cog1001 (22) and in cog1816 (23). A straightforward comparison of the amino acid sequences found within the proteins from cog3964 with the experimentally annotated adenine deaminases and dihydroorotases clearly indicates that the residues required for substrate recognition of adenine and/or dihydroorotate are not conserved. This observation suggests that either these annotations are clearly wrong for cog3964, or that a previously unrecognized novel constellation of amino acids has evolved for the deamination of adenine or the hydrolysis of dihydroorotate.
Figure 1.

Sequence similarity network for the organization of cog3964 within the amidohydrolase superfamily (18, 19). Each node in the diagram represents a sequence that has been annotated as belonging to cog 3964, and each edge represents the pairwise connection between two sequences at a BLAST E value or better than 10−70. Atu3266 and Oant2987 are found in group 6, and are depicted in purple. EF0837 is the locus tag for orthologous sequence in group 2. The structure in available in protein database, PDB id: 2ICS.
In this paper we report the three-dimensional crystal structure and substrate profile for two enzymes from cog3964; Atu3266 from Agrobacterium tumefaciens and Oant2987 from Ochrobactrum anthropic. Neither of these enzymes is able to catalyze the hydrolysis of dihydroorotate or the deamination of adenine. We have utilized a focused compound library and computational docking to discover that these two enzymes from cog3964 actually catalyze the hydrolysis of acetylated α-hydroxyl carboxylates as shown in Scheme 1. The best substrate identified to date is acetyl-R-mandelate. This compound is enzymatically hydrolyzed with a kcat/Km value of 2.8 × 105 M−1 s−1.
Scheme 1.

MATERIALS and METHODS
Materials
The genomic DNA from Ochrobactrum anthropi strain ATCC-49188 was purchased from the American Type Culture Collection (ATCC). DNA sequencing reactions were conducted at the Gene Technology Laboratory at Texas A&M University. The chemicals utilized in the expression, purification, and screening of Atu3266 and Oant2987 were obtained from Sigma-Aldrich, unless stated otherwise. The preparation of the acylated compounds tested for substrate activity is provided in the Supporting Information. The structures for many of these compounds are provided in Schemes 2 and 3. The pET-20b(+) expression vector and Rosetta-gami™ B(DE3)pLysS competent cells were obtained from Novagen. The acetate and formate detection kits were purchased from Megazyme. N-acetyl-DL-phenylglycine was purchased from Chem-Impex International. 3-Bromomandelic acid and 4-bromomandelic acid were purchased from Oakwood Products, Inc.
Scheme 2.

Scheme 3.

Gene Cloning
The gene for Oant2987 (gi|153010310) from Ochrobactrum anthropi was amplified by PCR using the primer pair 5′-ACAGGAGCCATATGATTTCCGGTGAACAGGCGAAGCCG-3′ containing an NdeI restriction site and 5′-ACGCGAATTCCCAGCGCCACGAATAGCCATGGCTATGGC-3′ having an EcoRI restriction site. Oant2987 was inserted into a pET-20b(+) vector that had been previously digested with NdeI and EcoRI. The gene for Atu3266 was initially cloned by the New York SGX Research Consortium for Structural Genomics from Agrobacterium tumefaciens (gi|159185666). The poor overexpression and solubility of this clone in BL21(DE3) cells led to the removal of the gene from its original TOPO-isomerase vector, pSGX3(BC), using the restriction sites CCGC↓GG and CA↓TATG of the construct and removing the gene by digestion with SacII and NdeI restriction enzymes. The gene was transferred to a pET-20b(+) vector previously digested with the same restriction enzymes. The modified clone contained a (His)6-C-terminal purification tag.
Expression and Purification of Oant2987 and Atu3266
The plasmid containing the gene for Oant2987 was transformed into BL21(DE3) and plated on LB agar-ampicillin plates. A single colony was inoculated into overnight cultures containing 5 mL of LB medium supplemented with 100 μg/mL of ampicillin. These cultures were utilized to make 1 L cultures containing the same concentration of ampicillin and grown at 30 °C in a shaker-incubator. After an OD600 of 0.3 was obtained, the cells were supplemented with 150 μM of 2,2′-bipyridyl to coordinate excess ferric iron from the medium. At an OD600 of 0.6, the cells were induced with 200 μM IPTG and the addition of 1.0 mM Zn(OAc)2. The temperature upon induction was reduced to 20 °C and the cultures were allowed to continue to grow for an additional 12 hours. The cells were harvested by centrifugation and frozen at −80 °C. The frozen cell pellet was thawed and re-suspended in 60 mL of 50 mM HEPES buffer, pH 7.6 (buffer A). The cells were supplemented with 10 μg/mL of phenylmethanesulfonyl fluoride (PMSF) and lysed by sonication at 0 °C. The supernatant solution was treated with 2% w/v protamine sulfate and centrifuged. The protein was precipitated with ammonium sulfate between 40% and 80% saturation. The protein pellet was resuspended with a minimum amount of buffer A and loaded onto a Superdex 200 gel filtration column. The fractions of interest were collected and subsequently loaded onto a Resource Q column. The protein was eluted from the column with a gradient of 20 mM HEPES buffer, pH 7.6, containing 1 M NaCl (buffer B). The purity of Oant2987 was greater than 95% based on SDS-PAGE.
The plasmid containing the gene for Atu3266 was transformed into Rosetta-gami™ B(DE3)pLysS electrocompetent cells. A single colony containing the gene of interest was inoculated into an overnight culture containing 5 mL LB medium, 100 μg/mL of ampicillin and 20 μg/mL of chloramphenicol. The 5 mL starter cultures were used to inoculate 1 L of LB medium. The cells were incubated at 30 °C in a shaker-incubator. When the OD600 reached 0.3, the cells were treated with 150 μM 2,2′-bipyridyl. The cells continued to grow to an OD600 of about 0.6, at which point 200 μM of IPTG and 1.0 mM ZnCl2 were added. The temperature was reduced to 20 °C and the cells were grown overnight for 16 hours. The cells where harvested by centrifugation at 8000g for 10 minutes and then frozen at −80 °C. The cell pellet was resuspended in binding buffer containing 20 mM HEPES, 500 mM NaCl and 5.0 mM imidazole at pH 7.6. The cells were supplemented with 10 μg/mL of PMSF and lysed by sonication at 0 °C. The cell debris was removed by centrifugation and then Atu3266 was purified using a His-tag affinity column. The cell lysate supernatant was filtered through a 0.2 μm cellulose acetate sterile filter and loaded onto Ni2+-NTA column equilibrated with binding buffer. The column was washed with binding buffer until the A280 remained constant and below an absorbance of 0.1. The protein of interest was eluted using a gradient of 0–500 mM imidazole in a buffer solution containing 10 mM HEPES, and 250 mM NaCl at pH 7.6. After dialysis, the protein was judged to be >95% pure by SDSPAGE.
Protein Concentration and Metal Analysis
Protein concentration was determined spectrophotometrically using a SPECTRAmax-384 PLUS UV-vis spectrophotometer. The concentration was obtained from the absorbance at 280 nm using the extinction coefficients determined from the amino acid sequence (web.expasy.org/protparam/). The extinction coefficients for Oant2987 and Atu3266 are 43,605 M−1 cm−1 and 43,969 M−1 cm−1, respectively (24). The metal content of the purified proteins was determined by inductively coupled plasma emission – mass spectrometry (ICP-MS) using a Perkin-Elmer Analyst 700 atomic absorption spectrometer. The samples were prepared by heating 1.0 μM enzyme with 1% (v/v) HNO3 for 30 minutes.
Synthesis of Dipeptide Libraries, N-Acyl Amino Acids and Methyl Phosphonates
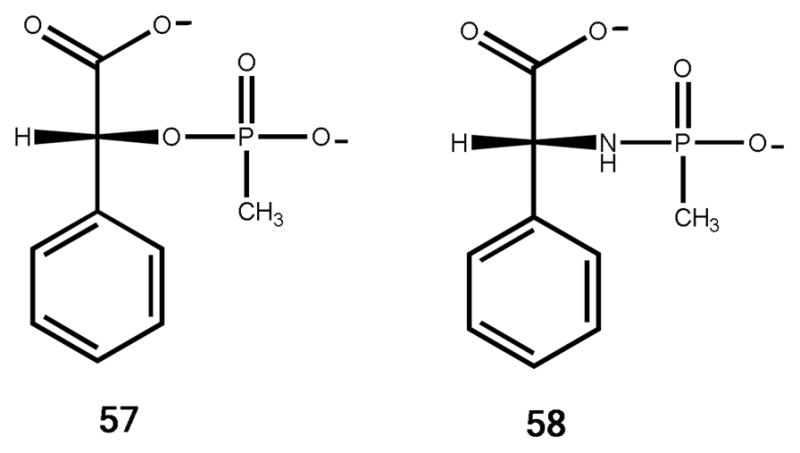
Syntheses of dipeptide libraries (L-Xaa-L-Xaa, D-Xaa-L-Xaa, and L-Xaa-D-Xaa), and N-acetyl, N-formyl, and N-succinyl derivatives of D- and L-amino acids were prepared as described previously (25, 26). The preparation of the O- and N-methyl phosphonate derivatives of R-mandelate (57) and, D-phenyl glycine (58) respectively, were prepared as previously described (25). The structures of these compounds are presented in Scheme 4; their composition was verified by mass spectrometry and NMR.
Scheme 4.
Screening of N-Acyl and Dipeptide Libraries
The preliminary substrate screening of Atu3266 and Oant2987 was initiated by mixing each protein with the N-modified libraries of the D- and L- amino acids (N-acetyl, N-formyl, and N-succinyl), and three dipeptide libraries (L-Xaa- L-Xaa, D-Xaa,-L-Xaa and L-Xaa-D-Xaa). The assays were conducted as previously described (26). Each assay was buffered with 20 mM HEPES, pH 7.6 and each library contained 17–19 modified amino acids (L- and D- cysteine were not included). Each assay contained ~100 μM of each component and 0–1000 nM of Atu3266 or Oant9287 was added to initiate the reaction. A negative control was prepared without the addition of either Atu3266 or Oant9287. The initial screening reactions were conducted at 30 °C for 15 hours. The formation of free amino acids was detected using a modified Cd-ninhydrin assay (27). Each 70 μL reaction mixture was quenched with 280 μL of ninhydrin reagent. The entire reaction mixture was developed by heating at 85 °C for 15 minutes and then cooled. A 250 μL aliquot was transferred to a 96-well UV-visible micro plate and the extent of total free amino acids was measured at 507 nm.
Screening Methods for Deacetylase Activity
All compounds having a hydrolysable acetyl moiety were screened using the acetic acid kit, KACETAF, from Megazyme®. The catalytic activities of Atu3266 and Oant2987 were monitored by the formation of acetate and the subsequent reduction of NAD+ by the coupled activity of acetyl-coenzyme A synthetase, citrate synthase and L-malate dehydrogenase. The reaction was monitored spectrophotometrically at 340 nm. The 250 μL reaction mixture contained 1.0 mM of the test compound, 20 mM HEPES, pH 7.6 and 75 μL of the coupling system containing 128 mM TEA buffer pH 8.4, 5.0 mM NAD+, 3.1 mM ATP, 3.2 mM MgCl2, 0.15 mM CoA, 4 U of L-malate dehydrogenase, 0.6 U of citrate synthase, and 0.3 U of acetyl CoA synthetase. Each compound was tested for ester hydrolysis at 30 °C and the reaction was initiated by the addition of 1.0 μM of Atu3266 or Oant2987.
Screening of O-Propionyl Compounds
The hydrolysis of R-3-hydroxy-2-(propionyloxy) propanoate (21) and R-2-(propionyloxy)-butanoate (22) was assessed using a pH-sensitive colorimetric assay (28). The hydrolysis of the ester bond releases a proton that was detected using a pH indicator dye, cresol purple. The screening reactions were carried out in 2.5 mM Bicine buffer, pH 8.3, containing 0.2 M NaCl and 1.0 mM of each compound with up to 1.0 μM enzyme. Each reaction contained 0.1 mM cresol purple in 1% DMSO and the change in absorbance was monitored at 577 nm. The extinction coefficient under these reaction conditions was determined to be 1.51 × 103 M−1 cm−1 using acetic acid as a titrant.
Data Analysis
The kinetic constants kcat, Km, and kcat/Km were determined for Atu3266 and Oant2987 for selected substrates by fitting the initial velocity data to equation 1. Competitive inhibition constants were determined using equation 2 for a competitive inhibitor. In these equations v is the initial velocity, Et is the enzyme concentration, kcat is the turnover number, A is the substrate concentration, Km is the Michaelis-Menten constant, I is the inhibitor concentration and Kis is the slope inhibition constant.
| (1) |
| (2) |
Crystallization and Structure Determination of Atu3266
Selenomethionine (SeMet) substituted Atu3266 from Agrobacterium tumefaciens with the 6x-His tag intact was crystallized by mixing 1.5 μL of protein (5 mg/mL in a buffer of 10 mM HEPES pH 7.5, 150 mM NaCl, 10 mM methionine, 10% glycerol and 5 mM DTT) with 1.5 μL of reservoir solution containing 0.1 M HEPES pH 7.5, 25% PEG3350, 0.2M ammonium sulfate and equilibrating against the same reservoir solution by the hanging drop vapor diffusion method. Crystals appeared after three days and were flashed-cooled in liquid nitrogen for data collection.
A native dataset and a complete MAD dataset from single crystals were collected at 100 K on beamline 31-ID at APS using a Mar CCD 225 detector. Crystals diffracted to 2.62 Å, and belong to the orthorhombic space group P212121 with 6 molecules in the asymmetric unit. Data were indexed, integrated and scaled using the program HKL2000 (29). Selenium sub-structure was determined using SHELXD (30). The phase refinement and density modifications were carried out using SHAARP and SOLOMON (31, 32). Model building was done using ARP/wARP (33). Further model building and refinement of the structure was carried out in iterative cycles using the program O and CNS 1.1 (34, 35). Extra residual density was observed near the NZ atom of Lys-175 and the lysine residue was modeled as a carboxylated lysine. In addition, two zinc ions and one imidazole were located per molecule and included in the later stages of refinement. The atomic coordinates and structure factors for the Atu3266 structure have been deposited in the Protein Data Bank under accession code 2OGJ. Crystal, diffraction data and refinement details are given in Table 1.
Table 1.
Data Collection and Refinement Statistics for Atu3266
| beamline | NSLS beamline X12C | ||
|---|---|---|---|
| Data Set - Unit Cell Parameters | |||
| a(Å) | 90.65 | ||
| b(Å) | 139.2 | ||
| c(Å) | 206.13 | ||
| α = β = γ = 90° | |||
| space group | P212121 | ||
| Data Collection Statistics | |||
| Peak | Inflection | Remote | |
| wavelength (Å) | 0.9793 | 0.9798 | 0.94 |
| resolution range (Å) | 50-2.6 | 50-2.8 | 50-2.8 |
| redundancy | 5.8 | 5.5 | 5.2 |
| Mean I/σ(I) | 14.5 | 13.2 | 12.8 |
| Rmergea | 0.121 | 0.125 | 0.118 |
| no. of unique reflections | 139906 | 138603 | 138965 |
| Phasing Statistics | |||
| phasing powerb (ano) | 0.21/0.28 | ||
| FOMc: (centric/acentric) | 0.85 | ||
| Refinement Statistics | |||
| resolution range (Å) | 2.62–39.7 | ||
| no. of reflections (work) | 139800 | ||
| no. of reflections (test) | 4173 | ||
| no. of protein atoms | 16450 | ||
| no. of heterogen atoms | 22 | ||
| no. of solvent atoms | 113 | ||
| Rfactord/Rfree | 0.256/0.302 | ||
| rmsd bond length (Å) | 0.016 | ||
| rmsd bond angles (°) | 1.6 | ||
| <B-values> (Å2) | 58.3 | ||
| PDB entry code | 2OGJ | ||
Rmerge = Σj | Ih − 〈Ih〉/ΣIh where 〈Ih〉 is the average intensity over symmetry equivalents.
As defined in SHARP.
FOM = figure of merit as defined by SHARP.
Rfactor = Σ | Fobs−Fcalc|/Σ|Fobs|; Rfree same as Rfactor but for test set.
Computational Docking
A non-redundant virtual library of compounds from the Kyoto Encyclopedia of Genes and Genomes (KEGG) was filtered based on possible amidohydrolase reactions and subsequently prepared as high-energy intermediates (HEI). The HEI compounds are generated by the addition of an activated water to the metabolite molecules from KEGG, which approximates of the transition state with variable substrate protonation states (9, 36). This library contains 6,440 unique metabolites that generated 57,672 HEI molecules. This virtual HEI library (HEI KEGG) was docked against Atu3266.
The Atu3266 structure was prepared for docking as previously described (37). All six chains from the asymmetric unit of Atu3266 (PDB: 2OGJ) were aligned against chain A, and the best rotamer positions for each amino acid were chosen based on the 2fofc density maps. For example, the position of residues 177, 267 and 268 near the active site were chosen based on chain D positions. Histidine protonation states were manually defined in the vicinity of the two zinc ions, directing polar hydrogens away from the binuclear metal center. All other polar hydrogens were automatically generated. The charges on the α- and β-zinc ions were assigned as 1.4 and 1.3 respectively (37), and the remaining charge difference was distributed to the metal ligating residues (His-77, His-79, His-175, and His-231 (0.25 each); Asp-291 (−0.9) and Kcx-175 (−0.8)) to give each metal an apparent charge of 2.0.
Molecules were docked into the active site of Atu3266 with DOCK3.6 (38, 39) (http://dock.compbio.ucsf.edu/). Docking was performed using receptor and ligand bin sizes of 0.4 Å, an overlap of 0.1–0.2 Å, a distance tolerance of 1.5 Å and label matching turned off. The docked molecules were subjected to 250 cycles of rigid-body minimization and were scored based on van der Waals, electrostatic and solvation terms. Each pose of the scored HEI metabolites was filtered, based on a maximum distance of 4 Å between the reactive center of the HEI molecule and the zinc metal center. Finally, the top 500 scored molecules were manually inspected to identify potential substrates. Some of the docked substrates were further subjected to minimization with the program SZYBKI (39); this was used to confirm substrate fit and to further investigate hydrogen bond networks.
Since many molecules were synthesized de novo for this project, they were not present in the KEGG library. To investigate the stereospecificity of the compounds hydrolyzed by Atu3266 and Oant2987, a dedicated library (HEI AC) containing all of the experimentally tested compounds (Schemes 2 and 3; compounds 1–56) was generated as previously described for the HEI KEGG library (9, 36). To further identify new ligands, the HEI AC library was expanded to contain other molecules with the acetylated α-hydroxyl carboxylate motif (smiles string O=C(O)C(R2)OC(=O)C]), and molecules with insertions around the chiral carbon (smiles string [O=C(O)XnC(R2)XnOC(=O)Xn]). These new compounds were docked and scored in the same manner as the HEI KEGG library molecules.
RESULTS
Purification of Atu3266 and Oant2987
Atu3266 was purified to homogeneity and the identity of this protein was confirmed by sequencing the first 8 amino acids from the N-terminus. Oant2987 was purified to homogeneity using gel filtration and anion exchange chromatography. Each protein was >95% pure based on SDS-PAGE analysis. ICP-MS confirmed the presence of 2.0 ± 0.2 equivalents of Zn per subunit of Atu3266 and 1.0 ± 0.1 of Zn per subunit of Oant2987 with less than 0.1 equivalents of iron and manganese. The addition of supplemental zinc to Oant2987 did not improve the catalytic hydrolysis of acetyl-R-mandelate.
Three-Dimensional Structure of Atu3266
The three-dimensional structure of Atu3266 was determined to a resolution of 2.62 Å (PDB id: 2OGJ). The six protomers in the asymmetric unit form a dimer of trimers as illustrated in Figure 2. The hexamer appears as two discs (made of three protomers) stacked against each other. Each protomer consists of two domains; a TIM barrel domain and a second domain consisting of two β-sheets formed by both N- and C-terminal residues as shown in Figure 3. The structure reveals a distorted (β/α)8-TIM barrel fold that is similar to other structurally characterized enzymes from the AHS (14). The active site is dominated by a binuclear divalent metal center that is reminiscent of the metal centers found in phosphotriesterase (40) and dihydroorotase (20) as illustrated in Figure 4. The structure of Atu3266 was determined in the presence of imidazole in the active site that is coordinated to the β-metal at a distance of 2.2 Å (not shown). The α-metal is coordinated by His-77 and His-79 at a distance of 2.2 and 2.4 Å, respectively. These residues are positioned at the C-terminal end of β-strand 1. This metal ion is also coordinated to Asp-291 from β-strand 8 and a carboxylated Lys-175 (Kcx-175) from β-strand 4. The β-metal is coordinated to the carboxylated lysine from β-strand 4, and to His-208 and His-231 from β-strands 5 and 6. A water molecule is at 3.1 Å from the β-metal ion. The two metal ions are 3.3 Å apart.
Figure 2.

Representation of the hexameric arrangement of subunits for the three-dimensional structure of Atu3266. Face-on (A), and edge-on (B) view of hexameric structure of 2OGJ.
Figure 3.

Ribbon representation of the three dimensional structure domain of Atu3266. The metal coordinating ligands are depicted in purple. The central β-barrel is shown in gray; the helices are shown in blue. In rose and tan color are the N- and C- terminal domains of the enzyme, respectively.
Figure 4.

Structural model for the active site of Atu3266 showing the residues that coordinate the binuclear metal center. The metal ligating residues are shown in gray, the binuclear metal center is shown in magenta, the coordinating water molecule to the β-metal is shown in red.
Substrate Specificity of Atu3266 and Oant2987
D- and L-Dihydroorotate (56) were the first two compounds to be tested as substrates for Atu3266, since this enzyme has been annotated in various databases as a dihydroorotase. However, no hydrolysis of these compounds could be observed and thus this annotation is not correct. This experiment was followed by the utilization of a focused library of N-acetyl, N-succinyl and N-formyl derivatives of the common D- and L- amino acids and multiple libraries of L-Xaa-L-Xaa, L-Xaa-D-Xaa, and D-Xaa-L-Xaa dipeptides. From the more than 1200 compounds tested in the initial screening only two compounds were found with very weak rates of hydrolysis. The values of kcat/Km for N-acetyl- D-serine (1) and N-acetyl-D-threonine (2) are 4.0 M−1 s−1 and 2.0 M−1 s−1 respectively. This finding prompted the separate synthesis of N-acetyl-D-cysteine (3) and N-formyl-D-serine (4), but these compounds were not detectable substrates for Atu3266. Methylating the α-carboxylate (compound 5) or phosphorylating the side chain hydroxyl (compound 6) of the weak substrate, N-acetyl-D-serine, abolished the catalytic activity. The N-carbamoyl (compound 7) and N-phosphoryl (compound 8) derivatives of D-serine were not catalytically active. However, the substitution of the α-amino group with an α-hydroxyl group, as in 2-acetyl-R-glycerate (9) resulted in a two order of magnitude improvement in kcat/Km, relative to N-acetyl-D-serine.
The dramatic increase in the rate of hydrolysis of 2-acetyl-R-glycerate (9) prompted the exploration of modifications to this sub-structure. However, 2-phospho-R-glycerate (10) and 3-phospho-2-acetyl-R-glycerate (11) were not hydrolyzed and no activity could be observed with 2-acetyl glycerol (12). Surprisingly, excision of the hydroxymethyl substituent to form acetyl glycolate (13) increased kcat/Km by a factor of 27, relative to compound 9. Further substitutions to the sub-structure of acetyl glycolate (13) did not result in any further improvements (compounds 14 to 28) in catalytic activity except for the addition of a phenyl group. Acetyl-R-mandelate (28) is hydrolyzed with a kcat/Km of 2.8 × 105 M−1 s−1. This is a factor of 20 better than acetyl glycolate (13) and nearly 5 orders of magnitude better than the initial hit, N-acetyl-D-serine (1). No activity was observed with phospho(enol)pyruvate (30), the amide of acetyl-R-mandelate (31), 2-phospho-R-mandelate (32), acetyl-D-phenyl glycine (33), the methyl ester of acetyl-R-mandelate (34), acetyl-S-mandelate (35), and 2-acetoxy-isobutyric acid (29). Further modifications to acetyl-R-mandelate did not improve the rate of hydrolysis (see compounds 36 to 50), relative to acetyl-R-mandelate (28). Other modifications, including p-nitrophenyl acetate (51), aspirin (52) and various lactones (53–55) were not active. The kinetic constants for the catalytically active substrates are provided in Table 2.
Table 2.
Kinetic parameters for Atu3266 and Oant2987
| Atu3266 | Oant2987 | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Substrate | kcat (s−1) | Km (mM) | kcat/Km (M−1s−1) | kcat (s−1) | Km (mM) | kcat/Km (M−1s−1) |
| 1 | n.a. | n.a. | 4 | n.c. | n.c. | n.c. |
| 2 | n.a. | n.a. | 2 | n.c. | n.c. | n.c. |
| 3 | <0.001 | n.a. | n.a. | <0.001 | n.a. | n.a. |
| 9 | 12 ± 1 | 25 ± 4 | (4.8 ± 0.1) × 102 | 0.6 ± 0.1 | 2.1 ± 0.5 | (3 ± 1) × 102 |
| 13 | 40 ± 3 | 3.1 ± 0.5 | (1.3 ± 0.2) × 104 | 4.2 ± 0.2 | 1.1 ± 0.2 | (4.2 ± 0.8) × 103 |
| 14 | 0.8 ± 0.1 | 0.5 ± 0.1 | (1.6 ± 0.2) × 103 | 0.4 ± 0.1 | 0.6 ± 0.1 | (7 ± 1) × 102 |
| 15 | 0.5 ± 0.1 | 10 ± 2 | 50 ± 10 | n.a. | n.a | 40 |
| 16 | 0.6 ± 0.1 | 9.5 ± 0.7 | 58 ± 6 | n.a | n.a. | 60 |
| 17 | 1.5 ± 0.1 | 7 ± 1 | (2.1 ± 0.3) × 102 | n.a. | n.a. | 200 |
| 18 | 3.1 ± 0.3 | 6 ± 1 | (5.0 ± 0.8) × 102 | 0.81 ± 0.08 | 1.2 ± 0.1 | (8.1 ± 0.8) × 103 |
| 19 | 1.1 ± 0.2 | 9.5 ± 3.0 | (1.2 ± 0.4) × 102 | 0.23 ± 0.02 | 1.4 ± 0.1 | (2.2 ± 0.2) × 102 |
| 20 | 0.41 ± 0.04 | 2.1± 0.5 | (2.0 ± 0.5) × 102 | 0.30 ± 0.02 | 0.71 ± 0.02 | (4.3 ± 0.4) × 102 |
| 21 | n.a. | n.a. | (1.1 ± 0.6) × 102 | n.a. | n.a. | 30 |
| 22 | n.a. | n.a. | (1.8 ± 0.6) × 102 | n.a. | n.a. | 30 |
| 23 | n.a. | n.a. | 50 ± 10 | n.c. | n.c. | n.c. |
| 24 | n.a. | n.a. | (2.4 ± 0.5) × 102 | 2.0 ± 0.1 | 2.0 ± 0.5 | (1.0 ± 0.2) × 103 |
| 25 | 0.10 ± 0.03 | 0.4 ± 0.1 | (2.5 ± 0.2) × 102 | <0.05 | n.a. | n.a. |
| 26 | 0.21 ± 0.01 | 0.3 ± 0.1 | (6.6 ± 1.0) × 102 | <0.02 | n.a. | n.a. |
| 27 | 2.3 ± 0.2 | 30 ± 10 | 67 ± 22 | n.a. | n.a. | 40 |
| 28 | 280 ± 5 | 1.0 ±0.1 | (2.8 ± 0.2) × 105 | 130 ± 1 | 0.7 ± 0.1 | (1.8 ± 0.2) × 105 |
| 36 | 34 ± 2 | 1.1 ± 0.1 | (3.4 ± 0.3) × 104 | 6.0 ± 0.2 | 0.21 ± 0.02 | (3.0 ± 0.3) × 104 |
| 37 | 70 ± 7 | 7 ± 1 | (1.0 ± 0.1) × 104 | 60 ± 4 | 3.4 ± 0.5 | (1.7 ± 0.2) × 104 |
| 38 | 43 ± 5 | 4 ± 1 | (1.1 ± 0.2) × 104 | 4.1 ± 0.3 | 0.5 ± 0.1 | (8.0 ± 0.8) × 103 |
| 39 | 30 ± 9 | 10 ± 3 | (3.0 ± 0.9) × 103 | 60 ± 6 | 18 ± 2 | (3.3 ± 0.3) × 103 |
| 40 | n.a | n.a | (3.0 ± 0.3) × 103 | 50 ± 5 | 5 ± 1 | (1.0 ± 0.2) × 104 |
| 41 | n.a | n.a | (5.0 ± 0.4) × 103 | 36 ± 5 | 8 ± 1 | (4.5 ± 0.5) × 103 |
| 42 | 190 ± 20 | 12 ± 3 | (1.6 ± 0.1) × 104 | 150 ± 10 | 3.0 ± 0.5 | (5 ± 1) × 104 |
| 43 | n.a | n.a | (2.2 ± 0.2) × 103 | 30 ± 2 | 4.0 ± 0.5 | (7.5 ± 0.9) × 103 |
| 44 | n.a | n.a | (1.0 ± 0.1) × 103 | 50 ± 1 | 5 ± 1 | (1.0 ± 0.2) × 104 |
| 45 | 10 ± 1 | 2.0 ± 0.2 | (5.0 ± 0.5) × 103 | 40 ± 5 | 11 ± 3 | (3.6 ± 0.9) × 103 |
| 46 | 7.0 ± 0.2 | 0.6 ± 0.1 | (1.0 ± 0.1) × 104 | 10 ± 1 | 0.8 ± 0.2 | (1.2 ± 0.3) × 104 |
| 47 | 10 ± 1 | 1.4 ± 0.3 | (7.0 ± 0.2) × 103 | 20 ± 2 | 2.0 ± 0.5 | (1.0 ± 0.2) × 104 |
| 48 | 50 ± 4 | 10 ± 1 | (5.1 ± 0.1) × 103 | 60 ± 6 | 9 ± 1 | (7.0 ± 0.7) × 103 |
| 49 | 90 ± 9 | 7 ± 1 | (1.3 ± 0.1) × 104 | 80 ± 6 | 3 ± 0.2 | (2.7 ± 0.2) × 104 |
| 50 | 17 ± 3 | 5 ± 1 | (3.5 ± 0.1) × 103 | 28 ± 10 | n.a. | (1 ± 0.3) × 103 |
n.c denotes a parameter that was not calculated, mainly because there was no detection of acetic acid by the coupling system. Whereas n.a. denotes a parameter that was not available, this was under the event that the substrate did not saturate the enzyme and the rates obtained were fit to a linear curve to obtain V/K for observed activity.
Inhibition by N-Methyl Phosphonate of D-phenyl Glycine
The N-methylphosphonate and O-methylphosphonate derivatives of D-phenyl glycine (57) and R-mandelate (56), respectively, were synthesized and tested as inhibitors for the hydrolysis of acetyl-R-mandelate (28) by Atu3266 and Oant2987. The O-methylphosphonate inhibitor of R-mandelate was unstable. However, the N-methylphosphonate analog of D-phenyl glycine (57) was found to be a competitive inhibitor versus acetyl-R-mandelate. A fit of the data to equation 2 gave an inhibition constant of 35 ± 2 μM for Atu3266 and 40 ± 2 μM for Oant2987.
Computational Docking to the Active Site of Atu3266
Molecules from the HEI KEGG library consisted of high-quality poses enriched for carbamoylated and acetylated amino acids. Thus, acetylated amino acids such as N-acetyl-L-lysine (rank #40), N-acetyl-D-methionine (rank #171), N-acetyl-D-cysteine (3) (rank #218), N-acetyl-L-leucine (rank #313), and N-acetyl-D-phenylalanine (48) (rank #440) (Figure S1) and other metabolites such as N-carbamoyl-L-aspartate, N-acetyl-D-glucosaminate and oxalureate were found at the top of the docking list. Further virtual docking experiments with the HEI AC library suggested a common binding pose for the compounds that contain the HEI acetylated α-hydroxyl carboxylate (Figure 5a). A detailed examination revealed that the acetylated α-hydroxyl carboxylates direct the ester group towards the metal cluster, where the carbonyl oxygen is positioned over the α- and β-zinc atoms and the transition state is stabilized by Arg-177 (Figure 5b). In this orientation, the methyl group of the acetyl moiety is surrounded by the hydrophobic pocket lined by Ile-87, Leu-140, Cys-142, and the plane of the imidazole side chain of His-79 and His-293. This hydrophobic pocket is large enough that it can accommodate an ethyl group such as of R-propionyloxy mandelate, ranked #241 (Figure S2i). The bridging oxygen of the ester moiety is oriented towards Asp-291, thus facilitating the enzymatic hydrolysis of the acetylated α-hydroxyl carboxylates.
Figure 5.

Modeled structure of docked acetyl-R-mandelate into the active site of Atu3266. Based on the docking results of the HEI library, (A) is a model of the ground state acetylated α-hydroxyl carboxylate substrates (green sticks) in the active site. The orange spheres illustrate the possible orientation of the R- and S- enantiomers. The active site is shown as a surface representation, hydrogen bonds between the carboxylate of the substrate and loop 267–269 are shown as dash lines. In this orientation the bridging oxygen of the ester points towards Asp-291 and the carbonyl oxygen stacks over the binuclear metal center (purple spheres), directing the methyl group of the acetate into a hydrophobic pocket lined by Ile-87, Leu-140, Cys-142, His-79 and His-293. (B) The HEI substrate, acetyl-R-mandelate (28) (cyan sticks) ranked #221, is shown in its best docked pose. Here the hydrogen on the bridging oxygen is 1.9 Å away from Asp-291. The HEI intermediate is stabilized by Arg-177. The carboxylate hydrogen bonds with loop 267–269 and the phenyl group is positioned between the cavity walls, allowing for stacking interactions with Asn-144.
The docking calculations indicate that the two remaining fragments of the molecule, the carboxylate and the mostly nonpolar substituent at the chiral carbon, are necessary for the correct binding and orientation of the substrates in the active site. Critical to the binding is the carboxylate, which forms a hydrogen bond network with the loop formed between β-strand 7 and α-helix 7 of the AHS domain. These hydrogen bond interactions include the amide nitrogens of Gly-267 and Ala-268, and the hydroxyl side chain of Ser-269. In some instances, an additional hydrogen bond is formed between the carboxylate of the substrate and the Nε hydrogen of His-293. The second and more variable portion of the substrates for Oant2987 and Atu3266, contain mostly nonpolar substitutions at the chiral α-carbon of the acetylated α-hydroxyl carboxylates. All molecules identified as substrates possess the R-stereochemistry at C2 of the acetylated α-hydroxyl carboxylates, including the best substrate acetyl-R-mandelate (28). The catalytically competent pose of the transition state mimic for acetyl-R-mandelate (28) identified from the HEI AC library had an energy score of −140.65. Thus, this molecule would have ranked #221 out of 57,672 HEI KEGG molecules, placing it in the 99.6th percentile. In this pose the aromatic phenyl ring moiety of acetyl-R-mandelate (28) provides pi-stacking interaction with the side chain nitrogen atom of Asn-144. The distance between these two components varies depending on the substrates docked; however, in the catalytically competent poses these molecules adopt conformations where the center of the ring is 2.8–3.5 Å away from the amino acid side chain (41).
With the HEI AC virtual library, we have been able to screen compounds with slightly longer backbones (insertions around the chiral carbon). Docking results suggest that larger molecules can be accommodated into the active site, such as O-acetyl-S-carnitine, (S)-3-acetoxy-4-(dimethylamino)butanoic acid, acetyl tropic acid and 3-carbamoyloxy-2-phenylpropionic acid (Figure S3a). Of these, (S)-3-acetoxy-4-(dimethylamino)butanoic acid (Figure S3b) and racemic acetyl tropic acid (Figure S3c) were tested as possible substrates for Atu3266. However, only acetyl tropic acid is active, albeit with a very poor kcat/Km of 200 M−1 s−1. This indicates that a small extension between the chiral carbon and the ester can be tolerated, but the simple acetylated hydroxyl carboxylate chemotype is preferred.
DISCUSSION
Three-Dimensional Structure of Atu3266
The three-dimensional structure of Atu3266 shows that this enzyme possesses a binuclear metal center, reminiscent of other enzymes in the amidohydrolase superfamily such as dihydroorotase, phosphotriesterase and urease (13). The buried α-metal is coordinated by His-77, His-79, Asp-291 and the carboxylated Lys-175. The more solvent exposed β-metal is coordinated to His-208, His-231, the carboxylated Lys-175 and a water molecule. The individual subunits of Atu3266 associate to form a hexameric oligomer.
Cog3964
There are 24 clusters of orthologous groups in the AHS and cog3964 is one of the smallest, with about 200 sequences (Figure 1). The most common annotation in NCBI for this cluster of enzymes is dihydroorotase, and in some cases, adenine deaminase. The basis for this annotation is not clear, but the experiments reported here demonstrate that this annotation is incorrect. When the E-value cutoff is set to 10−70, the sequence similarity network for cog3964 segregates into 8 groups with 3 or more sequences per group and Atu3266 and Oant2987 belong to Group 6 of cog3964. An amino acid sequence comparison between those enzymes in Group 6 of cog3964 and the structurally characterized dihydroorotases and adenine deaminases (not shown) demonstrates that the key residues that have been implicated in substrate binding and recognition in these enzymes are not present in either Atu3266 or Oant2987. This supports the conclusion that Atu3266 and Oant2987 do not catalyze the deamination of adenine or the hydrolysis of dihydroorotate.
Determination of Substrate Profile
The first indication of catalytic activity by Atu3266 was observed in a library of N-acetyl-D-amino acids. After each component of the library was tested independently, it was observed that N-acetyl-D-serine (4 M−1 s−1) and N-acetyl-D-threonine (2 M−1s−1) were the only compounds in the N-acetyl-D-Xaa library to be hydrolyzed. Screening for hydrolytic activity was also conducted using a comprehensive library of dipeptides; however, none of the dipeptide libraries showed detectable catalytic activity. Subsequently we focused on the activity obtained from N-acetyl-D-serine, and developed a library of compounds with various modifications including changes in stereochemistry at the central α-chiral carbon, the side chain and carboxylate functionalities. None of these changes generated a better substrate. However, when the amide linkage was changed to an ester, a 100-fold increase in activity was observed. Atu3266 hydrolyzes the ester linkage of acetyl-R-glycerate with a rate constant of 400 M−1s−1. When the hydroxymethyl group was removed and replaced with a hydrogen (acetyl glycolate) the rate of hydrolysis increased by an additional two orders of magnitude (1.3 × 104 M−1 s−1). The highest activity was achieved in the deacetylation of acetyl-R-mandelate with a rate constant of 2.8 × 105 M−1 s−1.
Additional modifications to the acetyl-R-mandelate substructure failed to improve the catalytic activity. Substitutions and modifications to the C1 carboxylate eliminated deacetylation activity. These alterations included methyl ester and amide formation, phosphorylation, and reduction to a hydroxy methyl group. At C2, the most beneficial modification was the change of the amide nitrogen to oxygen to form an ester. Modification of the acetyl group to a formyl, succinyl or carbamoyl group did not prove to be a better substituent than the acetyl group. The stereochemistry at C2 must be in the R-configuration. The acetyl group could be extended to a propionyl group but the rate of hydrolysis is reduced. The presence of the phenyl group at the side chain position proved to be the best substitution, but addition of functionality to the aromatic ring decreased the rate of hydrolysis.
The organisms containing Atu3266 and Oant2987 are devoid of other genes that are obvious candidate for the metabolism of mandelate. However, most of the organisms that contain a protein from cog3964 also have an adjacent gene that is an apparent homologue to selenocysteine synthase (SelA) that belongs to cog1921. The genes for two of the proteins (Atu3263 and Oant2990) were cloned, expressed, and the proteins purified to homogeneity (data not shown). The purified enzymes contained pyridoxal-5′-phosphate but we were unable to identify any catalytic activity for these proteins with a library of amino acids. The relationship between the proteins from cog1921 and cog 3964 is not obvious.
Comparisons to Other N-acetyl D-Amino Acid Deacetylases
Other enzymes within the amidohydrolase superfamily have been shown to catalyze the hydrolysis of N-acetyl-D-amino acid derivatives. For example, enzymes within cog3653 have been shown to be N-acetyl-D-amino acid deacetylases (26, 42–44). In cog3653 the binuclear metal center is not bridged by a carboxylated lysine residue and the α-carboxylate group of the substrate is ion paired and hydrogen bonded to conserved arginine and tyrosine residues (26). These motifs are not conserved in the enzymes of cog3964, where instead, the carboxylate group of the substrate is recognized by the backbone residues following β-strand 7.
At this point it is not clear as to whether the enzymes identified in this investigation from cog3964 are generic esterases, or whether there is a more specific substrate. Acetyl-R-mandelate has apparently not been identified as a physiological metabolite in bacteria. In those organisms that have been found to use mandelate as a carbon source, there is no indication of a requirement of a deacetylase for the hydrolysis of acetyl-R-mandelate (45).
Computational Docking for Identification of Substrate-Enzyme Interactions
Docking of the HEI KEGG virtual metabolite library to the Atu3266 structure was highly enriched for carbamoylated or acetylated amino acids (acetyl-Met, -Cys, -Phe, -Leu, and –Lys) (Figure S1). An examination of the KEGG database revealed that the database only included 10 acetylated amino acids (Met, Cys, Phe, Leu, Lys, His, Trp, Asp, Glu and Gln). This indicates a high degree of enrichment in the docking list for the N-acetyl amino acids as five of them were found in the top 0.76% of the overall pool of HEI KEGG compounds. As such, the initial docking experiments of the HEI KEGG library did not include the two catalytically competent acetylated amino acids, N-acetyl-D-serine (1) and N-acetyl-D-threonine (2). These were subsequently prepared and docked in the HEI AC library and obtained scores that would respectively rank them #321 and #265 out of 57,672 HEI KEGG molecules. The poses of the two amino acids are suboptimal as they either leave the side chain hydroxyl unsatisfied or allow it to partially interact with the carboxylate binding loop. However, other N-acetylated amino acids are found to optimally occupy the active site, as seen with N-acetyl-D-phenyl glycine (33) (Figure S4).
Based on analysis of the focused HEI AC library docking results, molecules that are turned over by the enzyme contain an acetylated α-hydroxyl carboxylate that positions the acetyl moiety over the zinc atoms, directing the methyl group into a small hydrophobic pocket. This pocket is large enough to accommodate ethyl groups as seen by the substrate R-propionyloxy mandelate (Figure S2i) and R-2-(propionyloxy)-butanoate (22)(Figure S2b). In addition, this catalytically competent pose orients the carboxylate of the substrate towards the loop formed by Gly-267, Ala-268 and Ser-269 (Figure 5a). In this pose any substitutions or bulky additions on the substrate carboxylate would change the electrostatics or cause steric clashes, precluding binding. For example, molecules with an addition of a methyl group to form an ester (34) or molecules with a substitution of the carboxylate for an organic amide (31), demonstrate no enzymatic activity.
Further docking indicates a preference for the R-enantiomer of the acetylated α-hydroxyl carboxylates, such as acetyl-R-mandelate (28). The R-enantiomers direct the hydrogen of the chiral carbon towards the floor of the active site cavity and provide ample space for the substituent (Figure 5b), while still orienting the bridging oxygen with ideal geometry and distance to Asp-291 thus facilitating catalysis. Based on the well-positioned docking poses of the substrates, we can infer from the HEI docked molecules that the ground state R-enantiomers could easily enter the active site in the extended conformation (Figure 5a). In this conformation, the ligands could easily be subjected to nucleophilic attack by an activated water molecule. Substrates that contain the R-enantiomer can easily accommodate bulky substituents, such as the phenyl ring of acetyl-R-mandelate (28), and even larger hydrophobic groups (37, 43, 46) (Figure S2d,g,h,k), which fit into the active site between the cavity walls. Furthermore, compounds with aromatic rings engage in perpendicular pi stacking interactions with the side chain nitrogen atom of Asn-144. This interaction is not present in the small branched or cyclohexane containing compounds (13–14, 18–19, 22, 24–27, 48–50), which may explain the difference in affinity parameters. This is illustrated by the different binding modes and interactions of R-2-acetoxybutanoic acid (18) and R-2-acetoxy-2-cyclohexylacetate acid (27) (Figure S2a,j). Only a few ionic interactions are observed for charged substituents on the acetylated α-hydroxyl carboxylates, these charged interactions are made with Asn-144 (Figure S2f,l) and Lys-236 (Figure S2c,e) for some of the substrates.
Strategy for Functional Annotation
The functional annotation of Atu3266 as an enzyme that was able to catalyze the hydrolysis of an ester linkage from substituted α-hydroxyl carboxylates with R-stereochemistry began with the identification of the weak substrate, N-acetyl D-serine. The combination of focused chemical libraries and computational docking to the three-dimensional crystal structure resulted in the identification of acetyl-R-mandelate as a substrate that was 5 orders of magnitude better than the initial hit. In addition to Oant2987 we predict that 7 other enzymes from Group 6 of cog3964 will have the same substrate profile as Atu3266. These proteins are listed in Table S1. The identification of function for Group 6 of cog3964 will facilitate the functional annotation for the additional groups of proteins in this COG. In preliminary experiments, a protein from Group 2 of cog3964 has been purified and crystallized (EF0837 from Enterococcus faecalis, gi|29375425, PDB id: 2ICS). This protein shares a 35% sequence identity with Atu3266 and catalyzes the hydrolysis of acetyl-R-mandelate with a value of kcat/Km that is 3-orders of magnitude smaller than the value observed for Atu3266 and Oant2987. These results suggest that the substrate profile for this enzyme will be different than for Atu3266. Efforts to identify the substrate for the enzymes of Group 2 are currently underway.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported in part by the Robert A. Welch Foundation (A-840) and the National Institutes of Health (GM 71790 and GM 74945).
We thank Dr. Peter Kolb and Dr. Ryan G. Coleman for technical discussions relating to computational techniques
Footnotes
Accession Codes
The X-ray coordinates and structure factors for Atu3266 have been deposited in the Protein Data Bank as entry 2OGJ.
The authors declare no competing financial interest.
The synthetic procedures for the preparation of the compounds used in this investigation and additional figures and tables are provided. This material is available free of charge at http://pubs.acs.org.
References
- 1.Roberts RJ. Identifying protein function - A call for community action. PLoS Biol. 2004;2:E42. doi: 10.1371/journal.pbio.0020042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rost B, Liu J, Nair R, Wrezeszczynski KO, Ofran Y. Automatic prediction of protein function. Cell Mol Life Sci. 2003;60:2637–2650. doi: 10.1007/s00018-003-3114-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Freidberg I. Automated protein function prediction – The genomic challenge. Brief Bioinform. 2006;7:225–242. doi: 10.1093/bib/bbl004. [DOI] [PubMed] [Google Scholar]
- 4.Glasner M, Gerlt JA, Babbitt PC. Evolution of enzyme superfamilies. Curr Opinion in Chem Biol. 2006;10:492–497. doi: 10.1016/j.cbpa.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 5.Newton RJ, Griffin LE, Bowles KM, Meile C, Gifford S, Givens CE, Howard EC, King E, Oakley CA, Risch CR, Rinta-Kanto J, Sharma S, Sun S, Varaljay V, Vila-Costa M, Westrich JR, Moran MA. Genome characteristics of a generalist marine bacterial lineage. ISME Journal: Multidisciplinary Journal of Microbial Ecology. 2010;4:784–798. doi: 10.1038/ismej.2009.150. [DOI] [PubMed] [Google Scholar]
- 6.Sweetlove LJ, Fernie AR. Regulation of metabolic networks: Understanding metabolic complexity in the systems biology era. New Phytol. 2005;168:9–24. doi: 10.1111/j.1469-8137.2005.01513.x. [DOI] [PubMed] [Google Scholar]
- 7.Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO. Assigning protein functions by comparative genome analysis: Protein phylogenetic profiles. Proc Natl Acad Sci. 1999;96:4285–4288. doi: 10.1073/pnas.96.8.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim S-H. Structural genomics of microbes: An objective. Curr Opinion in Biol. 2000;10:380–383. doi: 10.1016/s0959-440x(00)00100-7. [DOI] [PubMed] [Google Scholar]
- 9.Hermann JC, Ghanem E, Li Y, Raushel FM. Predicting substrates by docking high-energy intermediates to enzyme structures. J Am Chem Soc. 2006;128:15882–15891. doi: 10.1021/ja065860f. [DOI] [PubMed] [Google Scholar]
- 10.Kolb P, Ferreira RS, Irwin JJ, Shoichet BK. Docking and chemoinformatic screens for new ligands and targets. Curr Opinion in Biochem. 2009;20:429–436. doi: 10.1016/j.copbio.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Senisterra GA, Markin E, Yamazaki K, Hui R, Vedadi M, Awrey DE. Screening for ligands using a generic and high-throughput light-scattering-based assay. J Biomol Screen. 2006;11:940–948. doi: 10.1177/1087057106294699. [DOI] [PubMed] [Google Scholar]
- 12.Galperin MY, Koonin EV. From complete genome sequence to ‘complete’ understanding? Trends in biotech. 2010;28:398–406. doi: 10.1016/j.tibtech.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holm L, Sander C. An evolutionary treasure: Unification of a broad set of amidohydrolases related to urease. Proteins. 1997;28:72–82. [PubMed] [Google Scholar]
- 14.Seibert CM, Raushel FM. Structural and catalytic diversity within the amidohydrolase superfamily. Biochemistry. 2005;44:6383–6391. doi: 10.1021/bi047326v. [DOI] [PubMed] [Google Scholar]
- 15.Li T, Iwaki H, Fu R, Hasegawa Y, Zhang H, Liu A. α-amino-β-carboxymuconic-ε-semialdehyde decarboxylase (ACMSD) is a new member of the amidohydrolase superfamily. Biochemistry. 2006;45:6628–6634. doi: 10.1021/bi060108c. [DOI] [PubMed] [Google Scholar]
- 16.Hara H, Masai E, Katayama Y, Fukuda M. The 4-oxalomesaconate hydratase gene, involved in the protocatechuate 4,5-cleavage pathway, is essential to vanillate and syringate degradation in Sphingomonas paucimolbilis SYK-6- hydration reaction. J Bacteriol. 2000;182:6950–6957. doi: 10.1128/jb.182.24.6950-6957.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams L, Nguyen T, Si Y, Porter T, Raushel FM. Uronate isomerase: A nonhydrolytic member of the amidohydrolase superfamily with an ambivalent requirement for divalent metal ion. Biochemistry. 2006;45:7453–7462. doi: 10.1021/bi060531l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nuc Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Federova ND, Koonin EV. The COG database: New developments in phylogenetic classification of proteins from complete genomes. Nuc Acids Res. 2001;29:22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thoden JB, Phillips GN, Neal TM, Raushel FM, Holden HM. Molecular structure of dihydroorotase: A paradigm for catalysis through the use of a binuclear metal center. Biochemistry. 2001;40:6989–6997. doi: 10.1021/bi010682i. [DOI] [PubMed] [Google Scholar]
- 21.Porter TN, Li Y, Raushel FM. Mechanism of the dihydroorotase reaction. Biochemistry. 2004;43:16285–16292. doi: 10.1021/bi048308g. [DOI] [PubMed] [Google Scholar]
- 22.Kamat SS, Bagaria A, Kumaran D, Holmes-Hampton GP, Fan H, Sali A, Sauder MJ, Burley SK, Lindahl PA, Swaminathan S, Raushel FM. Catalytic mechanism and three-dimensional structure of adenine deaminase. Biochemistry. 2011;50:1917–1927. doi: 10.1021/bi101788n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goble AM, Zhang Z, Sauder MJ, Burley SK, Swaminathan S, Raushel FM. Pa0148 from Pseudomonas Aeruginosa catalyzes the deamination of adenine. Biochemistry. 2011;50:6589–6597. doi: 10.1021/bi200868u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient for a protein. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiang D, Patskovsky Y, Chengfu X, Meyer AJ, Sauder JM, Burley SK, Almo SC, Raushel FM. Functional identification of incorrectly annotated prolidases from the amidohydrolase superfamily of enzymes. Biochemistry. 2009;48:3730–3742. doi: 10.1021/bi900111q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cummings JA, Fedorov AA, Xu C, Brown S, Fedorov E, Babbitt PC, Almo SC, Raushel FM. Annotating enzymes of uncertain function: The deacylation of D-amino acids by members of the amidohydrolase superfamily. Biochemistry. 2009;48:6469–6481. doi: 10.1021/bi900661b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doi E, Shibata D, Matoba T. Modified colorimetric ninhydrin methods for peptidase assay. Anal Biochem. 1981;118:173–184. doi: 10.1016/0003-2697(81)90175-5. [DOI] [PubMed] [Google Scholar]
- 28.Chapman E, Wong CH. A pH sensitive colorimetric assay for the high-throughput screening of enzyme inhibitors and substrates: A case study using kinases. Bioorg Med Chem. 2002;10:551–555. doi: 10.1016/s0968-0896(01)00306-6. [DOI] [PubMed] [Google Scholar]
- 29.Otwinowski Z, Minor W. Processing of X-ray diffraction Data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 30.Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Cryst. 2002;D58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 31.CCP4 . CCP4 Suite: Programs for protein crystallography. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 32.De-La-Fortelle E, Bricogne G. Maximum-likelihood heavy atom parameter refinement in the MIR and MAD methods. Methods Enzymol. 1997;276:472–493. doi: 10.1016/S0076-6879(97)76073-7. [DOI] [PubMed] [Google Scholar]
- 33.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nature Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 34.Brunger AT, Adams PD, Clore GM, Delano WL, Gros P, Grosse-Kunstleve RW, Jiang FS, Kuszwewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Somonsom T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 35.Jones TA, Zou J-Y, Cowan SW, Kjeldgaard M. Improved methods in building protein models in electron density map and the location of errors in these models. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 36.Xiang DF, Kolg B, Fedorov AA, Meier MM, Fedorov LV, Nguyen TT, Sterner R, Almo SC, Shoichet BK, Raushel FM. Functional annotation and three-dimensional structure of Dr0930 from Deinococcus radiodurans, A close relative of phosphotriesterase in the amidohydrolase superfamily. Biochemistry. 2009;48:2237–2247. doi: 10.1021/bi802274f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Irwin JJ, Raushel FM, Shoichet BK. Virtual screening against Metalloenzymes for inhibitors and substrates. Biochemistry. 2005;44:12316–12328. doi: 10.1021/bi050801k. [DOI] [PubMed] [Google Scholar]
- 38.Irwin JJ, Shoichet BK, Mysinger MM, Huang N, Colizzi F, Wassam P, Cao Y. Automated docking screens: A feasibility study. J Med Chem. 2009;52:5712–5720. doi: 10.1021/jm9006966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wlodek S, Skillman AG, Nicholls A. Ligand entropy in gas-phase, upon salvation and protein complexation. Fast estimation of quasi-newton hessian. J Chem Theory Comput. 2010;7:2140–2152. doi: 10.1021/ct100095p. [DOI] [PubMed] [Google Scholar]
- 40.Benning MM, Kuo JM, Raushel FM, Holden HM. Three-dimensional structure of the binuclear center of phosphotriesterase. Biochemistry. 1995;34:7973–7978. doi: 10.1021/bi00025a002. [DOI] [PubMed] [Google Scholar]
- 41.Ma JC, Dougherty DA. The cation-pi interaction. Chem Rev. 1997;97:1303–1324. doi: 10.1021/cr9603744. [DOI] [PubMed] [Google Scholar]
- 42.Liaw S-H, Chen S-J, Ko T-P, Hsu C-S, Chen C-J, Wang AH-J, Tsai Y-C. Crystal structure of D-aminoacylase from Alcaligenes faecalis DA1. J Biol Chem. 2003;278:4957–4962. doi: 10.1074/jbc.M210795200. [DOI] [PubMed] [Google Scholar]
- 43.Lai W-L, Chou L-Y, Ting C-Y, Kirby R, Tsai Y-C, Wang AH-J, Liaw S-H. The functional role of the binuclear metal center in D-aminoacylase: one-metal activation and second-metal attenuation. J Biol Chem. 2004;279:13962–13967. doi: 10.1074/jbc.M308849200. [DOI] [PubMed] [Google Scholar]
- 44.Liu J, Asano Y, Ikoma K, Yamashita S, Hirose Y, Shimoyama T, Takahashi S, Nakayama T, Nishino T. Purification, characterization and primary structure of a novel N-acyl-D-amino acid amidohydrolase from Microbacterium natoriense TNJL143–2. J Biosci Bioeng. 2012 doi: 10.1016/j.jbiosc.2012.05015. [DOI] [PubMed] [Google Scholar]
- 45.Ju X, Yu H-L, Pan J, Wei D-Z, Xu J-H. Bioproduction of chiral mandelate by enantioselective deacylation of α-acetoxyphenylacetic acid using whole cells of newly isolated Pseudomonas sp. ECU1011. Appl Microbiol Biotechnol. 2010;86:83–91. doi: 10.1007/s00253-009-2286-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.