

Abstract
Hydroxylated analogues of the anticancer topoisomerase I (Top1) inhibitors indotecan (LMP400) and indimitecan (LMP76) have been prepared because: 1) a variety of potent Top1 poisons are known that contain strategically placed hydroxyl groups, which provides a clear rationale for incorporating them in the present case, and 2) the hydroxylated compounds could conceivably serve as synthetic standards for the identification of metabolites. Indeed, incubating LMP400 and LMP776 with human liver microsomes resulted in two major metabolites of each drug, which had HPLC retention times and mass fragmentation patterns identical to the synthetic standards. The hydroxylated indotecan and indimitecan metabolites and analogues were tested as Top1 poisons and for antiproliferative activity in a variety of human cancer cell cultures, and in general were found to be very potent. Differences in activity resulting from the placement of the hydroxyl group are explained by molecular modeling analyses.
Introduction
Topoisomerases are nature’s ubiquitous solution for managing the topology and torsional states of DNA. Topoisomerase I (Top1) is an essential enzyme that relaxes supercoiled DNA so that it may be replicated, transcribed, and repaired.1–4 The enzyme acts through a nucleophilic tyrosine residue (Tyr723), which nicks the phosphodiester backbone of double-stranded, supercoiled DNA and forms a transient “cleavage complex” in which the 3′ end of the broken DNA strand is covalently linked to the enzyme. Within this “cleavage complex”, the scissile (broken) strand undergoes “controlled rotation” around the unbroken strand, a process that relaxes the DNA. The catalytic cycle ends when the 5′ end of the scissile strand religates the DNA and the enzyme is released. If this cycle is inhibited, DNA damage ensues, which in turn activates DNA damage responses, leading to cell cycle arrest or the eventual triggering of pro-apoptotic cascades.1, 5–9
As Topl is overexpressed and DNA damage responses are defective in some human tumors, several Top1 inhibitors have been developed as chemotherapeutic agents.4, 10 Representative examples are shown in Figure 1. The alkaloid camptothecin (1)11 is not used clinically, but its semisynthetic derivatives topotecan (2) and irinotecan (3) are FDA-approved.1, 10, 12 These compounds act by intercalating between the base pairs in the cleavage complex and binding at the Top1-DNA interface,13 where they “poison” the complex (prevent DNA religation), resulting in persistent, covalent Top1-DNA lesions that are then converted into irreversible double-strand breaks when they collide with the advancing replication machinery, resulting in apoptosis.6–9 Although potent, camptothecin derivatives suffer from many shortcomings, including poor solubility, dose-limiting toxicity, pharmacokinetic limitations resulting from the instability of the E-ring lactone under physiological pH, and binding of the lactone hydrolysis product to plasma proteins.10, 14–16
Figure 1.

Representative Top1 Inhibitors.
The indenoisoquinolines were therefore developed as therapeutic alternatives. In 1998 a COMPARE analysis17, 18 was performed on NSC 314622 (4), which indicated that it may act in a manner similar to camptothecin and derivatives. Indeed, this compound was found to be a Top1 inhibitor.19 Since then, many optimization and SAR studies have produced potent indenoisoquinolines such as MJ-III-65 (5),20–24 which inhibit Top1 through an intercalation and interfacial mechanism similar to compound 1. Two of these compounds, indimitecan (LMP776, 6) and indotecan (LMP400, 7) were promoted into Phase I clinical trials at the National Cancer Institute.25 These compounds appear to be stable, and are powerful, cytotoxic Top1 poisons that induce long-lasting DNA breaks and overcome the drug resistance issues associated with the camptothecins.20, 26, 27
The metabolism of 6 and 7 is currently under investigation, which has led to the synthesis of potential metabolites to be used as synthetic standards for metabolism studies. As part of this study, structural analogues of the proposed metabolites are also being prepared and investigated for Top1 inhibitory activity. It was proposed that the indenoisoquinolines 6 and 7 could be metabolically labile at several positions (Figure 2). The methoxy groups of 6 and 7 are likely to be cleaved in vivo. O-Dealkylation, catalyzed chiefly by hepatic cytochrome P450 enzymes, is a common and well-precedented metabolic process that, for example, plays a significant role in human metabolism of the chemotherapeutic agents etoposide and teniposide,28 opiates and opiate antagonists,29, 30 and the topoisomerase inhibitors berberine and Genz-64482.31, 32
Figure 2.

Proposed metabolic pathways and potential metabolites of 6 and 7.
Methylenedioxy rings are also possible sites of metabolism (likely via oxidation). Both demethylenation (to yield catechols) and demethylenation/alkylation processes (to yield o-methoxyphenols) are observed in rodent and human metabolism of berberine,31 safrole and piperonal derivatives,33, 34 MDMA (“ecstasy”),35 and the designer drug MDPPA.36
Compounds 6 and 7 could also be substrates for metabolic ketone reductases. This phase I process is observed in mice for the related indenoisoquinoline oracin37 as well as for the indenoisoquinoline rexinoid AM6-36 in human hepatocytes.38 The synthesis of hydroxy and reduced analogues is also justified by literature that is rich with potent Top1 inhibitors bearing phenolic hydroxyls, including the active metabolite of 3 (SN-38), the alkaloid fagaronine,10 various other 10-and 11-hydroxycamptothecins,39, 40 indolocarbazoles,41 and the dual Top1/Top2 inhibitor TAS-103.42 Additionally, some 11-hydroxy (reduced) indenoisoquinolines have been synthesized and tested against Top1 with promising results,43, 44 and 9-hydroxyindenoisoquinolines have been assayed against Top2,45 but to our knowledge, no A- or D-ring hydroxyindenoisoquinolines have been assayed against Top1. To this end, the demethylenated/alkylated analogues 14a-b (Scheme 1) and regioisomers 22a-b (Scheme 2), the demethylated analogues 35a-b (Scheme 4) and regioisomers 44a-b (Scheme 5), the 11-hydroxyindenoisoquinolines 45a-b (Scheme 6), and the catechols 52a-b (Scheme 7) were synthesized and evaluated for Top1 inhibitory and antiproliferative activity and used as standards for metabolism studies.
Scheme 1a.

aReagents and conditions: (a) BnBr, DMF, K2CO3, r.t.; (b) 3-bromopropylamine HBr; (c) CHCl3, 10 °C - r.t.; (d) i. SOCl2, r.t., ii. AlCl3 (2 eq.), 1,2-dichloroethane, r.t.; (e) imidazole or morpholine, NaI, DMF, 70 °C.
Scheme 2a.

aReagents and conditions: (a) BnBr, DMF, K 2CO3, r.t.; (b) 3-bromopropylamine HBr, Et3N, Na2SO4, CHCl3, r.t.; (c) 11, CHCl3, 10 °C - r.t.; (d) SOCl2, r.t.; (e) imidazole or morpholine, NaI, DMF, 70 ° C; (f), HBr, AcOH, H2O, 55–70 °C.
Scheme 4a.

aReagents and conditions: (a) i. KOH, BnCl, EtOH, reflux, ii. KOH, H2O, reflux; (b) H2CO, H2O, HCl, AcOH, 45 °C; (c) i. KOH, H2O, r.t., ii. KMnO4, H2O, 0 °C-r.t., iii. EtOH, reflux; (d) AcCl, reflux; (e) 3-bromopropylamine HBr, Et 3N, Na2SO4, CHCl3, r.t.; (f) CHCl3, 0 ° C - r.t.; (g) i. SOCl2, r.t., ii. AlCl3 (2 eq.), DCE, 0 °C; (h), imidazole or morpholine, NaI, DMF, 60 °C.
Scheme 5a.

aReagents and conditions: (a) H2CO, H2O, HCl, AcOH, 120 °C-r.t.; (b) BnBr, K2CO3, acetone, 56 °C; (c) i. KOH, H2O, r.t., ii. KMnO4, H2O, 0 °C-r.t., iii. EtOH, reflux; (d) AcCl, reflux; (e) 3-bromopropylamine HBr, Et 3N, Na2SO4, CHCl3, r.t.; (f) CHCl3, 0 °C - r.t.; (g) SOCl2, r.t.,(h) AlCl3 (2 eq.), nitrobenzene, 90 °C; (i), imidazole or morpholine, NaI, DMF, 70 °C.
Scheme 6a.

aReagents and conditions: (a) NaBH4, MeOH, 0 °C.
Scheme 7a.

aReagents and conditions: (a) BnBr, DMF, r.t.−70 °C; (b) 3-bromopropylamine HBr, Et3N, Na2SO4, r.t.; (c) 11, CHCl3, 0 °C - r.t.; (d) SOCl2, −4 °C to r.t.; (e) imidazole or morpholine, NaI, dioxane, 65 °C; (f) 48% HBr-H2O, 70 °C (for 51a); (g) H2, Pd/C, MeOH-THF (for 51b).
Chemistry
Indenoisoquinolines have been prepared by many routes, including the condensation of primary amines with the appropriately substituted isochromenones,46 Suzuki-Miyaura cross-coupling followed by ring-closing metathesis,47 and through the oxidative cyclization of the cis acid condensation products formed by the reaction of homophthalic anhydrides and benzylidene Schiff bases.48 The last route was envisioned for the synthesis of hydroxyindenoisoquinolines as it has a high functional group tolerance and was previously used to synthesize 6 and 7.22 Due to the presence of harsh and oxidative intermediary steps, the phenol would have to be unmasked at a late stage in the synthesis, and preparation of the CD ring (indenone) system of 8-hydroxy-9-methoxyindenoisoquinolines (Scheme 1) thus began with benzylation of isovanillin (8) to yield the ether 9. Condensation of this aldehyde with 3-bromopropylamine afforded the Schiff base 10 in excellent yield. The isoquinolone (AB) ring system 12 was synthesized from the condensation of 10 with 4,5-dimethoxyhomophthalic anhydride (11),49 prepared by established literature procedures.50, 51 When conducted at low temperature, the reaction provided the thermodynamically less stable cis diastereomer 12 as a solid precipitate in good yield. Treatment of 12 with thionyl chloride at room temperature, followed by AlCl3 in dichloroethane, resulted in four reactions: acid chloride formation, Friedel-Crafts cyclization, dehydrogenation, and debenzylation, to yield the crude phenol 13. Reaction of 13 with imidazole or morpholine in the presence of sodium iodide in DMF yielded, respectively, the imidazolyl compound 14a or the morpholinyl compound 14b.
Analogously, 9-hydroxy-8-methoxyindenoisoquinolines were prepared from vanillin (15, Scheme 2). Benzylation (to yield 16) and treatment with 3-bromopropylamine afforded the Schiff base 17. Condensation with 11 likewise yielded the cis acid 18. Treatment with thionyl chloride led to the benzyl-protected phenol 19. Interestingly, adding two equivalents of AlCl3 did not produce a clean debenzylation as observed for the regioisomer and gave only very low yields of the phenol 20 (Scheme 3). Compound 19 was therefore elaborated with imidazole or morpholine to obtain the benzyl-protected phenols 21a and 21b, respectively. Unfortunately, many standard conditions for debenzylation failed, yielding either unchanged starting material or decomposition products. Aqueous hydrobromic acid in AcOH afforded the free phenols 22a and 22b, but the poor solubility of the starting materials often resulted in competing demethylation and oxidation processes and this method was low-yielding and hard to reproduce.
Scheme 3a.

aReagents and conditions: (a) PMB-Cl, DMF, K2CO3, 70 °C; (b) 3-bromopropylamine HBr, Et 3N, Na 2SO4, CHCl 3, r.t.; (c) 11, CHCl3, 0 °C - r.t.; (d) SOCl2, r.t.; (e) imidazole or morpholine, NaI, DMF, 70 °C or dioxane, reflux.
An alternative route (Scheme 3) was therefore devised. From compound 15, O-PMB-vanillin (23) was prepared and converted into the Schiff base 24. Condensation with compound 11 afforded the cis acid 25. Treatment of compound 25 with SOCl2 again performed four reactions (acid chloride formation, Friedel-Crafts acylation, dehydrogenation, and PMB cleavage) to yield 20, which could be obtained on multigram scale. Reaction of 20 with imidazole or morpholine and sodium iodide in DMF (the use of dioxane under argon resulted in improved yields) led to 22a and 22b in acceptable yields.
Several procedures were used to prepare A-ring demethylated analogues. A similar benzyl protecting group strategy was employed to prepare homophthalic anhydrides containing the desired protected phenols, which was necessary in order to avoid colored byproducts and other unidentified impurities formed from the unprotected phenols in the presence of the harsh acidic, basic, and oxidative conditions (Schemes 4 and 5). Therefore, to prepare 2-hydroxy-3-methoxyindenoisoquinolines, homoisovanillic acid (26, Scheme 4) was first benzylated to yield compound 27.52 Ortho-hydroxymethylation afforded the homophthalide 28,53 which was saponified and oxidized with KMnO4 to yield the diacid 29. The anhydride 30 was prepared by heating the diacid in refluxing acetyl chloride. Compound 30 was condensed with the Schiff base 32 derived from piperonal (31) to yield the cis acid 33 in excellent yield.21 The use of aluminum chloride resulted in debenzylation following the cyclization, but yields of the indenoisoquinoline 34 were very low (10–15%). A large amount of grayish insoluble decomposition product formed, presumably via competing demethylenation, oxidation, and complexation processes. Nonetheless, 34 could be treated with sodium iodide and imidazole or morpholine, and the analogues 35a and 35b were eventually obtained in pure form.
Figure 8.
11-Hydroxycamptothecin.
A similar route was used to prepare 3-hydroxy-2-methoxyindenoisoquinolines. Homovanillic acid (36, Scheme 5) was hydroxylmethylated to yield compound 37.54 Benzylation yielded 38 and a saponification/oxidation sequence was employed to prepare the homophthalic acid 39, which was then converted into the anhydride 40. Condensation with 32 afforded the cis acid 41. Treatment of 41 with SOCl2 produced the benzyl intermediate 42, but deprotection (here performed with AlCl3 in nitrobenzene) was also low yielding (~10%), as observed for the regioisomer (vide supra). The resulting phenol 43 was elaborated as earlier described to yield the analogues 44a and 44b.
11-Hydroxy (keto-reduced) indenoisoquinoline analogues 45a and 45b were prepared via sodium borohydride reduction of 6 and 7, respectively (Scheme 6). Finally, the catechols 52a and 52b (Scheme 7) were also prepared using the benzyl protecting group strategy. 3,4-Dihydroxybenzaldehyde (46) was dibenzylated to yield compound 47 and converted into the Schiff base 48. Condensation with anhydride 32 afforded the cis acid 49, which was cyclized in cold SOCl2 to furnish the intermediate indenoisoquinoline 50. Treatment with imidazole or morpholine as described earlier yielded the protected compounds 51a and 51b, respectively. Heating with aqueous hydrobromic acid affected the debenzylation of 51a (yielding the catechol 52a), and atmospheric pressure hydrogenation of 51b afforded the analogous compound 52b.
Metabolism Studies
Compounds 6 and 7 were incubated at 37 °C with pooled human liver microsomes in the presence of NADPH (full details in experimental section). After chilling and terminating the reactions, the samples were centrifuged, and the supernatatants were analyzed by LC-MS and LC-tandem electrospray mass spectrometry (LC-MS/MS). Indenoisoquinolines 14a-b, 22a-b, 35a-b, 44a-b, 45a-b, and 52a-b were used as synthetic standards.
In the case of both 6 and 7, positive-ion electrospray LC-MS detected two major metabolites in their protonated forms. The formation of these species required both liver microsomes and NADPH, since omission of either produced no detectable metabolites. In the case of 6, the most abundant metabolite M1 (Figure 3a and 3b) was detected at a retention time of 12.8 minutes. Accurate mass measurement provided an m/z value of 448.1508, which was within −0.2 ppm of the theoretical formula of C24H22N3O6. This formula was consistent with the loss of a methylene group, and LC MS/MS comparison with the synthetic standards provided confirmed this metabolite as catechol 52a. A second metabolite, M2, eluted at 13.2 minutes and produced an m/z value of 446.1361, which corresponded to the elemental composition C24H20N3O6 (2.0 ppm). This formula suggested the loss of a methyl group, and comparison with the standards identified this metabolite as the 3-desmethyl compound 44a.
Figure 3.

LC-MS retention times (a) and positive ion electrospray ion tandem mass spectra fragmentation patterns (b) for metabolites obtained upon incubation of 6 with human liver microsomes.
An identical analysis performed for compound 7 (Figure 4a and 4b) yielded similar results; the most abundant metabolite M1 [12.3 min; m/z 467.1823; C25H27N2O7 (1.1 ppm)] matched a synthetic standard of catechol 52b. Likewise, the structure of metabolite M2 [13.6 min; m/z 465.1654; C25H25N2O7 (−1.7 ppm)] was confirmed as 3-desmethyl-LMP400 (44b) by comparison of the retention times and fragmentation pattern of the metabolite with the synthetic standard.
Figure 4.

LC-MS retention times (a) and positive ion electrospray ion tandem mass spectra fragmentation patterns (b) for metabolites obtained upon incubation of 7 with human liver microsomes.
In both cases, these were the only metabolites detected, and no matches were found with the other standards (14a-b, 22a-b, and the regiomeric 2-desmethyl compounds 35a-b, or 45a-b), indicating that the predominant routes of human hepatic metabolism for 6 and 7 are likely demethylenation and 3-O-demethylation, as predicted by both the apparent metabolic lability of these sites (Figure 2) and extensive literature precedent. Catechol methylation, 2-O-demethyation, and ketone reduction do not occur.
Biological Evaluation of Hydroxyindenoisoquinolines
The indenoisoquinolines 14a-b, 22a-b, 35a-b, 44a-b, 45a-b, and 52a-b were tested for antiproliferative activity in the National Cancer Institute’s Developmental Therapeutics Assay (the “NCI-60”) against cell lines derived from a variety of human tumors (approximately 60 lines were used).55, 56 After an initial one-dose pre-screening assay at moderately high concentration (10−5 molar), selected compounds were tested at five concentrations ranging from 10−8 to 10−4 molar. Overall antiproliferative potential is quantified as a mean-graph midpoint (MGM). This value can be interpreted as a rough average GI50 value across the whole assay, where values that fall outside the tested concentration range (<10−8 or >10−4 M) are respectively assigned either 10−8 or 10−4 M. These MGM values, along with GI50 values from selected cell lines, are reported in Table 1. For completeness, the MGM values for 1, 4, 5, 6, and 7 are also given.
Table 1.
Antiproliferative Potencies and Topoisomerase I Inhibitory Activities of Hydroxyindenoisoquinolines.
| Compd | Cytotoxicity (GI50 in μM)a
|
MGMb | Top1 Cleavagec | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| lung HOP-62 | Colon HCT-116 | CNS SF-539 | melanoma UACC-62 | Ovarian OVCAR-3 | renal SN12C | prostate DU-145 | breast MCF-7 | |||
| 157 | 0.01 | 0.03 | 0.01 | 0.01 | 0.22 | 0.02 | 0.01 | 0.01 | 0.040±0.0187d | ++++ |
| 457 | 1.3 | 35 | 41 | 4.2 | 73 | 68 | 37 | 1.58 | 20.0 ± 14 | ++ |
| 546 | 0.02 | 0.10 | 0.04 | 0.03 | 0.5 | <0.01 | <0.01 | <0.01 | 0.21±0.19 | ++++ |
| 646 | <0.01 | <0.01 | 0.04 | <0.01 | 0.08 | <0.01 | <0.01 | 0.01 | 0.079±0.023 | +++++ |
| 746 | 1.78 | 1.15 | 0.04 | 0.03 | 74.1 | 0.813 | 0.155 | 0.37 | 4.64±1.25 | +++++ |
| 14a | >100 | >100 | − | 0.331 | >100 | − | >100 | 0.05 | 41.8±7.15 | ++(+) |
| 14b | 0.180 | 2.34 | 0.282 | 0.07 | 8.32 | 0.191 | 0.204 | 0.03 | 3.07±0.32 | +++ |
| 22a | <0.01 | <0.01 | <0.01 | <0.01 | 0.02 | <0.01 | <0.01 | <0.01 | 0.055±0.003 | ++++(+) |
| 22b | 0.02 | 0.03 | 0.02 | 0.02 | 0.03 | 0.02 | 0.02 | 0.02 | 0.087±0.063 | +++++ |
| 35a | <0.01 | 0.02 | <0.01 | 0.03 | 0.07 | <0.01 | <0.01 | − | 0.049 | +++++ |
| 35b | 0.257 | 0.279 | 0.335 | 0.282 | 0.871 | 0.195 | 0.257 | 0.144 | 0.412±0.005 | ++(+) |
| 44a | <0.01 | <0.01 | <0.01 | <0.01 | 0.03 | <0.01 | <0.01 | <0.01 | 0.043 | ++++ |
| 44b | 0.01 | 0.02 | <0.01 | <0.01 | 0.03 | <0.01 | <0.01 | <0.01 | 0.056 | +++(+) |
| 45a | 0.549 | 1.35 | 0.479 | 0.372 | 1.95 | 0.501 | 0.407 | 0.427 | 1.66 | ++ |
| 45b | 2.09 | 2.40 | 2.40 | 0.776 | 4.79 | 3.39 | 2.63 | 0.646 | 3.16 | ++ |
| 52a | 0.04 | 0.229 | 0.01 | <0.01 | 0.407 | 0.03 | 0.100 | 0.02 | 0.224 | ++ |
| 52b | 0.371 | 0.407 | 0.148 | 0.078 | 0.501 | 0.065 | 0.490 | 0.050 | 0.602 | ++++ |
The cytotoxicity GI50 values are the concentrations corresponding to 50% growth inhibition.
Mean graph midpoint for growth inhibition of all human cancer cell lines successfully tested, ranging from 10−8 to 10−4 molar.
Compound-induced DNA cleavage due to Top1 inhibition is graded by the following rubric relative to 1 μM camptothecin: 0, no inhibitory activity; +, between 20 and 50% activity; ++, between 50 and 75% activity; +++, between 75% and 95% activity; ++++, equipotent, +++++ more potent.
For MGM GI50 values in which a standard error appears, the GI50 values for individual cell lines are the average of two determinations; values without standard error are from one determination. The values for 1, 4, 5, 6, and 7 are from many determinations.
Top1 inhibition was graded by the ability of a compound to induce enzyme-linked DNA breakage, and is reported on a semi-quantitative scale relative to 1 μM camptothecin: 0, no inhibitory activity; +, between 20 and 50% activity; ++, between 50 and 75% activity; +++, between 75% and 95% activity; ++++, equipotent, +++++, more potent. Ambiguous scores (e.g., between two values) are designated with parentheses (e.g., ++(+) would be between ++ and +++). These values are also reported in Table 1.
A representative example of Top1-linked DNA cleavage by hydroxyindenoisoquinolines is shown in Figure 5. As observed in the figure and listed in Table 1, all compounds tested exhibited at least some Top1 inhibitory activity, with compounds 22a, 22b, and 35a possessing excellent inhibitory activity comparable to or greater than the clinical candidates 6 and 7. These compounds also have submicromolar antiproliferative potency (MGM values of 55 nM for 22a, 87 nM for 22b, and 49 nM for 35a), comparable to 6 (79 nM), and although they were weaker Top1 poisons, the confirmed metabolites 44a and 44b had MGM values of 43 and 56 nM, respectively, which could serve to prolong the antitumor effects of 6 and 7 in vivo. A standard COMPARE analysis17, 18 seeded with the GI50 data for 22a, 22b, and 35a displayed moderate to high correlations (Pearson coefficients of 0.65-0.8) with the cytotoxicity profile of topotecan (2), indicating that it is likely these compounds exert their cytotoxic or antiproliferative effects predominantly via Top1 inhibition. Generally, anti-Top1 activity and antiproliferative activity correlate well, with those compounds that are better Top1 inhibitors displaying higher potency in the NCI-60 screen.
Figure 5.

Top1-mediated DNA cleavage induced by indenoisoquinolines 22b, 14b, 35a, 44a, and 45a. Lane 1: DNA alone; lane 2: Top1 + DNA; lane 3: 1, 1 μM; lane 4: 5, 1 μM; lane 5–24: 22b, 14b, 35a, 44a, and 45a at 0.1, 1, 10 and 100 μM respectively from left to right. Numbers and arrows on the left indicate arbitrary cleavage site positions.
Interestingly, the D-ring hydroxyindenoisoquinolines display a striking correlation between regiochemistry, Top1 inhibition, and cytotoxicity. While the 9-hydroxy-8-methoxyindenoisoquinolines 22a and 22b are extremely potent, the regiomeric 8-hydroxy-9-methoxyindenoisoquinolines 14a and 14b possess approximately half the anti-Top1 activity and their MGM values are orders of magnitude greater than their isomeric counterparts. Similar regiochemical effects are reported for other classes of Top1 inhibitors (indenoisoquinolines, camptothecins, and aromathecins, among others).
What could be responsible for such a drastic effect? A molecular modeling and docking study was performed in an attempt to rationalize this disparity. Using GOLD, the minimized structures of morpholine 22b and its regioisomer 14b were docked into the cleavage site of a mutant, solvated Top1/topotecan crystal structure (PDB ID 1K4T) using the procedure previously described by Peterson et al..46 The minimized, highest-ranked GOLD pose for compound 22b is shown in Figure 6.
Figure 6.

Minimized, top-ranked GOLD pose of compound 22b in ternary complex with DNA and Top1, constructed in SYBYL. The ligand is colored in purple, surrounding structures are colored by atom, water molecules are depicted as red spheres, and hydrogens have been omitted. All distances are measured from heavy-atom to heavy-atom. The diagram is programmed for wall-eyed (relaxed) viewing.
A major commonality among Top1 poisons is that a large amount of steric bulk (e.g., multiple methoxy groups, halogens) projecting toward the nonscissile strand often has deleterious effects on potency in general.39, 40, 57 Indeed, crystal structures of several Top1 inhibitors58 reveal this region to be very “tight” sterically, in agreement with the observation of a low functional group tolerance. However, the 9-hydroxyl group of 22b appears to be quite well accommodated here. The indenoisoquinoline is calculated to bind in an intercalative mode identical to that observed in the crystal structure of a related Top1-DNA-indenoisoquinoline ternary complex,58 and the hydroxyl group is involved in water-mediated hydrogen bonding with Lys374. It is possible that with induced fit, this hydroxyl could also make contact with the nearby deoxyfuranose oxygen (here, 4.0 Å away), while the 8-methoxy group projects toward the major groove of the complex.
The highest ranked, energy minimized GOLD pose for compound 14b is shown in Figure 7. The nonscissile side appears to be significantly more crowded, as the 9-methoxy group now projects back toward the phosphodiester backbone, and the network of water-mediated contacts is absent. The 8-hydroxy group projects toward the solvent, where it hydrogen bonds with a nearby glutamate residue (Glu356). It is conceivable that this contact could stabilize the binding of 14b, as its activity is not abolished completely relative to 22b, and Glu356 is proposed to aid the binding of indolocarbazoles and compound 2.58 Nonetheless, there is a sizeable difference in the GoldScores: 67.9 for 14b vs. 81.6 for 22b, and the average difference in GoldScores between the top four poses for 22b and 14b is approximately sixteen points. As predicted by the model, the difference in the hydrogen bonding term of the GoldScore between the regioisomers is negligible (average of 10.3 for the top four poses of 14b; 12.1 for the top four of 22b), but the difference in the external van der Waals terms (a component of the fitness score that reflects a simple attractive/repulsive potential)59, 60 is significant: 56 (average 53.3) for 14b versus 65 (average 62.6) for 22b, likely symbolizing a “less favorable” or “more repulsive” fit for the 9-methoxy isomer. Interestingly, some 9-methoxyindenoisoquinolines are quite active against Top1, although most of these compounds also contain a 3-nitro group (which is well-documented to improve potency),23, 57 indicating that favorable A-ring electronics may somehow be enough to offset a negative steric effect.
Figure 7.

Minimized, top-ranked GOLD pose of compound 14b in ternary complex with DNA and Top1 (constructed in SYBYL). The ligand is colored in cyan, surrounding structures are colored by element, and hydrogens have been omitted. All distances are measured from heavy-atom to heavy-atom. The same water molecules from Figure 4 are shown as red spheres.
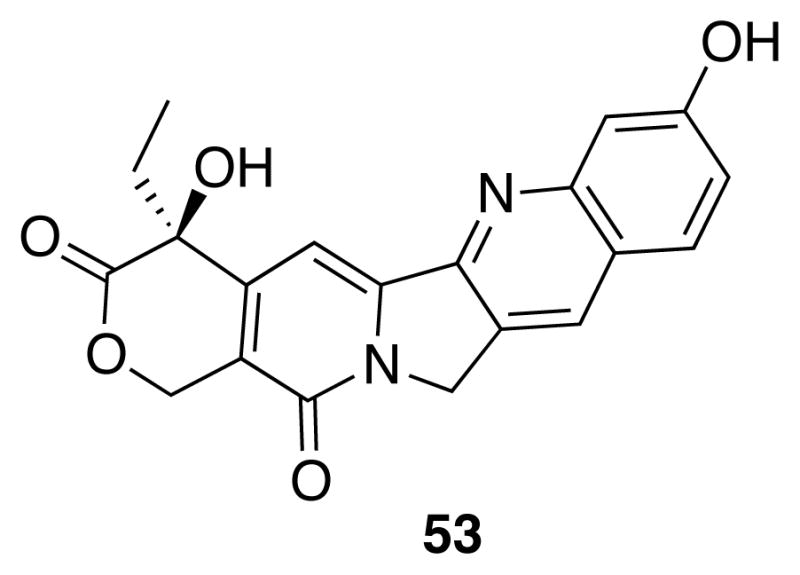
Additionally, perhaps the placement of 9-hydroxyl group is important as well. In a similar study of camptothecins performed by Wall et al.,40 most substitutions at position 11 of the camptothecin system attenuate the Top1 inhibitory activity (Morrell and others hypothesize that this position is roughly analogous to the indenoisoquinoline position 957, 61). One notable exception to this SAR is the active 11-hydroxycamptothecin (53, Figure 8), which would point its phenolic hydroxyl toward the nonscissile strand as well. Further support comes from a study of the regioisomers of hydroxylated indolocarbazoles performed by Zembower et al.62 Those compounds possessing a hydroxyl at position 9 (also roughly analogous to the numerically identical position of indenoisoquinolines) are the most potent!62
Interestingly, the regiochemical dependence is not observed on the A-ring side, and activity here is variable. The 2-hydroxyindenoisoquinoline 35a and 3-hydroxyindenoisoquinoline 44b both possess significant anti-Top1 activity, although some differences between the morpholines and imidazoles are observed. Assuming these compounds bind in ternary complex as 22b and 14b are proposed to, the indenoisoquinoline A-ring fits into the scissile-strand cavity. The steric tolerance of this spacious region is much higher and it may render regiochemical differences minimal to irrelevant. Docking supports this hypothesis as well: there is no difference (~0.03 points) between the GoldScores for the top-ranked poses of 35b and 44b (and the average scores for the top four poses are only ~3 points apart), and the van der Waals and hydrogen-bond contributions are approximately equal in both cases, indicating that the difference in Top1 activity between these compounds (lower than that observed between series 22 and series 14) may not be due to placement of the hydroxyl group. The literature supports this assumption as well: the benzonaphthyridine Top1 poison Genz-644282 is very similar in structure to 6 and 7, and its A-ring desemethyl metabolites do not display any correlation between regiochemistry and Top1 inhibition.32
The lowest activity observed was from the 11-hydroxy (reduced keto) analogues 45a and 45b. Although some potent 11-hydroxy and 11-alkoxyindenoisoquinolines have been reported, the molecular modeling studies indicated that they bind differently in the ternary cleavage complex, with an N-(para-methoxybenzyl) group instead of the indenoisoquinoline ring system intercalating between the base pairs. 43, 44 The fact that the indenoisoquinoline oracin is deactivated by ketone reduction is also consistent with the low potency of 45a and 45b.37 Overall, the evidence indicates that the ketone is important for optimal binding of the indenoisoquinoline in the DNA intercalative mode. The crystal structure of an indenoisoquinoline/DNA/Top1 ternary complex (PDB ID 1SC7)58 has demonstrated that the ketone hydrogen bonds to the minor groove residue Arg364 (proposed to aid in the stabilization of other bound inhibitors). The hydroxyl groups of 45a and 45b still can behave as hydrogen bond acceptors in our models (not shown), so lack of hydrogen bond-accepting capability per se cannot be responsible for the decrease in activity. Perhaps the reduction of the ketone changes the electronics of the system, interfering with π-π stacking, or the introduction of a tetrahedral carbon introduces nonplanarity that could disrupt intercalation.
The catechols 52a and 52b are both also active against Top1, although the imidazole 52a has only half the potency of the morpholine 52b. Significant differences between imidazole and morpholine substituents were observed for other pairs in this study (cf. 35a and 35b), although in the case of 35b vs. 35a the substituent effects on potency are opposite those observed with 52a vs. 52b.
Conclusions
A series of 8-, 9-, 2-, and 3-hydroxyindenoisoquinolines, 8,9-dihydroxyindenoisoquinolines, and reduced keto analogues of the clinical candidates 6 and 7 were designed based on the rationale that (a) they are potential metabolites of indotecan and indimitecan and could be useful as synthetic standards to compare with the actual metabolites, and (b) there are many examples of potent hydroxylated/phenolic Top1 poisons in the literature, so the hydroxyindenoisoquinolines might be of value as anticancer drugs. To this end, the indenoisoquinolines were prepared by the homopththalic anhydride/Schiff base condensation method49 and were assayed against human Top1 and a panel of human cancer cells. Interestingly, many of these hydroxylated indenoisoquinolines are extremely potent Top1 poisons, especially those bearing the 9-hydroxy-8-methoxy substitution pattern on the D ring. Molecular modeling supports the observed activity, especially when compared to the much less active 8-hydroxy-9-methoxyindenoisoquinolines. A less significant dependence of Top1 inhibitory potency on regiochemistry was observed for A-ring hydroxyindenoisoquinolines, a fact that is in agreement with our docking study. Additionally, a metabolism study indicates that several of the prepared hydroxyindenoisoquinolines are produced upon incubation of the parent drugs with human liver microsomes and NADPH. These data indicate that the primary routes of metabolism are 3-demethylation and cleavage of the 8,9-methylenedioxy group. Three additional points can be made about the potential future significance of this study: 1) the fact that the metabolites are biologically active can be expected to prolong the duration of antitumor activity expressed after drug administration; 2) the biologically active hydroxylated indenoisoquinolines provide convenient starting points for further drug development, especially with regard to prodrugs, since the phenolic hydroxyl groups provide convenient points for attachment of prodrug modules; and 3) the altered solubilities of the hydroxylated derivatives may offer advantages for IV formulation.
Experimental Section
General Procedures
Reagents and solvents were purchased from commercial vendors and were used without further purification. Melting points were determined in capillary tubes using a Mel-Temp apparatus and are not corrected. Infrared spectra were obtained as films on salt plates unless otherwise specified, using a Perkin-Elmer Spectrum One FT-IR spectrometer, and are baseline-corrected. 1H NMR spectra were obtained at 300 or 500 MHz, using a Bruker ARX300 and Bruker Avance 500 (QNP probe or TXI 5 mm/BBO probe). 13C NMR spectra were obtained at 125 or 75 MHz. Mass spectral analyses were performed at the Purdue University Campus-Wide Mass Spectrometry Center. ESIMS was performed using a FinniganMAT LCQ Classic mass spectrometer system. The electrospray ionization high resolution mass measurements were obtained in the peak matching mode using a FinniganMAT XL95 (FinniganMAT Corp., Bremen, Germany) mass spectrometer. The instrument was calibrated to a resolution on 10,000 with a 10% valley between peaks using the appropriate polypropylene glycol standards. EI/CIMS was performed using a Hewlett-Packard Engine or GCQ FinniganMAT mass spectrometer system. APCI-MS was performed using an Agilent 6320 Trap mass spectrometer. Combustion microanalyses were performed by Midwest Microlab LLC (Indianapolis, IN). Reported values are within 0.4% of calculated values. HPLC was performed using a Waters 1525 binary HPLC pump with a Waters 2487 Dual Absorbance Detector and an injection volume of 10 μL. A Sunrise C18 5μ 100 Å reverse-phase column, with dimensions of 15 cm × 4.6 mm (ES Industries), was used for all HPLC experiments. The intensity of the major peak in the analytical HPLC trace of each target compound was ≥95% that of the combined intensities of all of the peaks detected at 254 nm, and two different solvent systems were used to ascertain purity. Analytical thin-layer chromatography was performed on Baker-flex silica gel IB2-F plastic-backed TLC plates. Compounds were visualized with both short and long wavelength UV light. Silica gel flash chromatography was performed using 40–63 μm flash silica gel.
Benzylisovanillin (9)63
Isovanillin (8, 3.00 g, 19.7 mmol) was diluted with anhydrous DMF (50 mL). Benzyl bromide (3.47 g, 20.3 mmol) was added slowly, followed by K2CO3 (6.54 g, 47.3 mmol). The yellow mixture was rapidly stirred at room temperature for 2 h 15 min. The mixture was partitioned into an ether-water mixture (1:1, 100 mL of each) and stirred. The organic and aqueous layers were separated, and the aqueous layer was extracted with ether (1 × 50, 2 × 25 mL). The combined organic layers were washed with H2O (2 × 50 mL) and sat. aq NaCl (50 mL), dried over anhydrous sodium sulfate, and concentrated to yield a white microcrystalline solid (4.43 g, 93%) after washing with hexanes (50 mL) and re-filtering the filtrate: mp 55–58 °C (lit63 mp 61–62 °C). 1H NMR (CDCl3) δ 9.82 (s, 1 H), 7.49-7.32 (m, 6 H), 7.02 (d, J = 8.2 Hz, 1 H), 5.20 (s, 2 H), 3.97 (s, 3 H).
N-[3′-(Benzyloxy)-4′-methoxybenzylidene]-3-bromopropan-1-amine (10)
3-Bromopropylamine hydrobromide (2.08 g, 9.49 mmol) was diluted with CHCl3 (10 mL). Compound 9 (2.00 g, 8.26 mmol) was added slowly as a solution in CHCl3 (10 mL), and quantitatively transferred with 2 mL of the same solvent. Et3N (0.917 g, 9.08 mmol) was added slowly, upon which the solution became clear and colorless. Na2SO4 (1.00 g, 5.21 mmol) was added, and the mixture was stirred at room temperature for 24 h. Additional 3-bromopropylamine salt (0.905 g, 4.13 mmol), Et3N (0.500 g, 4.96 mmol) and approximately 0.500 g of Na2SO4 were added. After a total of 48 h, the mixture was washed with H2O (3 × 70 mL) and sat. aq NaCl (70 mL). The organic layer was dried over anhydrous sodium sulfate and concentrated to yield a dark yellow syrup (2.86 g, 96%). IR (film) 3291, 2932, 2837, 1645, 1601, 1511, 1431, 1265, 1137, 1024, 741, 697 cm−1; 1H NMR (CDCl3) δ 8.21 (s, 1 H), 7.49-7.31 (m, 6 H), 7.21 (dd, J = 2.1, 8.3 Hz, 1 H), 6.92 (d, J = 8.3 Hz, 1 H), 5.19 (s, 2 H), 3.93 (s, 3 H), 3.92 (s, 3 H), 3.73 (td, J = 1.0, 6.2 Hz, 2 H), 3.51 (t, J = 6.5 Hz, 2 H), 2.30-2.20 (m, 2 H); ESIMS m/z (rel. intensity) 362/364 (MH+, 97/100).
cis-[3-(Bromopropyl)amino)]-4-carboxy-3,4-dihydro-6,7-dimethoxy-3-(3-benzyloxy-4-methoxyphenyl)-1(2H)-isoquinolone (12)
Anhydride 11 (1.40 g, 6.30 mmol) was dissolved in CHCl3 (35 mL) and the resulting solution was cooled to 0 °C. A solution of compound 10 (2.40 g, 6.63 mmol) in cold CHCl3 (15 mL) was added slowly over 5 min and quantitatively transferred with CHCl3 (5 mL). Precipitate began forming after 30 min. The mixture was stirred for 2 h at 0 °C and then warmed to room temperature and stirred for an additional 2 h. The precipitate was filtered out and washed with CHCl3 (50 mL) to afford the title compound as a white solid (2.81 g 77%) after drying: mp 193–195 °C (dec). IR (film) 2933, 1733, 1619, 1597, 1573, 1515, 1169, 1027, 696 cm−1; 1H NMR (CDCl3-DMSO-d6) δ 7.53 (s, 1 H), 7.19-7.13 (m, 5 H), 7.05 (s, 1 H), 6.53 (s, 3 H), 4.83 (d, J = 6.4 Hz, 1 H), 4.79 (s, 2 H), 4.44 (d, J = 6.2 Hz, 1 H), 3.81 (s, 3 H), 3.81-3.70 (m, 1 H), 3.73 (s, 3 H), 3.64 (s, 3 H), 3.30-3.20 (m, 2 H), 2.95-2.90 (m, 1 H), 2.01-1.80 (m, 2 H); APCI-MS m/z (rel intensity) 540 (MH+ – CO2, 100). Anal. Calcd for C29H30BrNO7·H2O: C, 57.81; H, 5.35; N, 2.32. Found: C, 57.57; H, 4.99; N, 2.32.
6-(3-Bromopropyl)-5,6-dihydro-8-hydroxy-2,3,9-trimethoxy-5,11-dioxo-11H-indeno-[1,2-c]isoquinoline (13)
The cis acid 12 (1.00 g, 1.71 mmol) was diluted with SOCl2 (25 mL). The dark reddish-brown mixture was stirred at room temperature for 2 h and 20 min and was concentrated. The residue was dissolved in 1,2-dichloroethane (40 mL), and aluminum chloride (0.479 g, 3.6 mmol) was added. The mixture was stirred at room temperature for 20 h, and was quenched slowly by the addition of sat. aq NaHCO3 (40 mL) and H2O (50 mL). The aqueous and organic layers were separated, and the aqueous phase was extracted with CHCl3 (2 × 50 mL). The organic phase was washed with H2O (2 × 100 mL) and sat. aq NaCl (100 mL), dried over anhydrous sodium sulfate, and concentrated. The obtained residue was adsorbed onto SiO2 (~ 7 g), and purified by flash column chromatography (SiO2, ~ 50 g), eluting with a gradient of 0.25% MeOH in CHCl3 to 1% MeOH in CHCl3 to afford the crude product as a pinkish solid (0.414 g, 51%) after washing with 10% CHCl3 in ether (2 × 50 mL) and ether (50 mL) and drying: mp 248–252 °C. IR (film) 3296, 1635, 1554, 1471, 1396, 1396, 1030, 871, 762, 761 cm−1; 1H NMR (CDCl3) δ 8.04 (s, 1 H), 7.64 (s, 1 H), 7.31 (s, 1 H), 7.16 (s, 1 H), 7.95 (s, 1 H), 4.62 (t, J = 7.5 H, 2 H), 4.06 (s, 3 H), 3.99 (s, 6 H), 3.65 (t, J = 6.5 Hz, 1 H), 2.50-2.40 (m, 2 H); EIMS m/z (rel. intensity) 475 (M+, 100).
5,6-Dihydro-8-hydroxy-6-[3-(1H-imidazol-1-yl)propyl]-2,3,9-trimethoxy-5,11-dioxo-11H-indeno-[1,2-c]isoquinoline (14a)
Compound 13 (0.300 g, 0.632 mmol), imidazole (0.517 g, 7.59 mmol), and NaI (1.14 g, 0.7.59 mmol) were diluted with anhydrous DMF (90 mL) and the mixture was heated to 60 °C for 18 h. The mixture was cooled and poured into H2O (200 mL) and the solution was extracted with CHCl3 (2 × 100, 1 × 50 mL). The burgundy-colored organic layer was washed with H2O (5 × 200 mL) and sat. aq NH4Cl (200 mL). The organic phase was dried over anhydrous sodium sulfate, concentrated, and the residue was washed with ether (100 mL) and filtered. The precipitate was dissolved in CHCl3 (50 mL), and the solution was concentrated and adsorbed onto SiO2 (~7 g) and purified by flash column chromatography (SiO2, 50 g), eluting with a gradient of 1% MeOH in CHCl3 to 4% MeOH in CHCl3, to yield a fuchsia-colored solid (0.112 g, 38%) after suspending in DMF (10 mL) and precipitating with ether (150 mL) and washing with ether (100 mL) and drying: mp 265–266 °C (dec). IR (film) 3306, 2917, 1659, 1622, 1554, 1472, 1394, 1335, 1265, 1085, 1032, 762, 688 cm−1; 1H NMR (DMSO-d6) δ 10.10 (br s, 1 H), 7.84 (s, 1 H), 7.73 (s, 1 H), 7.44 (s, 1 H), 7.27 (s, 1 H), 7.04 (s, 1 H), 6.99 (s, 1 H), 6.92 (s, 1 H), 4.37 (t, J = 7.2 Hz, 2 H), 4.20 (t, J = 7.0 Hz, 2 H), 3.88 (s, 3 H), 3.85 (s, 3 H), 3.84 (s, 3 H), 2.25-2.20 (m, 2 H); ESIMS m/z (rel. intensity) 462 (MH+, 100). Purity was estimated to be ~100% by HPLC in both 90% MeOH – H2O and 85% MeOH – H2O. Anal. Calcd for C25H23N3O6·H2O: C, 62.62; H, 5.26; N, 8.76. Found: C, 62.40; H, 4.85; N, 8.57.
5,6-Dihydro-8-hydroxy-2,3,9-trimethoxy-6-[3-(N-morpholino)propyl]-5,11-dioxo-11H-indeno-[1,2-c]isoquinoline (14b)
Compound 13 (0.090 g, 0.190 mmol) and NaI (0.200 g, 1.32 mmol) were diluted with anhydrous DMF (20 mL). Morpholine (0.115 g, 1.32 mmol) was added and the mixture was heated to 70 °C for 18 h. The mixture was cooled and poured into H2O (100 mL) and the solution was extracted with CHCl3 (3 × 50 mL). The burgundy-colored organic layer was washed with H2O (4 × 100 mL). The organic layer was dried over anhydrous sodium sulfate, concentrated, and the resulting residue was adsorbed onto SiO2 (~ 3 g) and purified by flash column chromatography (SiO2, 26.0 g), eluting with a gradient of 1% MeOH in CHCl3 to 2.5 % MeOH in CHCl3 to yield a mauve-colored solid (0.033 g, 36%) after dissolving in CHCl3 (5 mL) and precipitating with ether (40 mL) and drying: mp 282–285 °C. IR (film) 2917, 1687, 1635, 1553, 1495, 1472, 1395, 1326, 1255, 1211, 1020, 867, 788, 757 cm−1; 1H NMR (DMSO-d6) δ 10.06 (br s, 1 H), 7.97 (s, 1 H), 7.46 (s, 1 H), 7.18 (s, 1 H), 7.06 (s, 1 H), 4.40-4.30 (m, 2 H), 3.89 (s, 3 H), 3.86 (s, 3 H), 3.84 (s, 3 H), 3.54 (s, 4 H), 2.50-2.44 (m, 2 H, partially obscured by solvent peak), 2.36 (s, 4 H), 2.00-2.90 (m, 2 H); ESIMS m/z (rel. intensity) 481 (MH+, 100). Purity was estimated to be 98.3% by HPLC in 95% MeOH – 5% H2O and 99% in 90% MeOH – 10% H2O. Anal. Calcd for C26H28N2O7·0.5 H2O: C, 63.79; H, 5.97; N, 5.72. Found: C, 63.88; H, 5.78; N, 5.69.
Benzylvanillin (16).64
Vanillin (15, 3.00 g, 19.7 mmol) was diluted with anhydrous DMF (50 mL). Benzyl bromide (3.47 g, 20.3 mmol) was added slowly, followed by K2CO3 (6.54 g, 47.3 mmol). The yellow mixture was rapidly stirred at room temperature for 2 h 20 min. The mixture was partitioned into an ether/water mixture (1:1, 100 mL of each) and stirred. The organic and aqueous layers were separated, and the aqueous layer was extracted with ether (2 × 25, 1 ×50 mL). The combined organic layers were washed with H2O (1 × 50, 1 × 30 mL) and sat. aq NaCl (50 mL), dried over anhydrous sodium sulfate, and concentrated to yield a white microcrystalline solid (4.51 g, 95%) after washing with hexanes (50 mL) and re-filtering the filtrate: mp 51–54 °C (lit63 mp 60–61 °C). 1H NMR (CDCl3) δ 9.84 (s, 1 H), 7.46-7.35 (m, 7 H), 7.00 (d, J = 8.2 Hz, 1 H), 5.25 (s, 2 H), 3.95 (s, 3 H).
N-[4′-(Benzyloxy)-3′-methoxybenzylidene]-3-bromopropan-1-amine (17)
3-Bromopropylamine hydrobromide (2.08 g, 9.49 mmol) was diluted with CHCl3 (10 mL). A solution of aldehyde 16 (2.00 g, 8.26 mmol) was added slowly as a solution in CHCl3 (12 mL) and transferred quantitatively with the same solvent (3 mL). Et3N (0.917 g, 9.08 mmol) was added, upon which the suspension became clear. Na2SO4 (anhydrous, approximately 2.00 g) was added, and the mixture was stirred at room temperature for 23 h. Additional 3-bromopropylamine hydrobromide (0.904 g, 4.13 mmol) and Et3N (0.500 g, 4.96 mmol) were added, and the mixture was stirred for a total of 44 h. The reaction mixture was diluted to a volume of 50 mL with CHCl3, and was washed with H2O (3 × 70 mL) and sat. aq NaCl (50 mL). The organic phase was dried over anhydrous sodium sulfate and concentrated to yield a yellow syrup (3.06 g, 100% with residual solvent). IR (film) 2935, 2840, 1645, 1600, 1585, 1511, 1455, 1419, 1267, 1231, 1138, 1033, 743, 697 cm−1; 1H NMR (CDCl3) δ 8.22 (s, 1 H), 7.45-7.31 (m, 6 H), 7.10 (dd, J = 8.2, 1.8 Hz, 1 H), 6.90 (d, J = 8.2 Hz, 1 H), 5.21 (s, 2 H), 3.95 (s, 3 H), 3.74 (t, J = 6.2 Hz, 2 H), 3.51 (t, J = 6.3 Hz, 2 H), 2.30-2.20 (m, 2 H); ESIMS m/z (rel. intensity) 362/364 (MH+, 100/95).
cis-3-(4-Benzyloxy-3-methoxyphenyl)-N-(3-bromopropyl)-4-carboxy-3,4-dihydro-6,7-dimethoxy-1(2H)-isoquinolone (18)
Compound 11 (1.93 g, 8.71 mmol) was diluted with CHCl3 (40 mL) and the mixture was cooled to 10 °C. The Schiff base 17 (3.00 g, 8.28 mmol) was diluted in CHCl3 (20 mL, cooled to 10 °C), and this solution was added slowly to the anhydride solution and transferred with 5 mL of the same solvent. The mixture was stirred at 10 °C for 30 min, and was then allowed to slowly warm to room temperature. After 3 h and 20 min, a precipitate had formed. This precipitate was collected and washed with CHCl3 (30 mL) to yield a white solid (2.86 g, 59%) after drying: mp 172–175 °C. IR (film) 2933, 1743, 1621, 1595, 1575, 1464, 1289, 1259, 1174, 1103, 1028, 908, 730, 697 cm−1; 1H NMR (CDCl3) δ 7.72 (s, 1 H), 7.38-7.29 (m, 5 H), 7.09 (s, 1 H), 6.71 (d, J = 8.9 Hz, 1 H), 6.60-6.57 (m, 2 H), 5.04 (s, 2 H), 5.00 (d, J = 6.2 Hz, 1 H), 4.70 (d, J = 6.2 Hz, 1 H), 4.03-3.98 (m, 1 H), 3.96 (s, 3 H), 3.88 (s, 3 H), 3.63 (s, 3 H), 3.22-3.41 (m, 2 H), 3.22-3.15 (m, 2 H), 2.28-2.21 (m, 1 H), 2.18-2.08 (m, 1 H); ESIMS m/z (rel. intensity) 584/585 (MH+, 100/94). Anal. Calcd for C29H30BrNO7: C, 59.60; H, 5.17; N, 2.40. Found: C, 59.49; H, 5.13; N, 2.34.
9-Benzyloxy-6-3-(bromopropyl)-5,6-dihydro-2,3,8-trimethoxy-5,11-dioxo-11H-indeno-[1,2-c]isoquinoline (19)
Compound 18 (1.00 g, 1.71 mmol) was diluted with SOCl2 (40 mL) and the reaction mixture was stirred at room temperature, upon which it became a bright reddish-purple color. TLC indicated completion after 4.5 h. The SOCl2 was evaporated, and the residue was dissolved in CHCl3 (40 mL) and quenched by the slow addition of sat. aq NaHCO3 (50 mL). The layers were separated, and the aqueous layer was extracted with CHCl3 (30 mL). The organic layers were washed with H2O (50 mL), and the aqueous layers were extracted with CHCl3 (20 mL). The organic layers were finally washed with sat. aq NaCl (50 mL), and the aqueous layers were again extracted with CHCl3 (20 mL). The combined organic layers were dried over anhydrous sodium sulfate, concentrated, and adsorbed onto SiO2 (~3.0 g). The residue was purified by flash column chromatography (SiO2, 37.2 g), eluting with CHCl3 to yield a purple solid (0.448 g, 45%) after dissolving in CHCl3 (30 mL) and precipitating with ether (200 mL). The precipitate was filtered and washed with ether (40 mL): mp 223–226 °C (dec). The 1H NMR indicated that the product contained some minor impurities and no further purification or characterization was performed; this crude material was used to prepare analogues. 1H NMR (CDCl3) δ 8.04 (s, 1 H), 7.62 (s, 1 H), 7.47-7.19 (m, 7 H), 5.23 (s, 2 H), 4.64 (t, J = 7.6 Hz, 2 H), 4.05 (s, 3 H), 4.03 (s, 3 H), 3.98 (s, 3 H), 3.71 (t, J = 6.0 Hz, 2 H), 2.40-2.40 (m, 2 H).
9-Benzyloxy-5,6-dihydro-6-[3-(1H-imidazol-1-yl)propyl]-2,3,8-trimethoxy-5,11-dioxo-11H-indeno-[1,2-c]isoquinoline (21a)
Compound 19 (0.200 g, 0.354 mmol), NaI (0.360 g, 2.40 mmol) and imidazole (0.163 g, 2.40 mmol) were diluted with anhydrous DMF (40 mL). The mixture was heated to 70 °C for 19 h and cooled. The mixture was poured into H2O (100 mL) and extracted with CHCl3 (3 × 50 mL). The solution was washed with H2O (1 × 200, 3 × 300 mL) and the aqueous layer was extracted with CHCl3 (30 mL). The combined organic layers were dried over anhydrous sodium sulfate, concentrated, and adsorbed onto SiO2 (~4.00 g). The residue was purified by flash column chromatography (SiO2, ~26.0 g), eluting with a gradient of 0.7% MeOH in CHCl3 to 2% MeOH in CHCl3 to yield a bright purple solid (0.103 g, 53%) after washing with 1% CHCl3 in ether (60 mL) and ether (20 mL), and drying: mp 236–240 °C. IR (film) 2918, 1690, 1546, 1554, 1495, 1303, 1260, 1212, 1025, 863, 786 cm−1; 1H NMR (CDCl3) δ 8.03 (s, 1 H), 7.64 (s, 1 H), 7.62 (s, 1 H), 7.46-7.33 (m, 5 H), 7.19 (s, 1 H), 7.11 (s, 1 H), 7.06 (s, 1 H), 6.79 (s, 1 H), 5.21 (s, 2 H), 4.58 (t, J = 6.4 Hz, 2 H), 4.24 (t, J = 6.7 Hz, 2 H), 4.05 (s, 3 H), 4.00 (s, 3 H), 3.86 (s, 3 H), 2.40-2.35 (m, 2 H); ESIMS m/z (rel. intensity) 552 (MH+, 100).
9-Benzyloxy-5,6-dihydro-2,3-8-trimethoxy-6-[3-(N-morpholino)propyl]-5,11-dioxo-11H-indeno-[1,2-c]isoquinoline (21b)
Compound 19 (0.100 g, 0.177 mmol) and NaI (0.178 g, 1.19 mmol) were diluted with anhydrous DMF (25 mL). Morpholine (0.104 g, 1.19 mmol) was added, and the mixture was heated to 70 °C for 2 h and then stirred for 17 h at room temperature. The mixture was poured into H2O (100 mL) and extracted with CHCl3 (3 × 50 mL). The organic phase was washed with H2O (4 × 200 mL), dried over anhydrous sodium sulfate, and adsorbed onto SiO2 (~3.0 g). The residue was purified by flash column chromatography (SiO2, ~30 g), eluting with a gradient of 0.2% MeOH in CHCl3 to 1% MeOH in CHCl3 to yield a purple-brown solid (0.070 g, 69%) after washing with 2% CHCl3 in ether (50 mL) and drying: mp 252–254 °C. IR (film) 2944, 1690, 1652, 1553, 1496, 1303, 1211, 1118, 1014, 918, 870, 786, 730 cm−1; 1H NMR (CDCl3) δ 8.04 (s, 1 H), 7.65 (s, 1 H), 7.47-7.34 (m, 5 H), 7.21 (s, 2 H), 7.11 (s, 1 H), 5.23 (s, 2 H), 4.58 (d, J = 7.1 Hz, 2 H), 4.05 (s, 1 H), 3.99 (s, 3 H), 3.98 (s, 3 H), 3.70 (t, J = 4.6 Hz, 4 H), 2.59 (t, J = 6.8 Hz, 2 H), 2.55 (br s, 4 H), 2.13-2.07 (m, 2 H); ESIMS m/z (rel. intensity) 571 (MH+, 100). Anal. Calcd for C33H34N2O6·H2O: C, 67.33; H, 6.16; N, 4.76. Found: C, 67.28; H, 5.81; N, 4.80.
5,6-Dihydro-9-hydroxy-6-[3-(1H-imidazol-1-yl)propyl]-2,3,8-trimethoxy-5,11-dioxo-11H-indeno-[1,2-c]isoquinoline (22a, Method 1)
Compound 21a (0.090 g, 0.163 mmol) was diluted in H2O (30 mL) and the mixture was sonicated to ensure a homogeneous suspension. A solution of HBr in AcOH (33%, 32 mL) was added slowly, and the mixture was heated at 65 °C for 2.5 h. The mixture was a clear, reddish-purple color. The mixture was concentrated, and H2O (30 mL) was added to the residue. Sat. aq NaHCO3 was added until the pH was neutral (approximately 6 mL were needed). The suspension was extracted with CHCl3 (3 × 50 mL), and the organic layers were washed with H2O (4 × 200 mL) and dried over anhydrous sodium sulfate. The solution was concentrated, adsorbed onto SiO2 (~4.0 g), and the residue was purified by flash column chromatography (SiO2, ~27 g), eluting with a gradient of 1.5% MeOH in CHCl3 to 4.5% MeOH in CHCl3 to yield the title compound as a chalky, mauve-colored solid (0.023 g, 30%) after washing with ether (30 mL) and drying: mp 254–257 °C (dec). IR (film) 2537, 1689, 1634, 1557, 1512, 1462, 1432, 1307, 1223, 1082, 1020, 872, 786 cm−1; 1H NMR (DMSO-d6) δ 10.00 (br s, 1 H), 7.89 (s, 1 H), 7.72 (s, 1 H), 7.49 (s, 1 H), 7.29 (s, 1 H), 7.00 (s, 1 H), 6.94 (s, 1 H), 6.90 (s, 1 H), 4.48 (t, J = 6.7 Hz, 2 H), 4.18 (t, J = 6.8 Hz, 2 H), 3.89 (s, 3 H), 3.87 (s, 3 H), 3.85 (s, 3 H), 2.27-2.23 (m, 2 H); ESIMS m/z (rel. intensity) 462 (MH+, 100). Purity was estimated to be 100% by HPLC in MeOH and 99.3% in 90% MeOH – 10% H2O. Anal. Calcd for C25H23N3O6·0.5 H2O: C, 63.82; H, 5.14; N, 8.93. Found: C, 63.70; H, 4.96; N, 8.56.
5,6-Dihydro-9-hydroxy-2,3,8-trimethoxy-6-[3-(N-morpholino)propyl]-5,11-dioxo-11H-indeno-[1,2-c]isoquinoline (22b, Method 1)
Compound 21b (0.074 g, 0.130 mmol) was diluted in H2O (60 mL) and the mixture was sonicated to ensure a homogeneous suspension. A solution of HBr in AcOH (33%, 60 mL) was added slowly, and the mixture was heated at 70 °C for 4 h. The reddish-brown solution was concentrated, and the residue was diluted with H2O (100 mL). Sat. aq NaHCO3 was added until the until gas evolution ceased (approximately 30 mL was required). The suspension was extracted with CHCl3 (2 × 50, 1 × 25 mL), and the organic layers were washed with H2O (2 × 150 mL) and sat. aq NaCl (150 mL) and dried over anhydrous sodium sulfate. The solution was concentrated, adsorbed onto SiO2 (~ 4 g) and the residue was purified by flash column chromatography (SiO2, ~ 25 g), eluting with a gradient of 1% MeOH in CHCl3 to 2.5% MeOH in CHCl3 to yield the title compound as a brown solid (0.035 g, 56%) after washing with 2% CHCl3 in ether (50 mL) and ether (10 mL) and drying: mp 258–260 °C (dec). Combining this with several reactions run in parallel and repurifying the resulting material twice by flash column chromatography (SiO2, ~25 g), eluting with a gradient of 0.5% MeOH in CHCl3, to 2.5% MeOH in CHCl3, afforded an analytically pure sample (0.040 g) after dissolving in tetrahydrofuran (10 mL) and precipitating with ether (50 mL). IR (film) 2917, 1692, 1650, 1552, 1496, 1394, 1261, 1210, 1116, 870, 786, 757 cm−1; 1H NMR (CDCl3) δ 8.05 (s 1 H), 7.64 (s, 1 H), 7.16 (s, 1 H), 7.02 (s, 1 H), 6.10 (br s, 1 H), 4.57 (t, J = 7.2 Hz, 2 H), 4.05 (s, 3 H), 4.01 (s, 3 H), 4.99 (s, 3 H), 3.70 (t, J = 4.5 Hz, 2 H), 2.60 (t, J = 7.1 Hz, 2 H), 2.40-2.50 (m, 4 H), 2.14-2.09 (m, 2 H); ESIMS m/z (rel. intensity) 481 (MH+, 100). Purity was estimated to be 97.5% by HPLC in 95% MeOH – 5% H2O and 97.7% in 90% MeOH – 10% H2O. Anal. Calcd for C26H28N2O7: C, 64.99; H, 5.87; N, 5.83. Found: C, 64.92; H, 5.86; N, 5.75.
O-(4-Methoxybenzyl)vanillin (23).65
Vanillin (15, 3.00 g, 19.7 mmol) was diluted in dry DMF (55 mL). PMB-Cl (3.18 g, 20.9 mmol) was added, followed by anhydrous potassium carbonate (6.53 g, 47.3 mmol). The yellow mixture was heated to 70 °C for 3 h and was then cooled and partitioned between ether and water (200 mL of each). The layers were separated, and the aqueous layer was extracted with ether (100 mL) and CHCl3 (1 × 100 mL, 2 × 50 mL). The combined organic extracts were washed with H2O (3 × 200 mL) and sat. aq NaCl (100 mL). The organic phase was dried over anhydrous sodium sulfate, filtered to remove particulate matter, and concentrated to yield a yellow residue. This residue was dissolved in CHCl3 (20 mL), and addition of hexanes (175 mL) resulted in the precipitation of a pale-yellow solid (4.82 g, 90%) which was washed with hexanes (25 mL) and dried: mp 101–104 °C (lit.65 mp 100–102 °C). 1H NMR (CDCl3) δ 9.39 (s, 1 H), 7.42-7.36 (m, 4 H), 7.02 (d, J = 8.0 Hz, 1 H), 6.93 (dd, J = 2.6, 6.7 Hz, 2 H), 5.18 (s, 2 H), 3.94 (s, 3 H), 3.81 (s, 3 H).
N-[4′-(4″-Methoxybenzyloxy)-3′-methoxybenzylidene]-3-bromopropan-1-amine (24)
3-Bromopropylamine HBr (1.85 g, 8.44 mmol) was diluted with CHCl3 (12 mL). Compound 23 (2.00 g, 7.34 mmol) was added as a solution in CHCl3 (15 mL) and was quantitatively transferred with CHCl3 (3 mL). Et3N (0.811 g, 8.7 mmol) was added slowly, which discharged the cloudiness of the reaction mixture, followed by anhydrous Na2SO4 (2.00 g, 14.1 mmol). The mixture was stirred at room temperature for 24 h and diluted to 70 mL with CHCl3. The mixture was washed with H2O (70 mL) and this aqueous phase was extracted with CHCl3 (50 mL). The combined organic layers were washed with H2O (2 × 50 mL) and sat. aq NaCl (70 mL), dried over anhydrous sodium sulfate, and concentrated to yield an orange oil (2.84 g, 99%) that solidified upon standing: mp 78–80 °C. IR (film) 2934, 2837, 1644, 1613, 1585, 1515, 1267, 1251, 1138, 1033 cm−1; 1H NMR (CDCl3) δ 8.22 (s, 1 H), 7.41-7.35 (m, 3 H), 7.11 (d, J = 8.0 Hz, 1 H), 6.92 (d, J = 8.1 Hz, 3 H), 5.13 (s, 2 H), 3.94 (s, 3 H), 3.81 (s, 3 H), 3.74 (t, J = 6.2 Hz, 2 H), 3.51 (d, J = 6.5 Hz, 2 H), 2.30-2.20 (m, 2 H); ESIMS m/z (rel. intensity) 392/394 (MH+, 100/97).
cis-3-(4-Methoxybenzyloxy-3-methoxyphenyl)-N-(3-bromopropyl)-4-carboxy-3,4-dihydro-6,7-dimethoxy-1(2H)-isoquinolone (25)
Anhydride 11 (1.03 g, 4.64 mmol) was diluted with CHCl3 (30 mL), and the solution was cooled to 0 °C. The Schiff base 24 (1.19 g, 4.88 mmol) was added slowly as a pre-cooled solution in CHCl3 (20 mL) and quantitatively transferred with CHCl3 (3 mL). The mixture was stirred at 0 °C for 2 h, followed by 2 h at room temperature, upon which a precipitate had formed. Hexanes (12 mL) were added, and the precipitate was collected, washed with 20% hexanes in CHCl3 (50 mL) and dried to yield the title compound as a white amorphous solid (2.22 g, 78%): mp 139–141 °C (dec). IR (film) 1996, 1722, 1619, 1592, 1573, 1251, 1172, 1138 cm−1; 1H NMR (CDCl3/DMSO-d6) δ 7.63 (s, 1 H), 7.24 (d, J = 8.6 Hz, 2 H), 7.15 (s, 1 H), 6.81 (d, J = 8.6 Hz, 2 H), 6.63-6.51 (m, 3 H), 4.95-4.89 (m, 3 H), 4.54 (d, J = 6.4 Hz, 1 H), 4.00-3.88 (m, 1 H), 3.88 (s, 3 H), 3.81 (s, 3 H), 3.72 (s, 3 H), 3.60 (s, 3 H), 3.41-3.34 (m, 2 H), 3.10-3.00 (m, 1 H), 2.20-2.00 (m, 2 H); ESIMS m/z (rel. intensity) 614/616 (MH+, 4.04/3.68), 534 (MH+ – HBr, 100).
3-(Bromopropyl)-5,6-dihydro-9-hydroxy-2,3,8-trimethoxy-5,11-dioxo-11H-indeno-[1,2-c]isoquinoline (20)
The cis acid 25 (4.71 g, 7.67 mmol) was diluted with SOCl2 (80 mL). The mixture was stirred at room temperature for 5 h. The dark burgundy mixture was then concentrated and the residue was diluted with CHCl3 (40 mL). Ether (250 mL) was added, and the precipitate was collected, washed with ether (100 mL), subjected to a second, identical precipitation (20 mL CHCl3, 200 mL ether, followed by washing with 100 mL ether), and dried to afford the crude product as a reddish-brown solid (2.48 g, 68%). This material was used without any further purification. 1H NMR (DMSO-d6) δ 10.00 (br s, 1 H), 7.86 (s, 1 H), 7.46 (s, 1 H), 7.10 (s, 1 H), 6.93 (s, 1 H), 4.52 (t, J = 8.5 Hz, 2 H), 3.91 (s, 3 H), 3.88 (s, 3 H), 3.84 (s, 3 H), 3.74 (t, J = 3.6 Hz, 2 H), 2.35-2.25 (m, 2 H).
5,6-Dihydro-9-hydroxy-6-[3-(1H-imidazol-1-yl)propyl]-2,3,8-trimethoxy-5,11-dioxo-11H-indeno-[1,2-c]isoquinoline (22a, Method 2)
Compound 20 (1.12 g, 2.35 mmol), imidazole (1.60 g, 23.5 mmol), and NaI (3.53 g, 23.5 mmol) were diluted with dioxane (300 mL). The mixture was heated under argon to reflux for 29 h. The mixture was cooled, concentrated, and the residue was dissolved in 5% MeOH in CHCl3 (200 mL). This solution was washed with H2O (300 mL), and the aqueous phase was exhaustively extracted with CHCl3 (~500 mL). The organic phase was washed with H2O (approximately 2 × 600 mL) and dried over anhydrous sodium sulfate. The solution was concentrated, adsorbed onto SiO2 (~10 g), and purified by flash column chromatography (SiO2, ~50 g), eluting with a gradient of 1.5% MeOH in CHCl3 to 5% MeOH in CHCl3 to yield a mauve-colored solid (0.474 g, 44%) after dissolving in CHCl3 (10 mL), precipitating with ether (300 mL), and washing with ether (50 mL). The analytical data for this compound are identical to that prepared by method 1.
5,6-Dihydro-9-hydroxy-2,3,8-trimethoxy-6-[3-(N-morpholino)propyl]-5,11-dioxo-11H-indeno-[1,2-c]isoquinoline (22b, Method 2)
Compound 20 (0.100 g, 0.211 mmol) and NaI (0.189 g, 1.27 mmol) were diluted with dry DMF (50 mL). The mixture was heated to 65 °C and morpholine (0.111 g, 1.27 mmol) was added. After 18 h at 65 °C, the mixture was cooled and poured into H2O (120 mL). The mixture was extracted with CHCl3 (3 × 75 mL) and the organic phase was washed with H2O (5 × 250 mL) and sat. aq NH4Cl (250 mL). The extract was dried over anhydrous sodium sulfate. The solution was concentrated, adsorbed onto SiO2 (~ 3 g), and the product purified by flash column chromatography (SiO2, ~20 g), eluting with a gradient of 0.5% MeOH in CHCl3 to 2% MeOH in CHCl3. The product was suspended in CHCl3 (3 mL), precipitated with ether (100 mL), and washed with ether (20 mL) to yield a brown solid (0.045 g, 44%). The analytical data for this compound are identical to that prepared by method 1.
O-Benzylhomoisovanillic Acid (27).52
KOH (3.93 g, 70.0 mmol) was diluted with absolute EtOH (100 mL) and the mixture was warmed to 55 °C, upon which the mixture became a clear solution. Homoisovanillic acid (26, 5.00 g, 27.4 mmol) was added in small portions, and the mixture was heated to gentle reflux under a continuous argon flow. Benzyl chloride (7.49 g, 60.0 mmol) was added as a solution in absolute EtOH (10 mL), dropwise, over 15 min, and was quantitatively transferred with EtOH (3 mL). The mixture was heated at reflux for 2 h following addition, upon which a cloudy precipitate formed. A solution of KOH (1 g) in H2O (20 mL) was added to the mixture, and reflux was continued for 20 min. The solution was cooled to room temperature and stirred for 1 h before it was concentrated to 1/3 of its original volume. The resulting residue was diluted with H2O (400 mL) and made acidic (pH = 1) by the addition of HCl (concd, 5 mL). The resulting precipitate was collected, dried, and recrystallized from 40% acetone in ether to yield the product as an off-white solid (5.68 g, 76%) after drying: mp 111–114 °C (lit63 mp 124–125 °C). 1H NMR (DMSO-d6) δ 12.4 (br s, 1 H), 7.45-7.32 (m, 5 H), 6.96 (s, 1 H), 6.91 (d, J = 9.6 Hz, 1 H), 6.80-6.79 (m, 1 H), 5.01 (s, 2 H), 3.72 (s, 3 H), 3.45 (s, 2 H).
6-(Benzyloxy)-7-methoxy-1H-isochromen-3(4H)-one (28).54
Compound 27 (7.00 g, 25.7 mmol) was diluted with glacial AcOH (90 mL). Formalin (37% formaldehyde in H2O, 24 mL) was added, followed by HCl (concd, 6 mL) The mixture was heated at 45 °C for 22 h, cooled, and poured into H2O (300 mL). The cloudy suspension was extracted with CHCl3 (3 × 100 mL). The organic layers were washed with sat. aq NaHCO3 (3 × 150 mL) and H2O (3 × 200 mL). The combined aqueous layers were extracted with CHCl3 (100 mL). The organic layers were dried over anhydrous sodium sulfate and concentrated. The resulting pale yellow oil was dissolved in CHCl3 (10 mL), and hexanes (100 mL) were added slowly to precipitate a white amorphous solid, which was collected by vacuum filtration. Re-filtration of the filtrate afforded additional product. A total of 5.00 g (69%) was obtained, containing a small amount of impurities: mp 105–110 °C (lit53 mp 122 °C). 1H NMR (CDCl3) δ 7.42-7.34 (m, 5 H), 6.76 (s, 1 H), 6.72 (s, 1 H), 5.25 (s, 2 H), 5.15 (s, 2 H), 3.89 (s, 3 H), 3.58 (s, 2 H).
4-(Benzyloxy)-2-(carboxymethyl)-5-methoxybenzoic Acid (29)
Compound 28 (5.00 g, 17.6 mmol) was diluted with a solution of KOH (2.47 g, 44.0 mmol) in H2O (250 mL). The mixture was stirred at room temperature for 17 h. The clear orange solution was cooled to 0 °C and KMnO4 (6.21 g, 40.0 mmol) was added in portions over 10 min. The mixture was stirred at 0 °C for 3 h, and then at room temperature for 48 h. Absolute EtOH (60 mL) was added, and the mixture was heated at 75 °C for 30 min. The mixture was filtered hot to remove inorganic materials, and the filter cake was washed with H2O (100 mL). The filtrate was concentrated to half its volume and was washed with EtOAc (2 × 150 mL). The aqueous layer was diluted with H2O to a volume of 300 mL, and, while stirring, was made acidic (pH = 1) by the addition of HCl (concd, 12 mL). The mixture was chilled to 0 °C for 3 h, and the resultant white precipitate was collected by vacuum filtration, dried, triturated with EtOAc-hexanes (70:30, 75 mL), and washed with EtOAc (30 mL) to afford an off-white solid (3.11 g, 56%): mp 199–200 °C. IR (neat) 3583, 3408, 2917, 1682, 1598, 1575, 1276, 1218, 1173, 1074, 666 cm−1; 1H NMR (DMSO-d6) δ 12.3 (br s, 1 H), 7.47-7.36 (m, 6 H), 7.07 (s, 1 H), 5.11 (s, 2 H), 3.86 (s, 2 H), 3.77 (s, 3 H). One of the carboxyl protons is not visible due to exchange with residual water. 13C NMR (DMSO d6, 125 MHz) δ 172.9, 168.0, 150.7, 147.4, 136.8, 131.3, 128.8, 128.4, 128.4, 122.5, 117.2, 114.1, 70.2, 55.9; ESIMS m/z (rel intensity) 339.2 (MNa+, 100); negative ion 315.1 [(M - H+)−, 100)]. Anal. Calcd for C17H16O6·0.5H2O: C, 62.77; H, 5.27. Found: C, 62.60; H, 4.99.
6-(Benzyloxy)-7-methoxy-4H-isochromene-1,3-dione (30)
Compound 29 (2.25 g, 7.11 mmol) was diluted with acetyl chloride (40 mL), and the mixture was heated at reflux for 2.5 h. The mixture was then concentrated to yield a yellow solid (2.12 g, 100%): mp 160–165 °C. IR (neat) 2939, 1741, 1515, 1277, 1224, 1028 cm−1; 1H NMR (CDCl3) δ 7.60 (s, 1 H), 7.42-7.26 (s, 5 H), 6.71 (s, 1 H), 5.24 (s, 2 H), 4.00 (s, 2 H), 3.96 (s, 3 H); 13C NMR (CDCl3, 125 MHz) δ 164.9, 160.6, 154.4, 149.7, 135.2, 128.8, 128.7, 128.4, 127.1, 113.6, 111.5, 110.3, 71.0, 56.2, 33.9; CIMS m/z (rel intensity) 299 (MH+, 40), 209 (100), 181 (100).
N-[3′4′-(Methylenedioxy)benzylidene]-3-bromopropan-1-amine (32).21
3-Bromopropylamine hydrobromide (3.35 g, 15.3 mmol) was diluted with CHCl3 (15 mL). Piperonal (31, 2.0 g, 13.3 mmol) was added slowly as a solution in CHCl3 (15 mL), and quantitatively transferred with 5 mL of the same solvent. Et3N (1.6 g, 16.0 mmol) was added slowly, upon which the solution became clear and colorless. Na2SO4 (3.00 g, 21.1 mmol) was added, and the mixture was stirred at room temperature for 27 h. The mixture was diluted with CHCl3 to a volume of 70 mL and washed with H2O (70 mL). The aqueous layer was extracted with CHCl3 (50 mL), and the combined organic layers were washed with H2O (2 × 70 mL) and sat. aq NaCl (70 mL). The organic layer was dried over anhydrous sodium sulfate and concentrated to yield a yellow oil (3.44 g, 96%). 1H NMR (CDCl3) δ 8.21 (s, 1 H), 7.34 (d, J = 0.9 Hz, 1 H), 7.13 (dd, J = 0.9, 7.9 Hz, 1 H), 6.85 (d, J = 7.9 Hz, 1 H), 6.01 (s, 2 H), 3.73 (t, J = 6.3 Hz, 2 H), 3.52 (t, J = 6.4 Hz, 2 H), 2.30-2.20 (m, 2 H).
cis-6-(Benzyloxy)-N-(3-bromopropyl)-4-carboxy-3,4-dihydro-7-methoxy-3-(3,4-methylenedioxyphenyl)-1(2H)-isoquinolone (33)
Compound 30 (2.09 g, 7.03 mmol) was dissolved in CHCl3 (65 mL), and the solution was cooled to 0 °C. A solution of Schiff base 32 (2.00 g, 7.40 mmol) in CHCl3 (15 mL) was cooled to 0 °C and added slowly via addition funnel to the anhydride. Within minutes, a yellow precipitate began to form. The mixture was stirred for 2 h at 0 °C and then warmed to room temperature, where it was stirred for 2 h. The precipitate was collected via vacuum filtration and washed with CHCl3 (100 mL) to yield the desired product as a cream-colored solid (2.63 g, 66%) after drying: mp 198–199 °C (dec). IR (film) 2972, 2937, 1742, 1592, 1570, 1259, 1226, 1168 cm−1; 1H NMR (DMSO-66) δ 12.96 (br s, 1 H), 8.31 (s, 1 H), 7.53-7.35 (s, 5 H), 7.26 (s, 1 H), 6.80 (d, J = 8 Hz, 1 H), 6.57 (dd, J = 1.4, 8.1 Hz, 2 H), 6.46 (s, 1 H), 5.95 (s, 2 H), 5.03-5.00 (m, 3 H), 4.69 (d, J = 6 Hz, 1 H), 3.88-3.82 (s, 1 H), 3.82 (s, 3 H), 3.56-3.50 (s, 2 H), 2.95-2.88 (m, 1 H), 2.14-1.96 (m, 2 H); 13C NMR (DMSO, 75 MHz) δ 171.5, 163.6, 151.4, 148.8, 148.0, 147.8, 137.4, 132.0, 129.4, 129.1, 127.9, 122.6, 122.4, 113.2, 110.9, 108.9, 108.6, 102.1, 71.0, 62.4, 56.4, 48.3, 45.5, 33.4, 31.9; ESIMS m/z (rel intensity) 488.5 (MH+ – HBr, 100). Anal Calcd for C28H26BrNO7·H2O: C, 57.35; H, 4.81; N, 2.39. Found: C, 57.34; H, 4.46; N, 2.42.
6-(3-Bromopropyl)-5,6-dihydro-2-hydroxy-3-methoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (34)
Compound 33 (1.35 g, 2.38 mmol) was diluted with SOCl2 (30 mL). The mixture was stirred at room temperature for 3 h and 40 min. The dark red mixture was concentrated and the residue was suspended in 1,2-dichloroethane (200 mL). Aluminum chloride (0.475 g, 0.357 mmol) was added, and the mixture was stirred at 0 °C for 10 min. Additional AlCl3 (0.237 g, 1.78 mmol) was then added, and the mixture was sonicated for 1.5 h at 0 °C. The mixture was poured into a mixture of ice and sat. aq NaHCO3 (200 mL). The mixture was extracted with CHCl3 (4 × 150 mL) and the organic layer was filtered to remove aluminum salts. The organic layer was washed with H2O (3 × 200 mL), dried over anhydrous sodium sulfate, concentrated, and adsorbed onto SiO2 (~5 g). The residue was purified by flash column chromatography (SiO2, ~40 g), eluting with CHCl3, to yield a purple-grey solid (0.108 g, 10%) after suspending in CHCl3 (10 mL), precipitating with ether (100 mL) and washing with ether (20 mL): mp 276–278 °C. IR (film) 3368, 2918, 1681, 1652, 1615, 1491, 1432, 1389, 1304, 1267, 1033 cm−1; 1H NMR (DMSO-66) δ 10.44 (s, 1 H), 7.81 (s, 1 H), 7.47 (s, 1 H), 7.35 (s, 1 H), 7.05 (s, 1 H), 6.17 (s, 2 H), 4.50 (t, J = 6.9 Hz, 2 H), 3.84 (s, 3 H), 3.73 (t, J = 6.5 Hz, 2 H), 2.30-2.20 (m, 2 H); ESIMS m/z (rel intensity) 458/460 (MH+, 9/8.5), 378 (MH+ – HBr, 100).
5,6-Dihydro-2-hydroxy-6-3-(1H-imidazol-1-yl)propyl)-3-methoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (35a)
Compound 34 (0.212 g, 0.463 mmol), imidazole (0.315 g, 4.63 mmol), and NaI (0.693 g, 4.63 mmol) were diluted with dry DMF (60 mL) and the mixture was heated at 60 °C for 20 h. Additional imidazole (0.200 g, 2.94 mmol) was added and the mixture was heated for an additional 40 min. The mixture was cooled and poured into H2O. CHCl3 (200 mL) was added, and an insoluble suspension formed. This suspension was saved, and the remaining aqueous layer was extracted with CHCl3 (2 × 200 mL). These layers were washed with H2O (4 × 400 mL), sat aq NH4Cl (200 mL), dried over anhydrous sodium sulfate, and concentrated. The residue was washed with ether (50 mL), dissolved in CHCl3 (50 mL), concentrated, and adsorbed onto SiO2 (3 g). This residue was purified by flash column chromatography (SiO2, 60 g), eluting with a gradient of 0.5% MeOH in CHCl3 to 7% MeOH in CHCl3, to afford a purple solid. Additional product was obtained by drying the CHCl3-insoluble suspension over anhydrous sodium sulfate and concentrating the mixture. This solid was washed with H2O (80 mL), and purified by dissolving in DMF (5 mL) and precipitating with ether (50 mL). The solid (0.054 g, 26%) was washed with 10% CHCl3 in ether (50 mL) and ether (20 mL) and dried. To produce an analytically pure sample (0.034 g), the solid was adsorbed onto SiO2 (3 g) and purified by flash column chromatography (SiO2, 20 g), eluting with a gradient of 2% MeOH in CHCl3 to 5% MeOH in CHCl3 and then washing the product with 10% CHCl3 in ether (20 mL) and ether (20 mL): mp 269–272 °C (dec). IR (film) 3391, 3117, 2916, 1691, 1637, 1605, 1488, 1304, 1225, 1091, 1028 cm−1; 1H NMR (DMSO-d6) δ 10.46 (br s, 1 H), 7.81 (s, 1 H), 7.76 (s, 1 H), 7.47 (s, 1 H), 7.27 (s, 1 H), 7.04 (s, 1 H), 6.98 (s, 1 H), 6.92 (s, 1 H), 6.16 (s, 2 H), 4.40-4.30 (m, 2 H), 4.29 (t, J = 6.1 Hz, 2 H), 2.20-2.10 (m, 2 H); ESIMS m/z (rel intensity) 446.3 (MH+, 100); HRESIMS m/z 446.1347 (MH+), calcd for C24H20N3O6 446.1352. Anal Calcd for C24H19N3O4·1.25 H2O: C, 61.60; H, 4.63; N, 8.98. Found: C, 61.67; H, 4.27; N, 8.79.
5,6-Dihydro-2-hydroxy-3-methoxy-8,9-methylenedioxy-6-3-[(morpholino)propyl)]-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (35b)
Compound 34 (0.080 g, 0.17 mmol) was dissolved in anhydrous DMF (10 mL), and NaI (0.314 g, 2.09 mmol) and morpholine (0.18 mL, 2.06 mmol) were added. The mixture was heated to 80 °C for 26 h under argon. After the evaporation of the solvent, the residue was partitioned between chloroform (60 mL) and water (30 mL). The organic layer was washed with water (30 mL) and purified by silica gel column chromatography (chloroform-MeOH, 10:0.5), which afforded the product (0.020 g, 25%): mp 228–230 °C. IR (neat) 3583, 2918, 2852, 1701, 1634, 1610, 1470, 1389, 1270, 1115, 1033, 666 cm−1; 1H NMR (CDCl3, 300 MHz) δ 7.90 (s, 1 H), 7.46 (s, 1 H), 7.16 (s, 1 H), 6.97 (s, 1 H), 6.05 (s, 2 H), 4.61 (t, J = 7.2 Hz, 2 H), 4.05 (s, 3 H), 3.70-3.63 (m, 5 H), 2.51 (t, J = 6.8 Hz, 2 H), 2.43 (br, 4 H), 1.96-1.91 (m, 2 H); 13C NMR (CDCl3, 125 MHz) δ 187.3, 160.7, 152.5, 150.5, 148.5, 146.2, 138.5, 131.7, 126.9, 124.9, 124.6, 122.2, 109.3, 107.3, 105.8, 103.9, 102.1, 66.8, 56.2, 53.4, 41.0, 25.5; ESIMS m/z (rel intensity) 465.1 (MH+, 100); negative ion 463.2 [(M - H+) −, 100]; HRESIMS m/z 465.1659 (MH+), calcd for C25H25N2O7 465.1662. Anal. Calcd for C25H24N2O7: C, 64.65; H, 5.21; N, 6.03. Found: C, 64.69; H, 5.22; N, 6.18.
7-Hydroxy-6-methoxyisochroman-3-one (37).54
Glacial AcOH (25 mL) was added to 36 (0.96 g, 5.3 mmol). The solution was heated to 90 °C, and concd HCl (0.9 mL) and formalin (37% formaldehyde in H2O, 0.9 mL) were added rapidly, and then heating was stopped immediately. After 1.5 h at room temperature, ice (20 g) and H2O (20 mL) were added to the solution. The organic material was extracted with CHCl3 (4 × 50 mL), washed with sat. aq NaHCO3 (2 × 40 mL) and sat. aq NaCl (50 mL), and the solvent was evaporated to yield the crude product (0.81 g). Recrystallization from CH2Cl2-ether gave pure 37 as a white powder (0.36 g, 35%): mp 178–179 °C (lit.54 174–177 °C). 1H NMR (DMSO-d6, 300 MHz) δ 9.05 (s, 1 H), 6.88 (s, 1 H), 6.75 (s, 1 H), 5.18 (s, 2 H), 3.74 (s, 3 H), 3.62 (s, 2 H).
7-(Benzyloxy)-6-methoxyisochroman-3-one (38)
A mixture of 37 (0.160 g, 0.82 mmol), anhydrous K2CO3 (500 mg, 3.62 mmol), and benzyl bromide (0.25 mL, 2.08 mmol) in dry acetone was heated at 56 °C for 4 h. The inorganic compounds were filtered off and washed with acetone. Evaporation of the filtrate afforded a residue that was partitioned between CHCl3 (30 mL) and H2O (30 mL). The organic layer was dried over anhydrous sodium sulfate, concentrated, and the residue was purified by flash column chromatography (SiO2, ~25 g), eluting with a gradient of 10% EtOAc-hexanes to 25% EtOAc-hexanes, to yield the product (0.132 g, 57%) as a white powder: mp 129–130 °C (lit.54 137–138 °C). 1H NMR (CDCl3, 500 MHz) δ 7.31-7.43 (m, 5 H), 6.74 (s, 1 H), 6.73 (s, 1 H), 5.19 (s, 2 H), 5.14 (s, 2 H), 3.89 (s, 3 H), 3.62 (s, 2 H).
5-(Benzyloxy)-2-(carboxymethyl)-4-methoxybenzoic Acid (39)
A solution of compound 38 (2.00 g, 7.00 mmol) and KOH (2 g, 36.0 mmol) in H2O (60 mL) was stirred at 0 °C and KMnO4 (3.30 g, 20.1 mmol) was added slowly. After allowing the mixture to stand for 24 h at room temperature, excess KMnO4 was destroyed by addition of EtOH (20 mL) and by heating at 60–70 °C for 0.5 h. The solution was filtered and concentrated to a small volume under reduced pressure. Acidification with concd HCl (to pH = 2) gave a precipitate that was collected. Recrystallization from acetone gave the pure product (0.675 g, 31%) as a yellow powder: mp 236–237 °C (lit.66 238 °C). 1H NMR (DMSO-d6, 300 MHz) δ 12.35 (s, 2 H), 7.50 (s, 1 H), 7.33-7.42 (m, 5 H), 6.93 (s, 1 H), 5.08 (s, 2 H), 3.83 (s, 2 H), 3.80 (s, 3 H).
7-(Benzyloxy)-6-methoxyisochroman-1,3-dione (40)
Acetyl chloride (4.0 mL) was added to 39 (0.175 g, 0.550 mmol). The solution was heated at reflux for 2 h and was concentrated to yield an oily residue. Recrystallization from CHCl3-ether gave pure 40 as a yellow powder (0.157 g, 91%): mp 107–109 °C (dec). IR (film) 2940, 1710, 1601, 1517, 1272, 1221, 1015 cm−1; 1H NMR (CDCl3, 300 MHz) δ 7.63 (s, 1 H), 7.26-7.47 (m, 5 H), 6.70 (s, 1 H), 5.18 (s, 2 H), 4.05 (s, 2 H), 3.96 (s, 3 H); CIMS m/z (rel intensity) 299 (MH+, 100).
cis-7-(Benzyloxy)-N-(3-bromopropyl)-4-carboxy-3,4-dihydro-6-methoxy-3-(3,4-methylenedioxyphenyl)-1(2H)-isoquinolone (41)
A solution of the anhydride 40 (0.276 g, 0.92 mmol) dissolved in CHCl3 (3 mL) was added dropwise to a solution of the Schiff base 32 (0.191 g, 0.92 mmol) in CHCl3 (3 mL). After 24 h, hexanes (5 mL) were added to the mixture, and the precipitate that formed was filtered off and washed with a mixture of ethyl acetate (5 mL) and CHCl3 (5 mL) to afford an impure mixture containing the desired product. This mixture was purified by flash column chromatography (SiO2, ~25 g), eluting with a gradient of 10% MeOH in CHCl3 to yield the product as a yellow powder (0.188 g, 36%): mp 195–197 °C (dec). IR (film) 2942, 1712, 1677, 1601, 1578, 1526, 1220, 1178 cm−1; 1H NMR (DMSO, 300 MHz) δ 7.78 (s, 1 H), 7.32-7.50 (m, 5 H), 7.08 (s, 1 H), 6.60-6.65 (m, 2 H), 6.48 (s, 1 H), 5.90 (s, 2 H), 5.22 (s, 2 H), 4.97-4.99 (d, J = 6.00 Hz, 1 H), 4.68-4.71 (d, J = 6.48 Hz, 1 H), 3.95-3.97 (m, 1 H), 3.88 (s, 3 H), 3.45 (m, 2 H), 3.11-3.13 (m, 1 H), 2.13-2.24 (m, 2 H); ESIMS m/z (rel intensity) 488.3 (MH+ – HBr, 100).
6-(3-Bromopropyl)-5,6-dihydro-3-benzyloxy-2-methoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (42)
The cis acid 41 (1.00 g, 1.76 mmol) was diluted in SOCl2 (45 mL) and the mixture was stirred at room temperature for 20 h. The solvent was removed under reduced pressure, and the organic material was extracted with chloroform (3 × 20 mL), washed with sat. aq NaHCO3 (2 × 20 mL) and sat. aq NaCl (20 mL), and then concentrated. The obtained residue was adsorbed onto SiO2 (~ 10 g) and purified by flash column chromatography (SiO2, ~30 g), eluting with 1.25% MeOH in CHCl3, to yield the solid product (0.404 g, 42%): mp 234–236 °C. IR (film) 3421, 1698, 1644, 1483, 1309, 1282, 1036, 698 cm−1; 1H NMR (CDCl3, 300 MHz) δ 8.04 (s, 1 H), 7.70 (s, 1 H), 7.29-7.50 (m, 5 H), 7.27 (s, 1 H), 7.07 (s, 1 H), 6.09 (s, 2 H), 5.24 (s, 2 H), 4.55-4.60 (m, 2 H), 4.04 (s, 3 H), 3.78-3.82 (t, J = 6.0 Hz, 2 H), 2.33-2.38 (m, 2 H); ESIMS m/z (rel intensity) 468.5 (MH+-HBr, 100).
6-(3-Bromopropyl)-5,6-dihydro-3-hydroxy-2-methoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (43)
The benzylated indenoisoquinoline 42 (1.73 g, 3.16 mmol) was dissolved in nitrobenzene (30 mL) and AlCl3 (0.839 g, 6.32 mmol) was added. The mixture was stirred at room temperature for 15 min and then heated at 90 °C for 45 min. The mixture was cooled and poured into a mixture of ice and sat. aq NaHCO3 (80 mL). The mixture was extracted with CHCl3 (4 × 100 mL) and the organic layer was filtered to remove aluminum salts. The organic layer was washed with H2O (3 × 100 mL), dried over anhydrous sodium sulfate, concentrated, and adsorbed onto SiO2 (~5 g). The residue was purified by flash column chromatography (SiO2, ~50 g), eluting with a gradient of 0.25% MeOH in CHCl3 to 1% MeOH in CHCl3, to afford the crude product as a pinkish solid (0.230 g, 16%): mp 226–228 °C (dec). IR (film) 3446, 2346, 1698, 1638, 1483, 1394, 1310, 1215, 1034 cm−1; 1H NMR (DMSO, 300 MHz) δ 9.86 (s, 1 H), 7.85 (s, 1 H), 7.46 (s, 1 H), 7.35 (s, 1 H), 7.05 (s, 1 H), 6.17 (s, 2 H), 4.48 (m, 2 H), 3.89 (s, 3 H), 3.82-3.87 (t, J = 6.18 Hz, 2 H), 2.19 (m, 2 H); ESIMS m/z (rel intensity) 458/460 (MH+, 28/27).
5,6-Dihydro-3-hydroxy-6-3-(1H-imidazol-1-yl)propyl)-2-methoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (44a)
Compound 43 (0.210 g, 0.46 mmol) and NaI (0.82 g, 5.52 mmol) were diluted with anhydrous DMF (15 mL). Imidazole (0.375 g, 5.52 mmol) was added and the mixture was heated to 70 °C for 24 h under an argon atmosphere. Ice (30 g) was added and the solution was extracted with CHCl3 (5 × 50 mL). The organic layer was washed with sat. NaHCO3 (3 × 30 mL), water (3 × 30 mL) and dried over anhydrous sodium sulfate. The solution was concentrated, adsorbed onto SiO2 (~5 g), and purified by flash column chromatography (SiO2, ~60 g), eluting with a gradient of 1% MeOH in CHCl3, to 5% MeOH in CHCl3, to yield the product as a dark brown solid (0.045 g, 28%): mp >350 °C. IR (film) 3446, 1693, 1642, 1484, 1431, 1394, 1309, 1225, 1033, 659 cm−1; 1H NMR (DMSO, 300 MHz) 9.88 (s, 1 H), 7.88 (m, 2 H), 7.48 (s, 1 H), 7.32 (s, 1 H), 7.09 (s, 1 H), 7.05 (s, 1 H), 6.99 (s, 1 H), 6.17 (s, 2 H), 4. 38 (m, 2 H), 4.17 (m, 2 H), 3.89 (s, 1 H), 2.18 (m, 2 H); ESIMS m/z (rel intensity) 446.5 (MH+, 100); HRESIMS m/z 446.1355 (MH+), calcd for C24H19N3O6 446.1352. Purity was estimated to be 95.2% by HPLC in both 95% MeOH – H2O and 85% MeOH – H2O.
5,6-Dihydro-3-hydroxy-2-methoxy-8,9-methylenedioxy-6-3-[(morpholino)propyl)]-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (44b)
Compound 43 (1.23 g, 2.74 mmol) and NaI (4.93 g, 32.88 mmol) were diluted with anhydrous DMF (40 mL). Morpholine (2.87 mL, 32.88 mmol) was added and the mixture was heated to 65 °C for 22 h under an argon atmosphere. The mixture was cooled, poured into H2O (200 mL), and extracted with CHCl3 (4 × 100 mL). The organic phase was washed with H2O (200 mL) and sat. aq NaHCO3 (200 mL) and was dried over anhydrous sodium sulfate. The solution was concentrated, adsorbed onto SiO2 (~5 g), and purified by flash column chromatography (SiO2, ~60 g), eluting with a gradient of 1% MeOH in CHCl3 to 5% MeOH in CHCl3, to yield the title compound as a purple solid (0.481 g, 38%): mp 321–323 °C (dec). IR (film) 3244, 2962, 2811, 1687, 1655, 1503, 1286, 1111, 1035, 861 cm−1; 1H NMR (CDCl3, 300 MHz) δ 8.05 (s, 1 H), 7.75 (s, 1 H), 7.44 (s, 1 H), 6.91 (s, 1 H), 6.09 (s, 2 H), 4.47-4.52 (m, 2 H), 4.09 (s, 3 H), 3.74-3.77 (m, 4 H), 2.52-2.56 (m, 6 H), 2.00-2.04 (m, 2 H); ESIMS m/z (rel intensity) 465.2 (MH+, 100); HRESIMS m/z 465.1665 (MH+), calcd for C25H24N2O7 465.1662. Anal. Calcd for C25H24N2O7H2O: C, 62.23; H, 5.43; N, 5.81; Found: C, 62.14; H, 5.00; N, 5.70.
5,6-Dihydro-11-hydroxy-6-(3-imidazolylpropyl)-2,3-methylenedioxy--5-oxo-11H-indeno[ 1,2-c]isoquinoline (45a)
Indenoisoquinoline 6 (0.170 g, 0.370 mmol) was dissolved in MeOH (50 mL), NaBH4 (0.025 g, 0.740 mmol) was added at 0 °C, and the reaction mixture was stirred at room temperature for 3 h. The mixture was concentrated, CHCl3 (150 mL) was added, and the mixture was washed with water (2 × 100 mL). The CHCl3 was removed on a rotary evaporator and the crude mixture was purified by flash column chromatography (SiO2), eluting with 10% MeOH in CHCl3, to yield the title compound (0.110 g, 64%) as white solid: mp >280 °C. IR (KBr) 3583, 1635, 1610, 1481, 1292, 1258, 1034, 750, 666 cm−1; 1H NMR (CDCl3 + CD3OD, 300 MHz) δ 7.49 (s, 1 H), 7.43 (s, 1 H), 7.24 (s, 1 H), 7.03 (s, 1 H), 6.91 (s, 1 H), 6.87 (s, 1 H), 6.55 (s, 1 H), 5.90 (d, J = 1.6 Hz, 2 H), 5.25 (s, 1 H), 4.40 (m, 1 H), 4.21 (m, 1 H), 4.04 (m, 2 H), 3.89 (s, 3 H), 3.80 (s, 3 H), 2.13 (m, 2 H); 13C NMR (CDCl3 + CD3OD, 75 MHz) δ 162.7, 153.7, 148.6, 148.2, 147.5, 142.4, 138.0, 136.6, 129.8, 128.6, 128.1, 120.2, 118.8, 116.8, 107.8, 106.2, 103.0, 101.78, 101.72, 71.1, 55.8, 55.6, 44.5, 40.8, 30.9; ESIMS (m/z, relative intensity) 462 (MH+, 100); HRESIMS m/z 462.1670 (MH+), calcd for C25H23N3O6 462.1665. Purity by HPLC was estimated to be 99.4% in 85% MeOH – H2O and 99.5% in 90% MeOH – H2O.
5,6-Dihydro-11-hydroxy-2,3-methylenedioxy-6-(3-morpholinylpropyl)-5-oxo-11H-indeno[ 1,2-c]isoquinoline (45b)
Indenoisoquinoline 7 (0.1 g, 0.209 mmol) was dissolved in MeOH (30 mL), NaBH4 (0.014 g, 0.418 mmol) was added at 0 °C and reaction mixture was stirred at room temperature for 3 h. The mixture was concentrated, CHCl3 was added (100 mL), and the solution was washed with water (2 × 50 mL). The CHCl3 was removed on a rotary evaporator and the crude mixture was purified by flash column chromatography (SiO2), eluting with 5% MeOH in CHCl3 to give the title compound (0.55 g, 55%) as white solid: mp 228–229 °C. IR (KBr) 3325, 2956, 1610, 1629, 1553, 1483, 1294, 1257, 1037, 665 cm−1; 1H NMR (CDCl3, 300 MHz) δ 7.33 (s, 1 H), 7.23 (s, 1 H), 7.12 (s, 1 H), 7.09 (s, 1 H), 6.06 (s, 1 H), 6.02 (s, 1 H), 5.29 (s, 1 H), 4.31 (m, 1 H), 4.05 (s, 3 H), 3.72 (m, 4 H), 3.63 (s, 3 H), 3.42 (m, 1 H), 2.40 (m, 6 H), 1.76 (m, 2 H); 13C NMR (CDCl3, 75 MHz) δ 162.3, 153.5, 148.2, 147.4, 142.9, 138.2, 129.3, 128.7, 119.6, 116.9, 107.9, 106.5, 102.9, 102.8, 101.7, 77.2, 71.6, 66.7, 56.2, 56.1, 55.4, 54.0, 42.5, 25.9; ESIMS (m/z, relative intensity) 481 (MH+, 100); HRESIMS m/z 481.1972 (MH+), calcd for C26H28N2O7 481.1975. Purity by HPLC was estimated to be 98.3% in 95% MeOH – H2O and 97.2% in 40% MeOH – H2O
3,4-Dibenzyloxybenzaldehyde (47).67
Compound 46 (3.00 g, 21.7 mmol) was diluted with dry DMF (50 mL). Benzyl bromide (7.65 g, 44.7 mmol) was added slowly, followed by anhydrous K2CO3 (9.60 g, 69.4 mmol). The mixture was stirred at room temperature for 2 h. Additional K2CO3 (2.4 g, 17.3 mmol) was added, and the mixture was heated to 70 °C for 30 min and then cooled to room temperature. The mixture was partitioned between H2O and ether (120 mL each). The organic layer was separated, and the water layer was extracted with ether (3 × 50 mL). The pooled organic layers were washed with H2O (2 × 50 mL) and sat. aq. NaCl (50 mL). The pale, straw-colored extracts were dried over anhydrous sodium sulfate and concentrated to yield a cream-colored solid after washing with hexanes (75 mL) and drying: mp 87–88.5 °C (lit67 mp 85–87 °C). 1H NMR (CDCl3) δ 9.81 (s, 1 H), 7.49-7.31 (m, 12 H), 7.04 (d, J = 8.3 Hz, 1 H), 5.27 (s, 2 H), 5.22 (s, 2 H).
N-[3′,4′-(Dibenzyloxy)benzylidene]-3-bromopropan-1-amine (48)
3-Bromopropylamine hydrobromide (0.791 g, 3.61 mmol) was diluted with CHCl3 (5 mL). Compound 47 (1.00 g, 3.14 mmol) was added slowly as a solution in CHCl3 (6 mL), and quantitatively transferred with 3 mL of the same solvent. Et3N (0.348 g, 3.45 mmol) was added slowly, upon which the solution became clear and colorless. Na2SO4 (1.20 g, 6.24 mmol) was added, and the mixture was stirred at room temperature for 22 h. The mixture was diluted to a volume of 30 mL with CHCl3, and was washed with H2O (3 × 40 mL) and sat. aq. NaCl (40 mL). The organic layer was dried over anhydrous sodium sulfate and concentrated to yield a dark yellow syrup (1.36 g, 99%). IR (film) 3031, 2840, 1344, 1600, 1582, 1509, 1454, 1432, 1383, 1268, 1135, 1022, 735, 691 cm−1; 1H NMR (CDCl3) δ 8.20 (s, 1 H), 7.50-7.33 (m, 11 H), 7.20 (dd, J = 1.6, 8.3 Hz, 1 H), 6.96 (d, J = 8.3 Hz, 1 H), 5.21 (s, 4 H), 3.73 (td, J = 1.2, 6.2 Hz, 2 H), 3.51 (t, J = 6.6 Hz, 2 H), 2.30-2.22 (m, 2 H); ESIMS m/z (rel intensity) 438/440 (MH+, 100/92).
cis-[3′,4′-(Dibenzyloxy)phenyl]-N-3-(bromopropyl)]-4-carboxy-3,4-dihydro-6,7-dimethoxy-3-1(2H)-isoquinolone (49)
Anhydride 11 (1.0 g, 4.50 mmol) was diluted with CHCl3 (30 mL), and the solution was cooled to 0 °C. The Schiff base 48 (1.97 g, 4.50 mmol) was added slowly as a pre-cooled solution in CHCl3 (20 mL) and quantitatively transferred with CHCl3 (3 mL). The mixture was stirred at 0 °C for 2 h, followed by 10 h at room temperature, upon which a precipitate had formed. Hexanes (50 mL) were added, and the precipitate was collected, washed with 20% hexanes in CHCl3 (50 mL) and dried to yield the title compound as a light yellow amorphous solid (2.34 g, 79%): mp 168–169 °C (dec). IR (KBr) 3583, 2937, 1736, 1597, 1513, 1426, 1286, 1265, 1138, 1023, 665 cm−1; 1H NMR (CDCl3 + DMSO-d6, 300 MHz) δ 7.53 (s, 1 H), 7.30-7.20 (m, 10 H), 7.10 (s, 1 H), 6.70 (s, 1 H), 6.63 (s, 1 H), 4.94 (s, 2 H), 4.90 (d, J = 7.2 Hz, 1 H), 4.83 (s, 2 H), 4.50 (d, J = 7.4 Hz, 1 H) 3.83 (s, 3 H), 3.76 (s, 3 H), 3.33 (m, 2 H), 2.93 (m, 2 H), 2.00 (m, 2 H). ); ESIMS m/z (rel intensity) 660/662 (MH+, 100/97). Anal. Calcd for C35H34BrNO: C, 63.64; H, 5.19; N, 2.12. Found: C, 63.46; H, 5.21; N, 2.12.
8,9-Dibenzyloxy-6-(3-bromopropyl)-5,6-dihydro-2,3-dimethoxy-5,11-dioxo-11H-indeno-[ 1,2-c]isoquinoline (50)
Compound 49 (1.00 g, 1.51 mmol) was cooled to −4 °C and then diluted with SOCl2 (25 mL) and stirred for 3 h. After that the reaction mixture was stirred at room temperature for additional 4 h, upon which it had become a bright reddish-purple color. The SOCl2 was evaporated, the residue was dissolved in CHCl3 (40 mL), and the solution was neutralized by the slow addition of sat. aq NaHCO3 (40 mL). The layers were separated, and the aqueous layer was extracted with CHCl3 (30 mL). The organic layers were washed with H2O (50 mL) and sat. aq NaCl (50 mL). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by flash column chromatography (SiO2), eluting with CHCl3 to yield a reddish-purple solid (0.610 g, 63%): mp 214–215 °C. IR (KBr) 2939, 1688, 1643, 1613, 1591, 1552, 1494, 1432, 1301, 1265, 1207, 1015, 737, 665 cm−1; 1H NMR (CDCl3, 300 MHz) δ 7.99 (s, 1 H), 7.58 (s, 1 H), 7.46-7.29 (m, 10 H), 7.20 (s, 1 H), 7.18 (s, 1 H), 5.26 (s, 2 H), 5.22 (s, 2 H), 4.51 (t, J = 7.3 Hz, 2 H), 4.02 (s, 3 H), 3.95 (s, 3 H), 3.58 (t, J = 6.2 Hz, 2 H), 2.33 (m, 2 H); EIMS m/z (rel intensity) 639/641 (M+, 2/2).
8,9-Dibenzyloxy-5,6-dihydro-6-[3-(1H-imidazol-1-yl)propyl]-2,3-dimethoxy-5,11-dioxo-11H-indeno-[1,2-c]isoquinoline (51a)
Compound 50 (0.500 g, 0.782 mmol) was dissolved in dioxane (75 mL), NaI (0.971 g, 6.52 mmol) and imidazole (0.443 g, 6.52 mmol) were added at room temperature, and the reaction mixture was heated at 70 °C for 20 h. The reaction mixture was cooled to room temperature and concentrated, H2O (75 mL) was added, and the mixture was extracted with CHCl3 (2 × 100 mL). The combined organic layer was washed with sat. aq. NaCl (50 mL). The organic layer was concentrated and the crude mixture was purified by silica gel flash column chromatography, eluting with 2% MeOH in CHCl3, to yield the product (0.320 g, 67%) as purple solid: mp 235–236 °C. IR (KBr) 2961, 1689, 1648, 1589, 1553, 1493, 1433, 1398, 1300, 1206, 1021, 747 cm−1; 1H NMR (CDCl3, 300 MHz) δ 7.97 (s, 1 H), 7.57 (s, 1 H), 7.51 (s, 1 H), 7.47-7.33 (m, 10 H), 7.20 (s, 1 H), 7.06 (s, 1 H), 6.95 (s, 1 H), 6.72 (s, 1 H), 5.23 (s, 2 H), 5.19 (s, 2 H), 4.35 (t, J = 6.3 Hz, 2 H), 4.02 (s, 3 H), 3.96 (m, 2 H), 3.94 (s, 3 H), 1.98 (m, 2 H); ESIMS m/z (rel intensity) 628 (MH+, 100), 1255 (2MH+, 26). Anal. Calcd for C38H33N3O6: C, 72.71; H, 5.30; N, 6.69. Found: C, 72.30; H, 5.29; N, 6.62.
8,9-Dibenzyloxy-5,6-dihydro-2,3-dimethoxy-6-[3-(N-morpholino)propyl-5,11-dioxo-11H-indeno-[1,2-c]isoquinoline (51b)
Compound 50 (0.250 g, 0.392 mmol) was dissolved in dioxane (50 mL), NaI (0.466 g, 3.92 mmol) and morpholine (0.271 g, 3.12 mmol) were added at room temperature, and the reaction mixture was heated at 65 °C for 20 h. The reaction mixture was cooled to room temperature and concentrated, H2O (75 mL) was added and the mixture was extracted with CHCl3 (2 × 100 mL). The combined organic layer was washed with sat. aq. NaCl (50 mL). The organic layer was concentrated and the crude mixture was purified by flash column chromatography (SiO2), eluting with 1% MeOH in CHCl3, to yield the product (0.150 g, 60%) as brown solid: mp 208–210 °C. IR (KBr) 2827, 1676, 1649, 1612, 1591, 1552, 1496, 1380, 1300, 1254, 1206, 1117, 870, 697 cm−1; 1H NMR (CDCl3, 300 MHz) δ 8.01 (s, 1 H), 7.60 (s, 1 H), 7.46 (m, 10 H), 7.22 (s, 1 H), 7.03 (s, 1 H), 5.24 (s, 1 H), 5.23 (s, 1 H), 4.39 (m, 2 H), 4.03 (s, 3 H), 3.95 (s, 3 H), 3.62 (m, 4 H), 2.41 (m, 6 H), 1.87 (m, 2 H); ESIMS m/z (rel intensity) 647 (MH+, 100). Anal. Calcd for C39H38N2O7·0.75 H2O: C, 70.95; H, 6.03; N, 4.24. Found: C, 71.04; H, 5.86; N, 4.23
6-(3-(1H-Imidazol-1-yl)propyl)-8,9-dihydroxy-2,3-dimethoxy-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (52a)
Compound 51a (0.100 g, 0.159 mmol) was diluted in 48% aq HBr (25 mL) and the solution was heated at 70 °C for 15 h. After the mixture was cooled to room temperature, chloroform and acetone (10 mL each) were added to the reaction mixture and removed on the rotary evaporator three times. The leftover solid in water was cooled to 0 °C and then filtered off. The solid was washed with 10% MeOH in chloroform (25 mL) and with acetone (50 mL) to afford catechol 52a (0.068 g, 81%) as light brown solid: mp 273–274 °C. IR (KBr) 3230, 1688, 1628, 1577, 1557, 1425, 1303, 1247, 865, 782, 665 cm−1; 1H NMR (DMSO-d6, 300 MHz) δ 9.14 (s, 1 H), 7.91 (s, 1 H), 7.84 (s, 1 H), 7.67 (s, 1 H), 7.47(s, 1 H), 7.07 (s, 1 H), 6.94 (s, 1 H), 4.42 (m, 4 H), 3.88 (s, 3 H), 3.84 (s, 3 H), 2.33 (m, 2 H); 13C NMR (DMSO-d6, 125 MHz) δ 190.1, 161.6, 154.4, 154.3, 149.0, 148.3, 146.5, 135.5, 128.3, 127.8, 126.6, 121.9, 119.8, 115.7, 112.8, 111.6, 107.7, 106.2, 102.1, 55.6, 55.4, 46.5, 40.8, 29.9; ESIMS m/z (rel intensity) 448 (MH+, 100); HRESIMS m/z 448.1514 (MH+), calcd for C24H22N3O6 448.1509. Purity was estimated to be 98.1% by HPLC in 85% MeOH/1% TFA – 15% H2O and 98.3% in 70% MeOH/1% TFA – 30% H2O.
8,9-Dihydroxy-2,3-dimethoxy-6-(3-morpholinopropyl)-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (52b)
Compound 51b (0.125 g, 0.195 mmol) was dissolved in mixture of MeOH and THF (20 and 10 mL) and hydrogenated (with a balloon) in the presence of 10% Pd-C (10 mg) for 24 h. The Pd-C was filtered off and the filtrate was concentrated and purified by flash column chromatography (SiO2), eluting with 7% MeOH in CHCl3, to provide the product 52b (0.041 g, 47%) as brown solid: mp 285–287 °C. IR (KBr) 3217, 2927, 1645, 1614, 1591, 1550, 1520, 1471, 1354, 1322, 1253, 1216, 870, 785, 657 cm−1; 1H NMR (CDCl3 + CD3OD, 300 MHz) δ 7.88 (s, 1 H), 7.49 (s, 1 H), 7.01 (s, 1 H), 6.90 (s, 1 H), 4.36 (m, 2 H), 3.94 (s, 1 H), 3.88 (s, 1 H), 3.68 (t, J = 4.4 Hz, 2 H), 2.51 (m, 6 H), 2.02 (m, 2 H); 13C NMR (DMSO-d6, 125 MHz) δ 190.2, 162.0, 154.8, 154.5, 149.4, 148.7, 146.8, 128.6, 128.2, 126.9, 116.1, 113.1, 111.9, 108.1, 106.5, 102.4, 63.7, 55.9, 55.8, 53.7, 51.4, 41.1, 23.9; ESIMS m/z (rel intensity) 467 (MH+, 100); HRESIMS m/z 467.1824 (MH+), calcd for C25H27N4O7 467.1818; purity was estimated to be 97.7% by HPLC in 85% MeOH/1% TFA – 15% H2O and 98.8% in 70% MeOH/1% TF– 30% H2O.
Metabolism of LMP400 and LMP776 by Human Liver Microsomes.38
Pooled human liver microsomes were purchased from Life Technologies (Grand Island, NY). Other chemicals were purchased from Sigma-Aldrich (St. Louis, MO). All organic solvents were HPLC grade or higher and were purchased from Fisher Scientific (Hanover Park, IL).
Compound 7 (LMP400) or 6 (LMP776) (10 μM) was incubated at 37 °C with pooled human liver microsomes (15 donors, mixed gender) containing 1 mg/mL of microsomal protein and 50 mM phosphate buffer at pH 7.4 in a total volume of 200 μL. After a 5 min preincubation, 1 mM NADPH was added to initiate reaction, and the mixture was incubated for an additional 60 min. The reaction was stopped by chilling the mixture on ice and by addition of 20 μL of ice-cold acetonitrile/water/formic acid (86:10:4, v/v/v) to precipitate proteins. Samples were centrifuged, supernatants were removed, evaporated to dryness under nitrogen, and the residues were dissolved in the mobile phase prior to analysis using liquid chromatography-mass spectrometry (LC-MS) and LC-tandem mass spectrometry (LC-MS/MS). Control incubations were identical except for the elimination of microsomal protein or NADPH.
LC-MS and LC-MS/MS
Analyses of LMP400 or LMP776 and their metabolites were carried out using a Waters (Milford, MA) 2690 HPLC system equipped with a Waters Xterra 2.1×100 C18 column. The solvent system consisted of a linear gradient from 0.1% formic acid in water to methanol as follows: 20% to 45% methanol over 10 min, 45% to 90% methanol over 1 min and isocratic 90% methanol for another 2 min. The column was equilibrated with 20% methanol for at least 10 min between analyses. The flow rate was 0.2 mL/min, and the column temperature was 33 °C. The HPLC was interfaced with a high resolution Waters Q-TOF Synapt hybrid quadrupole/time-of-flight mass spectrometer, and positive ion electrospray was used for sample ionization. For accurate mass measurements, leucine enkephalin ([M+H]+ of m/z 556.2771) was introduced post-column as a lock mass. The mass accuracy obtained was < 5 ppm. Data were acquired from m/z 100–700. Tandem mass spectra were acquired at a collision energy of 20 eV using argon as the collision gas at a pressure of 2.0×10−5 mbar.
Topoisomerase I-Mediated DNA Cleavage Reactions
Human recombinant Top1 was purified from Baculovirus as previously described.68 DNA cleavage reactions were prepared as previously reported26 with the exception of the DNA substrate.69 Briefly, a 117-bp DNA oligonucleotide (Integrated DNA Technologies) encompassing the previously identified Top1 cleavage sites in the 161-bp fragment from pBluescript SK(−) phagemid DNA was employed. This 117-bp oligonucleotide contains a single 5′-cytosine overhang, which was 3′-end labeled by fill-in reaction with [α-32P]-dGTP in React 2 buffer (50 mM Tris-HCl, pH 8.0, 100 mM MgCl2, 50 mM NaCl) with 0.5 units of DNA polymerase I (Klenow fragment, New England BioLabs). Unincorporated 32P-dGTP was removed using mini Quick Spin DNA columns (Roche, Indianapolis, IN), and the eluate containing the 3′-end-labeled DNA substrate was collected. Approximately 2 nM of radiolabeled DNA substrate was incubated with recombinant Top1 in 10 μL of reaction buffer [10 mM Tris-HCl (pH 7.5), 50 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, and 15 μg/mL BSA] at room temperature for 20 min in the presence of various concentrations of compounds. The reactions were terminated by adding SDS (0.5% final concentration) followed by the addition of two volumes of loading dye (80% formamide, 10 mM sodium hydroxide, 1 mM sodium EDTA, 0.1% xylene cyanol, and 0.1% bromophenol blue). Aliquots of each reaction were subjected to 20% denaturing PAGE. Gels were dried and visualized by using a Phosphoimager and ImageQuant software (Molecular Dynamics). For simplicity, cleavage sites were numbered as previously described in the 161-bp fragment.68
Docking and Modeling Studies
The Top1 crystal structure for docking was prepared and the docking protocol was validated as previously described.46 The ternary complex centroid coordinates for docking were defined using the ligand in the Top1/topotecan crystal structure (PDB ID 1K4T) as the center of the binding pocket (x = 21.3419, y = −3.9888, z = 28.2163). The ligand was then deleted. Indenoisoquinolines to be modeled were constructed in SYBYL. Atom types were assigned using SYBYL atom typing, hydrogens were added, and the ligands were minimized by conjugate gradient method using the MMFF94s force field with MMFF94 charges, a distance-dependent dielectric function, and a 0.01 kcal/mol*Å energy gradient convergence criterion. Each ligand was docked into the mutant crystal structure using GOLD 3.2 using default parameters and the coordinates defined by the crystal structure as described above. The top four poses for each ligand were examined (as lower-ranked poses with much lower GoldScores were sometimes “flipped” or rotated binding modes). The highest-ranked poses for these ligands were merged into the crystal structure, and the entire complex was subsequently subjected to minimization using a standard Powell method, the MMFF94s force field and MMFF94 charges, a distance-dependant dielectric function, and a 0.05 kcal/mol*Å energy gradient convergence criterion. During the minimization, the ligand and a 7 Å sphere surrounding the ligands were allowed to move, while the structures outside this sphere were frozen in an aggregate.
Acknowledgments
This work was facilitated by the National Institutes of Health (NIH) through support with Research Grant UO1 CA89566. M.A.C. thanks Dr. Karl Wood for consultation and valuable discussion. Joe Fornefeld of Midwest Microlab and Dr. Martín Conda-Sheridan (Purdue) provided additional assistance. This project has also been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Abbreviations Used
- APCI
atmospheric pressure chemical ionization
- GOLD
Genetic Optimisation for Ligand Docking (program)
- MDMA
3,4-methylenedioxy-N-methylamphetamine
- MDPPA
3′,4′-methylenedioxy-alpha-pyrrolidinopropiophenone
- MGM
mean-graph midpoint
- MMFF94
Merck Molecular Force Field 94
- NCI-60
National Cancer Institute 60-cell line screening assay
- Top1
topoisomerase 1
Footnotes
Notes: The authors declare no competing financial interests.
References
- 1.Pommier E. Topoisomerase I Inhibitors: Camptothecins and Beyond. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 2.Stewart L, Redinbo MR, Qiu X, Hol WGJ, Champoux JJ. A Model for the Mechanism of Human Topoisomerase I. Science. 1998;279:1534–1541. doi: 10.1126/science.279.5356.1534. [DOI] [PubMed] [Google Scholar]
- 3.Wang JC. Cellular Roles of DNA Topoisomerases: A Molecular Perspective. Nat Rev Mol Cell Biol. 2002;3:430–440. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 4.Pommier Y. DNA Topoisomerase I Inhibitors: Chemistry, Biology, and Interfacial Inhibition. Chem Rev. 2009;109:2894–2902. doi: 10.1021/cr900097c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pommier Y, Barcelo JA, Rao VA, Sordet O, Jobson AG, Thibaut L, Miao ZH, Seiler JA, Zhang H, Marchand C, Agama K, Nitiss JL, Redon C. Repair of Topoisomerase I-Mediated DNA Damage. In: Moldave K, editor. Prog Nucleic Acid Res Mol Biol. Vol. 81. 2006. p. 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bailly C. Topoisomerase I Poisons and Suppressors as Anticancer Drugs. Curr Med Chem. 2000;7:39–58. doi: 10.2174/0929867003375489. [DOI] [PubMed] [Google Scholar]
- 7.Hsiang YH, Hertzberg R, Hecht S, Liu LF. Camptothecin Induces Protein-linked DNA Breaks via Mammalian DNA Topoisomerase I. J Biol Chem. 1985;260:14873–14878. [PubMed] [Google Scholar]
- 8.Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB, Stewart L. The Mechanism of Topoisomerase I Poisoning by a Camptothecin Analogue. Proc Natl Acad Sci US A. 2002;99:15387–15392. doi: 10.1073/pnas.242259599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sordet O, Khan QA, Kohn KW, Pommier Y. Apoptosis Induced by Topoisomerase Inhibitors. Curr Med Chem - Anticancer Agents. 2003;3:271–290. doi: 10.2174/1568011033482378. [DOI] [PubMed] [Google Scholar]
- 10.Teicher B. Next Generation of Topoisomerase I Inhibitors: Rationale and Biomarker Strategies. Biochem Pharmacol. 2008;75:1262–1271. doi: 10.1016/j.bcp.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 11.Wall ME, Wani MC, Cook CE, Palmer KH, McPhail AT, Sim GA. Plant Antitumor Agents. I. The Isolation and Structure of Camptothecin, a Novel Alkaloidal Leukemia and Tumor Inhibitor from Camptotheca acuminata. J Am Chem Soc. 1966;88:3888–3890. [Google Scholar]
- 12.Thomas CJ, Rahier NJ, Hecht SM. Camptothecin: Current Perspectives. Bioorg Med Chem. 2004;12:1585–1604. doi: 10.1016/j.bmc.2003.11.036. [DOI] [PubMed] [Google Scholar]
- 13.Pommier Y, Marchand C. Interfacial Inhibitors: Targeting Macromolecular Complexes. Nat Rev Drug Discovery. 2012;11:25–36. doi: 10.1038/nrd3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schaeppi U, Fleischman RW, Cooney DA. Toxicity of Camptothecin (NSC-100880) Cancer Chemother Rep, Part 3. 1974;5:25–36. [PubMed] [Google Scholar]
- 15.Luzzio MJ, Besterman JM, Emerson DL, Evans MG, Lackey K, Leitner PL, McIntyre G, Morton B, Myers PL, Peel M, Sisco JM, Sternbach DD, Tong WQ, Truesdale A, Uehling DE, Vuong A, Yates J. Synthesis and Antitumor-Activity of Novel Water-Soluble Derivatives of Camptothecin as Specific Inhibitors of Topoisomerase-I. J Med Chem. 1995;38:395–401. doi: 10.1021/jm00003a001. [DOI] [PubMed] [Google Scholar]
- 16.Mi Z, Burke TG. Differential Interactions of Camptothecin Lactone and Carboxylate Forms with Human Blood Components. Biochemistry. 1994;33:10325–10336. doi: 10.1021/bi00200a013. [DOI] [PubMed] [Google Scholar]
- 17.Paull KD, Hamel E, Malspeis L. Prediction of Biochemical Mechanism of Action from the In Vitro Antitumor Screen of the National Cancer Institute. In: Foye WO, editor. Cancer Chemotherapeutic Agents. American Chemical Society; Washington, DC: 1995. pp. 9–45. [Google Scholar]
- 18.Paull KD, Shoemaker RH, Hodes L, Monks A, Scudiero DA, Rubinstein L, Plowman J, Boyd MR. Display and Analysis of Patterns of Differential Activity of Drugs Against Human Tumor Cell Lines: Development of Mean Graph and COMPARE Algorithm. J Natl Cancer Inst. 1989;81:1088–1092. doi: 10.1093/jnci/81.14.1088. [DOI] [PubMed] [Google Scholar]
- 19.Kohlhagen G, Paull K, Cushman M, Nagafuji P, Pommier Y. Protein-Linked DNA Strand Breaks Induced by NSC 314622, a Novel Noncamptothecin Topoisomerase I Poison. Mol Pharmacol. 1998;54:50–58. doi: 10.1124/mol.54.1.50. [DOI] [PubMed] [Google Scholar]
- 20.Pommier Y, Cushman M. The Indenoisoquinoline Noncamptothecin Topoisomerase I Inhibitors: Update and Perspectives. Mol Cancer Ther. 2009;8:1008–1014. doi: 10.1158/1535-7163.MCT-08-0706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagarajan M, Morrell A, Fort BC, Meckley MR, Antony S, Kohlhagen G, Pommier Y, Cushman M. Synthesis and Anticancer Activity of Simplified Indenoisoquinoline Topoisomerase I Inhibitors Lacking Substituents on the Aromatic Rings. J Med Chem. 2004;47:5651–5661. doi: 10.1021/jm040025z. [DOI] [PubMed] [Google Scholar]
- 22.Nagarajan M, Morrell A, Ioanoviciu A, Antony S, Kohlhagen G, Agama K, Hollingshead M, Pommier Y, Cushman M. Synthesis and Evaluation of Indenoisoquinoline Topoisomerase I Inhibitors Substituted with Nitrogen Heterocycles. J Med Chem. 2006;49:6283–6289. doi: 10.1021/jm060564z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morrell A, Antony S, Kohlhagen G, Pommier Y, Cushman M. Synthesis of Nitrated Indenoisoquinolines as Topoisomerase I Inhibitors. Bioorg Med Chem Lett. 2004;14:3659–3663. doi: 10.1016/j.bmcl.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 24.Antony S, Jayaraman M, Laco G, Kohlhagen G, Kohn KW, Cushman M, Pommier Y. Differential Induction of Topoisomerase I-DNA Cleavage Complexes by the Indenoisoquinoline MJ-III-65 (NSC 706744) and Camptothecin: Base Sequence Analysis and Activity against Camptothecin-Resistant Topoisomerase I. Cancer Res. 2003;63:7428–7435. [PubMed] [Google Scholar]
- 25. [accessed February 17, 2012];A Phase I Study of Indenoisoquinolines LMP400 and LMP776 in Adults With Relapsed Solid Tumors and Lymphomas. http://clinicaltrials.gov/ct2/show/study/NCT01051635.
- 26.Antony S, Agama KK, Miao ZH, Takagi K, Wright MH, Robles AI, Varticovski L, Nagarajan M, Morrell A, Cushman M, Pommier Y. Novel Indenoisoquinolines NSC 725776 and NSC 724998 Produce Persistent Topolsomerase I Cleavage Complexes and Overcome Multidrug Resistance. Cancer Res. 2007;67:10397–10405. doi: 10.1158/0008-5472.CAN-07-0938. [DOI] [PubMed] [Google Scholar]
- 27.Chrencik JE, Staker BL, Burgin AB, Pourquier P, Pommier Y, Stewart L, Redinbo MR. Mechanisms of Camptothecin Resistance by Human Topoisomerase I Mutations. J Mol Biol. 2004;339:773–784. doi: 10.1016/j.jmb.2004.03.077. [DOI] [PubMed] [Google Scholar]
- 28.Relling MV, Nemec J, Schuetz EG, Schuetz JD, Gonzalez FJ, Korzekwa KR. O-Demethylation of Epipodophyllotoxins is Catalyzed by Human Cytochrome P450 3A4. Mol Pharmacol. 1994;45:352–358. [PubMed] [Google Scholar]
- 29.Küpfer A, Schmid B, Pfaff G. Pharmacogenetics of Dextromethorphan O-Demethylation in Man. Xenobiotica. 1986;16:421–433. doi: 10.3109/00498258609050249. [DOI] [PubMed] [Google Scholar]
- 30.Paar WD, Frankus P, Dengler HJ. The Metabolism of Tramadol by Human Liver Microsomes. Clin Investig. 1992;70:708–710. doi: 10.1007/BF00180294. [DOI] [PubMed] [Google Scholar]
- 31.Qiu F, Zhu ZY, Kang N, Piao SJ, Qin GY, Yao XS. Isolation and Identification of Urinary Metabolites of Berberine in Rats and Humans. Drug Metab Dispos. 2008;36:2159–2165. doi: 10.1124/dmd.108.021659. [DOI] [PubMed] [Google Scholar]
- 32.Sooryakumar D, Dexheimer TS, Teicher BA, Pommier Y. Molecular and Cellular Pharmacology of the Novel Noncamptothecin Topoisomerase I Inhibitor Genz-644282. Mol Cancer Ther. 2011;10:1490–1499. doi: 10.1158/1535-7163.MCT-10-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Benedetti MS, Malnoe A, Broillet AL. Absorption, Metabolism, and Excretion of Safrole in the Rat and Man. Toxicology. 1977;7:69–83. doi: 10.1016/0300-483x(77)90039-7. [DOI] [PubMed] [Google Scholar]
- 34.Klungsoyr J, Scheline RR. Metabolism of Piperonal and Piperonyl Alcohol in the Rat with Special Reference to the Scission of the Methylenedioxy Group. Acta Pharm Suec. 1984;21:67–72. [PubMed] [Google Scholar]
- 35.Lim HK, Foltz RL. In Vitro and In Vivo Metabolism of 3,4-(Methylenedioxy)amphetamine in the Rat: Identification of Metabolites Using an Ion Trap Detector. Chem Res Toxicol. 1988;1:370–378. doi: 10.1021/tx00006a008. [DOI] [PubMed] [Google Scholar]
- 36.Springer D, Fritschi G, Maurer HH. Metabolism and Toxicological Detection of the New Designer Drug 3 ′,4 ′-Methylenedioxy-alpha-pyrrolidinopropiophenone Studied in Urine Using Gas Chromatography-Mass Spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2003;793:377–388. doi: 10.1016/s1570-0232(03)00350-7. [DOI] [PubMed] [Google Scholar]
- 37.Novotna R, Wsol V, Xiong G, Maser E. Inactivation of the Anticancer Drugs Doxorubicin and Oracin by Aldo-keto Rductase (AKR) 1C3. Toxicol Lett. 2008;181:1–6. doi: 10.1016/j.toxlet.2008.06.858. [DOI] [PubMed] [Google Scholar]
- 38.Chen L, Conda-Sheridan M, Reddy PVN, Morrell A, Park EJ, Kondratyuk TP, Pezzuto JM, van Breemen R, Cushman M. Identification, Synthesis, and Biological Evaluation of the Metabolites of 3-Amino-6-(3′-aminopropyl)-5H-indeno[1,2-c]isoquinoline-5,11-(6H)dione (AM6–36), a Promising Rexinoid Lead Compound for the Development of Cancer Chemotherapeutic and Chemopreventive Agents. J Med Chem. 2012;55:5965–5981. doi: 10.1021/jm3006806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wall ME, Wani MC, Nicholas AW, Manikumar G, Tele C, Moore L, Truesdale A, Leitner P, Besterman JM. Plant Antitumor Agents. 30. Synthesis and Structure-Activity of Novel Camptothecin Analogs. J Med Chem. 1993;36:2689–2700. doi: 10.1021/jm00070a013. [DOI] [PubMed] [Google Scholar]
- 40.Wani MC, Nicholas AW, Manikumar G, Wall ME. Plant Antitumor Agents. 25. Total Synthesis and Antileukemic Activity of Ring A Substituted Camptothecin Analogues. Structure-Activity Correlations. J Med Chem. 1987;30:1774–1779. doi: 10.1021/jm00393a016. [DOI] [PubMed] [Google Scholar]
- 41.Bailly C, Riou JF, Colson P, Houssier C, RodriguesPereira E, Prudhomme M. DNA Cleavage by Topoisomerase I in the Presence of Indolocarbazole Derivatives of Rebeccamycin. Biochemistry. 1997;36:3917–3929. doi: 10.1021/bi9624898. [DOI] [PubMed] [Google Scholar]
- 42.Fortune JM, Velea L, Graves DE, Utsugi T, Yamada Y, Osheroff N. DNA Topoisomerases as Targets for the Anticancer Drug TAS-103: DNA Interactions and Topoisomerase Catalytic Inhibition. Biochemistry. 1999;38:15580–15586. doi: 10.1021/bi991792g. [DOI] [PubMed] [Google Scholar]
- 43.Van HTM, Le QM, Lee KY, Lee ES, Kwon Y, Kim TS, Le TN, Lee SH, Cho WJ. Convenient Synthesis of Indeno[1,2c]isoquinolines as Constrained Forms of 3-Arylisoquinolines and Docking Study of a Topoisomerase I Inhibitor into DNA-Topoisomerase I Complex. Bioorg Med Chem Lett. 2007;17:5763–5767. doi: 10.1016/j.bmcl.2007.08.062. [DOI] [PubMed] [Google Scholar]
- 44.Khadka DB, Cho WJ. 3-Arylisoquinolines as Novel Topoisomerase I Inhibitors. Bioorg Med Chem. 2011;19:724–734. doi: 10.1016/j.bmc.2010.10.057. [DOI] [PubMed] [Google Scholar]
- 45.Ryckebusch A, Garcin D, Lansiaux A, Goossens JF, Baldeyrou B, Houssin R, Bailly C, Henichart JP. Synthesis, Cytotoxicity, DNA Interaction, and Topoisomerase II Inhibition Properties of Novel Indeno[2,1-c]quinolin-7-one and indeno[1,2-c]isoquinolin-5,11-dione Derivatives. J Med Chem. 2008;51:3617–3629. doi: 10.1021/jm800017u. [DOI] [PubMed] [Google Scholar]
- 46.Peterson KE, Cinelli MA, Morrell AE, Mehta A, Dexheimer TS, Agama K, Antony S, Pommier Y, Cushman M. Alcohol-, Diol-, and Carbohydrate-Substituted Indenoisoquinolines as Topoisomerase I Inhibitors: Investigating the Relationships Involving Stereochemistry, Hydrogen Bonding, and Biological Activity. J Med Chem. 2011;54:4937–4953. doi: 10.1021/jm101338z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lebrun S, Couture A, Deniau E, Grandclaudon P. Suzuki-Miyaura Cross-Coupling and Ring-closing Metathesis: a Strategic Combination to the Synthesis of Indeno[1,2-c]isoquinolin-5,11-diones. Tetrahedron Lett. 2011;52:1481–1484. [Google Scholar]
- 48.Cushman M, Gentry J, Dekow FW. Condensation of Imines with Homophthalic Anhydrides. A Convergent Synthesis of cis- and trans-13-Methyltetrahydroprotoberberines. J Org Chem. 1977;42:1111–1116. doi: 10.1021/jo00427a001. [DOI] [PubMed] [Google Scholar]
- 49.Cushman M, Cheng L. Stereoselective Oxidation by Thionyl Chloride Leading to the Indeno[1,2-c]isoquinoline System. J Org Chem. 1978;43:3781–3783. [Google Scholar]
- 50.Finkelstein J, Brossi A. 6.7-Dimethoxy-3-isochromanone. Org Syn. 1988;6:471. [Google Scholar]
- 51.Rastogi SN, Bindra JS, Anand N. α-Methyldopa in a Rigid Framework: 3-Amino-3-methyl-6,7-dihydroxy-3,4-dihydroxycoumarins and 2-Amino-6,7-dihydroxytetralin-2-carboxylic Acids. Indian J Chem. 1971;9:1175–1182. [Google Scholar]
- 52.Nonni AJ, Dence CW. The Synthesis of 1.2-Diarylpropane-1,3-diols via the Ivanoff Reaction. J Wood Chem Technol. 1982;2:161–168. [Google Scholar]
- 53.Patra A, Ghosh G, Mukhopadhyay PK. Studies on Some Azlactones and Isochromanones. Synthesis and Spectral Characteristics. J Indian Chem Soc. 1987;64:414–417. [Google Scholar]
- 54.Spangler RJ, Beckmann BG, Kim JH. A New Synthesis of Benzocyclobutenes. Thermal and Electron Impact Induced Decomposition of 3-Isochromanones. J Org Chem. 1977;42:2989–2996. [Google Scholar]
- 55.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. J Natl Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 56.Boyd MR, Paull KD. Some Practical Considerations and Applications of the National Cancer Institute In Vitro Anticancer Drug Discovery Screen. Drug Development Res. 1995;34:91–109. [Google Scholar]
- 57.Morrell A, Placzek M, Parmley S, Grella B, Antony S, Pommier Y, Cushman M. Optimization of the Indenone Ring of Indenoisoquinoline Topoisomerase I Inhibitors. J Med Chem. 2007;50:4388–4404. doi: 10.1021/jm070307+. [DOI] [PubMed] [Google Scholar]
- 58.Staker BL, Feese MD, Cushman M, Pommier Y, Zembower D, Stewart L, Burgin AB. Structures of Three Classes of Anticancer Agents Bound to the Human Topoisomerase I-DNA Covalent Complex. J Med Chem. 2005;48:2336–2345. doi: 10.1021/jm049146p. [DOI] [PubMed] [Google Scholar]
- 59.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and Validation of a Genetic Algorithm for Flexible Docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 60.Jones G, Willett P, Glen RC. Molecular Recognition of Receptor Sites Using a Genetic Algorithm with a Description Desolvation. J Mol Biol. 1995;245:43–53. doi: 10.1016/s0022-2836(95)80037-9. [DOI] [PubMed] [Google Scholar]
- 61.Cinelli MA, Morrell AE, Dexheimer TS, Agama K, Agrawal S, Pommier Y, Cushman M. The Structure-Activity Relationships of A-Ring-Substituted Aromathecin Topoisomerase I Inhibitors Strongly Support a Camptothecin-like Binding Mode. Bioorg Med Chem. 2010;18:5535–5552. doi: 10.1016/j.bmc.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zembower DE, Zhang HP, Lineswala JP, Kuffel MJ, Aytes SA, Ames MM. Indolocarbazole Poisons of Human Topoisomerase I: Regioisomeric Analogues of ED-110. Bioorg Med Chem Lett. 1999;9:145–150. doi: 10.1016/s0960-894x(98)00710-0. [DOI] [PubMed] [Google Scholar]
- 63.Duclos RI, Tung JS, Rapoport H. A High-Yield Modification of he Pschorr Phenanthene Synthesis. J Org Chem. 1984;49:5243–5246. [Google Scholar]
- 64.Guthrie DA, Frank AW, Purves CB. Studies in the Polyoxyphenol Series. XI. Synthesis of Papaverine and Papaveraldine by the Pomerinz-Fritsch Method. Can J Chem. 1955;33:729–742. [Google Scholar]
- 65.Ploypradith P, Cheryklin P, Niyomtham N, Bertoni DR, Ruchirawat S. Solid-Supported Acids as Mild and Versatile Reagents for the Deprotection of Aromatic Ethers. Organic Lett. 2007;9:2637–2640. doi: 10.1021/ol070781+. [DOI] [PubMed] [Google Scholar]
- 66.Prasad D, Chaudhury DN. Synthesis of 3,4-Dihydro-7-hydroxy-6-methoxyisocoumarin. J Indian Chem Soc. 1962;39:672–676. [Google Scholar]
- 67.Bolek DJ, Guetschow M. Preparation of 4,6,3′,4′-Tetrasubstituted Aurones via Aluminum Oxide-Catalyzed Condensation. J Heterocycl Chem. 2005;42:1399–1403. [Google Scholar]
- 68.Pourquier P, Ueng LM, Fertala J, Wang D, Park HJ, Essigmann JM, Bjornsti MA, Pommier Y. Induction of Reversible Complexes Between Eukaryotic DNA Topoisomerase I and DNA-containing Oxidative Base Damages. 7,8-Dihydro-8-Oxoguanine and 5-Hydroxycytosine. J Biol Chem. 1999;274:8516–8523. doi: 10.1074/jbc.274.13.8516. [DOI] [PubMed] [Google Scholar]
- 69.Dexheimer TS, Pommier Y. DNA Cleavage Assay for the Identification of Topoisomerase I Inhibitors. Nat Protocol. 2008;3:1736–1750. doi: 10.1038/nprot.2008.174. [DOI] [PMC free article] [PubMed] [Google Scholar]