

Abstract
Human immunodeficiency virus type 2 (HIV-2) emerged in West Africa and has spread further to countries that share socio-historical ties with this region. However, viral origins and dispersal patterns at a global scale remain poorly understood. Here, we adopt a Bayesian phylogeographic approach to investigate the spatial dynamics of HIV-2 group A (HIV-2A) using a collection of 320 partial pol and 248 partial env sequences sampled throughout 19 countries worldwide. We extend phylogenetic diffusion models that simultaneously draw information from multiple loci to estimate location states throughout distinct phylogenies and explicitly attempt to incorporate human migratory fluxes. Our study highlights that Guinea-Bissau, together with Côte d’Ivoire and Senegal, have acted as the main viral sources in the early stages of the epidemic. We show that convenience sampling can obfuscate the estimation of the spatial root of HIV-2A. We explicitly attempt to circumvent this by incorporating rate priors that reflect the ratio of human flow from and to West Africa. We recover four main routes of HIV-2A dispersal that are laid out along colonial ties: Guinea-Bissau and Cape Verde to Portugal, Côte d’Ivoire and Senegal to France. Within Europe, we find strong support for epidemiological linkage from Portugal to Luxembourg and to the UK. We demonstrate that probabilistic models can uncover global patterns of HIV-2A dispersal providing sampling bias is taken into account and we provide a scenario for the international spread of this virus.
Introduction
Human immunodeficiency virus type 2 (HIV-2) is a rapidly evolving lentivirus with zoonotic origins in sooty mangabey monkeys (Cercocebus atys atys) that inhabit the coastal West African forests (Hahn et al., 2000). To date, eight distinct HIV-2 groups have been identified (HIV-2A–H), each originating from independent cross-species transmission events from sooty mangabeys naturally infected with simian immunodeficiency virus (SIVsmm) (Damond et al., 2004; Hahn et al., 2000). In several populations from West Africa, hunting, butchering bushmeat (LeBreton et al., 2007; Wolfe et al., 2005) and keeping these primates as pets (Chen et al., 1996) has been a common local practice, providing ample opportunity for cross-species transmission to humans. While HIV-2 groups C–H were only detected in one (Brennan et al., 1997; Chen et al., 1996, 1997; Gao et al., 1992) or two (Smith et al., 2008) individuals, HIV-2 groups A and B have established considerable spread in the human population, causing a total of 1–2 million cases in West Africa (UNAIDS, 2006).
A recent molecular survey detected SIVsmm strains that were closely related to both HIV-2A and HIV-2B groups in faecal samples from free-ranging sooty mangabeys in the Taï forest in Côte d’Ivoire (Santiago et al., 2005). While HIV-2B is less prevalent and mostly restricted to Côte d’Ivoire and Ghana (Pieniazek et al., 1999; Takehisa et al., 1997), serological surveys seem to suggest that Guinea-Bissau has been the clinical epicentre for the HIV-2A epidemic (da Silva et al., 2008; Poulsen et al., 1989). This group is also present in other West African countries and Nigeria (De Cock et al., 1990; Djomand et al., 1995; Esu-Williams et al., 1997; Mabey et al., 1988; Peeters et al., 1998; Ruelle et al., 2007; Takehisa et al., 1997; Wilkins et al., 1991; Zeh et al., 2005). Moreover, it has also spread to countries sharing socio-historical links with this region, particularly to Portugal and France (Smallman-Raynor & Cliff, 1991), but also to other European countries (Costarelli et al., 2008; Kvinesdal et al., 1992; Newmark, 1988; Treviño et al., 2011), the Americas (Ayanian et al., 1989; Neumann et al., 1989; Pieniazek et al., 1991; Popovsky et al., 1990), India (Grez et al., 1994) and South Korea (Nam et al., 2006). Most HIV-2 cases outside West Africa can be traced to immigration from high-prevalence countries (Smallman-Raynor & Cliff, 1991). However, the origins and global dispersal pathways, and whether there is on-going viral dispersal of HIV-2A outside West Africa are questions that remain elusive.
To gain insight into the spatiotemporal dynamics of pathogens, molecular epidemiology frequently resorts to the historical information embedded in molecular phylogenies along with the geographical distributions of the contemporaneous gene sequences (Holmes, 2008; Lemey et al., 2009; Nielsen & Beaumont, 2009; Pybus & Rambaut, 2009). Combined with models that accommodate nucleotide sequences with known sampling location, a recent phylogeographic framework infers ancestral locations at the nodes of time-measured phylogenetic trees (Lemey et al., 2009). Since many possible location exchanges are not expected to have occurred in reality, attempting to estimate rates of dispersal among all pairs of locations may suffer from overparameterization. To avoid this, a Bayesian stochastic search variable selection (BSSVS) procedure has been proposed to capitalize on those rates that contribute to the dispersal history with considerable support (Lemey et al., 2009).
In an effort to uncover the origins and spatiotemporal diffusion of HIV-2 group A, we analyse all available pol and env sequences for which time and location of sampling are known. Because separate gene analyses reveal a strong impact of sampling heterogeneity on the estimation of the ancestral root location, we adopt a flexible asymmetrical diffusion approach that draws inference from multiple independent loci simultaneously and uses prior specifications to incorporate immigration fluxes. To determine the patterns of HIV-2 dispersal at a global scale, we performed a Bayesian phylogeographic analysis of HIV-2 sequence data by incorporating isolates from eight West African countries, Spain, France, Belgium, Luxembourg, Sweden, India, South Korea and Brazil and previously unpublished data from Portugal, UK and Italy.
Results
Temporal and geographical signal of the HIV-2A global env and pol datasets
Our analysis of the temporal signal corroborates that faster evolving genes contain more temporal information for molecular clock calibration (Table 1) (Wertheim & Worobey, 2009). Particularly, the env dataset exhibited stronger temporal signal [ln Bayes Factor (BF) = 12.9 favouring a model that incorporates sampling dates over a model that ignores sampling dates, see Methods] compared with the pol dataset (ln BF = 2.5), supporting that HIV-2 group A originated around 1938 [95 % Bayesian credible intervals (BCI) 1928, 1947]. This is in line with previous estimates obtained using larger alignments (Lemey et al., 2003; Wertheim & Worobey, 2009). We revisited the effective population size dynamics of HIV-2A through time by applying a relaxed lognormal molecular clock model with a flexible Bayesian skyride demographic prior (Minin et al., 2008) to the most informative dataset. After an initial period of constant growth, the number of HIV-2A infections grew exponentially between 1950 and 1980 as proposed previously (Lemey et al., 2003). However, by extending the sampling time range in comparison with previous analysis (Lemey et al., 2003), our data reveals a levelling off in the effective population size of HIV-2 from the early 1990s onwards (Fig. 1). The change in HIV-2A effective population size dynamics towards the present is consistent with serological investigations (da Silva et al., 2008; Månsson et al., 2009; Poulsen et al., 1997; van Tienen et al., 2009), which have even demonstrated a declining HIV-2 incidence. Finally, we analyse the degree to which geographical locations correlate with shared ancestry (Parker et al., 2008). Each sequence was assigned to the country of sampling and the association index (AI) and parsimony score (PS) values, which measure the degree of association between phylogeny and sampling location were computed. Our analysis of the geographical signal for each dataset (Table 1) revealed high AI and PS values, suggesting relatively extensive viral migration dynamics among localities. Nevertheless, the association between phylogenetic clustering and geographical location was higher than expected by chance (P<0.0001).
Table 1. Temporal and geographical signal of the HIV-2A datasets used in this study.
| Global env | Global pol | |
| Temporal signal* | ||
| ln ML dated tips model (H1) | −19244.4 | −11131.0 |
| ln ML undated (contemporaneous) tips (H0) | −19257.3 | −11128.5 |
| ln BF (H1 vs H0)† | 12.9 | 2.5 |
| Geographical signal† | ||
| Association index (AI)‡ (95 % CI§) | 12.04 (11.04–12.97) | 21.94 (20.64–23.19) |
| Parsimony score (PS)‡ (95 % CI§) | 109.61 (103–117) | 194.89 (186–203) |
ln marginal likelihood (ML) and Bayes factor (BF) test.
ln BFs >1 indicates that model H1 is more strongly supported by the data than model H0.
All statistics resulted in a P-value <0.001.
Confidence interval.
Fig. 1.
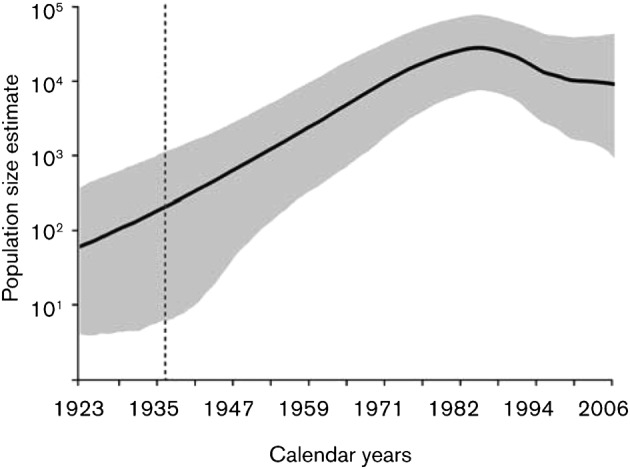
The historical population dynamics of HIV-2A. Bayesian skyride analysis depicting the mean changes in effective population size (Net) through time with 95 % BCI for the most informative dataset (global env).
Spatial origins and global diffusion of HIV-2A
To infer the spatial diffusion of HIV-2A at a global scale we included sequence data from a total of 19 countries (Supplementary Fig. S1, available in JGV Online). Our data collection included 70 new pol sequences from Portugal thus significantly contributing to the collection of HIV-2 nucleotide data from this country, which has experienced the highest prevalence of HIV-2 outside of West Africa (Gomes et al., 2003). We used a Bayesian phylogeographic approach in which the among-location exchange rate matrix between the two different genomic regions is shared (Lemey et al., 2009). Based on the standard asymmetrical discrete model in which transition rates were assumed to be equal a priori between, from and to West African locations, the most probable ancestral root location fluctuated between Caió [a rural location in Guinea-Bissau (Ricard et al., 1994)] and Portugal for individual or shared analyses for pol and env (Fig. 2). This may not be surprising as Portugal shares the same HIV-2A diversity with Guinea-Bissau due to historical migration, so a larger sample from Portugal may yield support for this location as the origin of HIV-2A. Because the standard phylogeographic continuous-time Markov chain (CTMC) implementation reveals a strong impact of the sampling heterogeneity on the estimation of the ancestral root location, we further specified priors that attempt to mimic the differences in human migration from and to West Africa. Particularly, we performed different analyses specifying a range of location exchange priors that favoured viral migration from West Africa to elsewhere by factors of 10 : 1, 100 : 1 and 1000 : 1 (see Methods for details).
Fig. 2.

Ancestral root state reconstructions for the global HIV-2A datasets. The analyses were performed separately (a and b) and together (c and d) for each locus and for the shared analyses (c and d).
The individual analysis for each locus places Caió at the root of the HIV-2A trees using the less informative 10 : 1 prior specification [ancestral root state probabilities of 0.97 and 0.98 for pol and env, respectively, Fig. 2(a) and (b)]. We obtained similar results when sharing the rate matrix between pol and env for the 10 : 1, 100 : 1 and 1000 : 1 priors [ancestral root state probabilities of 0.54, 0.70 and 0.94; 0.97, 0.91 and 0.93, respectively, for pol and env, Fig. 2(c) and (d)]. Using the 10 : 1 prior in the shared model, the ancestral reconstruction of the root in pol placed 46 % of the posterior mass in Côte d’Ivoire. Importantly, this model extension protects to a large extent against sampling bias, resulting from a higher number of non-African to African sequences and efficiently combines the spatial information embedded in both datasets.
Fig. 3 shows the maximum conservative clade credibility (MCC) trees for the shared model using the 1000 : 1 prior specification in which branches were coloured according to the most probable location and branch width proportional to location state posterior probability. Identical MCC topologies were obtained using the separated analyses for the 10 : 1, 100 : 1 and 1000 : 1 priors and the shared model analysis using the 1000 : 1 prior (not shown). Our data suggest that Guinea-Bissau, Senegal and Côte d’Ivoire acted as key hubs of viral migration in the early stage of the epidemic. Within West Africa, sequences from Senegal, Côte d’Ivoire, Gambia and Burkina Faso seem to be descendents from lineages in Caió, although sequences from Gambia are also found interspersed with sequences from Senegal. All Nigerian sequences form a monophyletic cluster [posterior support 0.99, Fig. 3(a)] that can be dated back to around 1967 (95 % BCI: 1955, 1977).
Fig. 3.

MCC genealogies for the global HIV-2A datasets. The pol (n = 320) and env (n = 248) phylogenies shown in (a) and (b), respectively, were generated using a multilocus Bayesian Markov chain Montc Carlo (MCMC) analysis under the asymmetrical diffusion model using the 1000 : 1 rate prior setup. Branch lengths are shown in units of time and branch widths are proportional to location state probability. Each colour represents the most probable location state of the descendant node.
Our phylogeographic analyses of the global datasets further allowed us to pinpoint Guinea-Bissau, Cape Verde, Senegal and Côte d’Ivoire as the main sources of extra-regional viral migration, seeding the virus to Portugal and France, the two first targets of extra-regional viral dispersion from West Africa (Fig. 3). Portuguese sequences show no obvious geographical clustering and were often interspersed among sequences from Caió, Bissau, Luxembourg, France and the UK (Fig. 3a). We demonstrate clusters composed of 2–6 sequences from Portugal, introduced from Guinea-Bissau, and clusters composed of 2–5 sequences from France, mostly introduced from Côte d’Ivoire, suggesting on-going transmission of HIV-2A in these countries, although at a limited level. All Indian HIV-2A env sequences formed a monophyletic cluster (posterior probability 0.73), possibly derived from Guinea-Bissau (location state posterior probability 0.98), with a divergence time around 1964 (95 % BCI: 1951, 1974) (Fig. 3b), more then 20 years before the first reports of HIV-2 in India (Grez et al., 1994).
Significant pathways of global HIV-2 dispersal
Mapping well-supported rates of HIV-2 dispersal at a global scale provides an explicit visualization of historical migration patterns of the virus. Fig. 4 shows the most significant epidemiological links obtained by sharing the rate matrix between the two loci and by combining the analyses for the different location exchange priors in order to summarize the most robust findings. An interactive visualization of the BF links generated with SPREAD software (Bielejec et al., 2011) is available as Supplementary Fig. S2 (available in JGV Online).
Fig. 4.

BF test for significant epidemiological non-zero rates. Epidemiological links with BF support greater than 15 were obtained by combining the results from three different prior specifications (1 : 10, 1 : 100 and 1 : 1000). The yellow–red gradient represents the relative strength by which the rates are supported. All locations involved in significant epidemiological linkage are depicted, except South Korea that was omitted for purpose of clarity (a KML interactive summary is available as Supplementary Fig. S2).
Within West Africa we obtained strong support for epidemiological links between geographically close localities such as Caió to Bissau (BF>1000) and Senegal (BF = 983.2) and Gambia to Senegal (BF = 107.3) (Table 2). Interestingly, our analysis further recovered epidemiological links between Bissau and Nigeria (BF = 49.6), Côte d’Ivoire and Gambia (BF = 32.8) and between Cape Verde and Côte d’Ivoire (BF = 27.3) and Cape Verde and Caió (BF = 16.4).
Table 2. Most significant epidemiological linkage of HIV-2 group A global dispersal under an asymmetrical model of viral diffusion (BF>15).
| From | To | BF |
| Bissau | Portugal | >1000 |
| Caió | Guinea-Bissau | >1000 |
| Caió | Senegal | 983.2 |
| Côte d’Ivoire | France | 193.7 |
| Gambia | Senegal | 107.3 |
| Cape Verde | Portugal | 96.3 |
| Guinea-Bissau | Nigeria | 49.6 |
| Senegal | France | 46.3 |
| Portugal | Luxembourg | 43.1 |
| Côte d’Ivoire | Gambia | 32.8 |
| Portugal | UK | 29.2 |
| Cape Verde | Côte d’Ivoire | 27.3 |
| Guinea-Bissau | South Korea | 26.4 |
| Gambia | Caió | 17.6 |
| Cape Verde | Caió | 16.4 |
With respect to migration of HIV-2 out of West Africa, we obtained overwhelming support for epidemiological linkage from Bissau and Cape Verde to Portugal (BF>1000 and BF = 96.3, respectively) and statistically significant linkage from Côte d’Ivoire and Senegal to France (BF = 193.7 and BF = 46.3). Essentially, the extra-regional links identified by the BSSVS approach are remarkably consistent with known historical ties. Moreover, sequences from South Korea formed two clusters with 2 and 6 sequences, consistent with previous findings (Nam et al., 2006), and the BSSVS analysis shows support for an epidemiological link with Bissau (BF = 26.4). The two introductions of HIV-2A in South Korea probably represent isolated events and do not necessarily have to directly involve Guinea-Bissau (Nam et al., 2006), but ultimately they find their most likely origin in Guinea-Bissau. Finally, in terms of migration events within Europe, we found significant support for viral migration from Portugal to Luxembourg (BF = 43.1) and the UK (BF = 29.2).
Discussion
In an attempt to disentangle the diffusion patterns that gave rise to the global contemporaneous distribution of HIV-2A, we performed a fully probabilistic phylogeographic reconstruction of the historical dispersal patterns of this virus. We have taken advantage of the spatial detail embedded in the genetic data by using an asymmetrical diffusion model in which rates of location exchange are shared between two loci, and we reduce the impact of spatial sampling bias by specifying priors on viral transition rates that reflect human migratory flux from West Africa to elsewhere. Despite these precautions, it remains difficult to accurately estimate ancestral root locations based on convenience sampling, which can result in datasets that are not representative for the entire population. Our statistical analyses based on pol and env data sampled from 19 countries worldwide show that the spatial structure of this virus is rather fluid at an international scale and corroborate that Guinea-Bissau and Cape Verde, together with Côte d’Ivoire and Senegal, acted as viral source populations in the early epidemic history, from where the virus, later on, spread at an extra-regional scale to countries sharing socio-historical ties. Within West Africa, most viral flows seem to have occurred between neighbouring countries, while in Europe viral migration seems to resemble Portuguese flows towards emigration hubs (Garcia et al., 2000).
Despite the relatively low degree of association between the phylogenies and localities indicating extensive viral migration among localities, we still detect significant spatial substructure in the HIV-2 phylogenies. Consistently, a close inspection of viral flow from Bissau to Portugal, and from Côte d’Ivoire and Senegal to France reveals a scenario of multiple introductions, resulting in relatively small clusters of transmission. That the virus was initially established in Portugal and France and further spread to other countries within Europe is also in agreement with HIV-2 being found in European and not only in West African immigrants (Brücker et al., 1987; Damond et al., 2001; Soriano et al., 2000; Valadas et al., 2009). Notably, we obtained strong support for viral diffusion between Portugal and the UK and Portugal and Luxembourg. In the early 1980s, Portuguese migrants constituted 26 % of the total population living in Luxembourg (Garcia et al., 2000) and viral migration between these countries has also been depicted for HIV-1 subtype B (Paraskevis et al., 2009). It is likely that the virus could also have spread from Portugal to other Portuguese migration centres such as Switzerland, Belgium and Germany (Smallman-Raynor & Cliff, 1991), but these either resulted in insignificant onwards transmission or they have not been identified due to a lack of representative data.
We do not attribute considerable importance to the evidence supporting a spatial root of HIV-2A in Guinea-Bissau – even though in line with serological evidence (da Silva et al., 2008; Poulsen et al., 1993) – over a root in Côte d’Ivoire, where the closest SIVsmm have been found (Santiago et al., 2005), because this could also be the result of a heterogeneous sampling scheme. The global pol dataset comprised a higher number of sequences from Côte d’Ivoire, the 10 : 1 and 100 : 1 prior specifications led to posterior masses of 0.45 and 0.20 for this country being at the root of the HIV-2A phylogenies. Therefore, we cannot rule out the possibility that the convenience sampling utilized here is biased against a spatial origin in Côte d’Ivoire (Santiago et al., 2005; de Sousa et al., 2010). Our prior specifications mainly altered support from Portugal to Caió because sequences from these locations tend to cluster together. Also, the earliest indications of HIV-2-positive samples in Côte d’Ivoire date back to the 1960s (Le Guenno, 1989; Kawamura et al., 1989). Clearly, more data are needed to establish which location gave rise to the emergence of the HIV-2A epidemic. The closely related SIVsmm lineages in Côte d’Ivoire arguably provide the most compelling evidence for pinpointing the cross-species transmissions (Santiago et al., 2005). However, in analogy with HIV-1, for which cross-species transmission most likely occurred in southern Cameroon but epidemic establishment may have been in the Democratic Republic of Congo (Keele et al., 2006), HIV-2A may have been introduced into Guinea-Bissau early after cross-species transmission. Human migration between Guinea-Bissau and Côte d’Ivoire (separated by over 1 400 km) could be accomplished through boat connections that linked the most populated ports and through Gambia and Senegal that together with Côte d’Ivoire recruited the highest proportion of migrants in this region during this period (Supplementary Fig. S3, available in JGV Online). Interestingly, our findings concerning epidemiological linkage between these locations are in line with this hypothesis. It was proposed that the emergence of epidemic HIV-1 and HIV-2 was favoured by commercial sex work in nascent colonial cities, and the resulting epidemics of genital ulcer diseases, factors that were relatively important in Ivorian cities, in the 1930s and 1940s. It is further possible that the less prevalent practise of circumcision in both Côte d’Ivoire and the northern half of Guinea-Bissau, compared to other West African countries in the mid 20th century, may have promoted the initial establishment of this virus in West Africa (de Sousa et al., 2010). Other studies emphasize parenteral risk factors to explain both the initial adaptation of HIV-2 to humans (Marx et al., 2001), and the exceptional prevalence this virus attained in Guinea-Bissau (Pépin et al., 2006). In addition, the independence war between Guinea-Bissau and Portugal might have further contributed to shape the spread of HIV-2 within Guinea-Bissau and outside West Africa (Lemey et al., 2003). Finally, despite the low prevalence of HIV-2 in Liberia (Gao et al., 1992), Sierra Leone (Chen et al., 1997) and Guinea-Conakry (Lakiss, 1991), the absence of nucleotide data does not allow us to exclude the possibility of an HIV-2 group A origin in these countries.
The discrete Bayesian phylogeographic approach based on asymmetrical diffusion models provides a powerful framework for hypothesis testing. However, the availability of sufficient time-stamped and geo-referenced data to build representative datasets may considerably hamper its applications. The convenience sampling for pol and env datasets was skewed in terms of sequences per location and may therefore constitute only a poor representation of the HIV-2 genetic diversity in each location. Nevertheless, we extend an approach to recover spatial detail from the largest global datasets of HIV-2 sequences collected to date that may prove useful for future applications. Particularly, we took several precautions to counteract the impact of sampling bias and improve the level of accuracy of spatial inference. First, we used a recently developed discrete asymmetrical approach that allows location exchange rates to vary according to directionality (Edwards et al., 2011), which provides a more realistic description of the diffusion process. Second, we extended an approach that allows phylogeographic inference to draw information simultaneously from different loci. This approach allows genealogical parameters from different loci to be estimated independently, while sharing the location exchange matrix between the loci (Lemey et al., 2009). Finally, we informed the directionality of gene flow by specifying a range of priors on the transition rates that attempt to mimic the ratio of human fluxes from and to West Africa. Although it is clear that the number of international migrants from West Africa has fluctuated over the time of observation (OECD, 2011) and that accurate migration numbers over time might be difficult to assess, this approach prevents us from drawing conclusions that would be overly affected by sampling heterogeneity. European locations such as Portugal were more represented than their former West African colonies, and many independent introductions to Portugal ensured that they harbour a similar HIV-2 genetic diversity as the West African source populations. In such cases, the mere overrepresentation of Portugal may bias the ancestral location estimates. Our study pinpoints Guinea-Bissau and Portugal’s contribution to the current distribution of the HIV-2A global epidemic and we cannot exclude that this reflects a bias towards the number of sequences used from these countries in our analyses. However, in line with human population movements having a strong impact in HIV-2 dispersal within and outside West Africa, it is interesting to note that the highest emigration rates among the countries affected by HIV-2 were from Guinea-Bissau, Cape Verde and Portugal (Supplementary Fig. S3). Despite the fact that the compilation of larger and representative HIV-2 datasets may be a daunting task, in particular because plasma viral loads are undetectable for the majority of the infected individuals (da Silva et al., 2008; Popper et al., 1999), epidemiological inference would benefit from broader and more comprehensively sampled viral genetic data.
Overall, our study sheds light on the global spatiotemporal dynamics of HIV-2 and highlights the limitations of applying phylogeographic reconstructions to convenience sampling. Further understanding of the key source areas of viral dispersal and the factors underlying the emergence and spread of fast evolving viruses can help directing epidemiological surveillance efforts. This is a critical area for future research that may ultimately contribute to improve public health measures against infectious diseases.
Methods
Portuguese HIV-2 group A isolates.
An in-house PCR method was used to amplify a 1280 bp HIV-2 pol gene fragment encompassing the entire protease (PR) and part of the reverse transcriptase (RT) (positions 2517–3748 HIV-2 BEN) from plasma samples obtained from 70 Portuguese patients as described in Soares et al., 2011. We used the bioMérieux’s EasyMAG automatic extraction procedure to extract viral RNA from 1 ml plasma samples. Viral RNA was retro-transcribed and amplified using Access RT-PCR Core Reagents kit (Merck) and outer primers JA218 [(+1859) 5′-GAAAGAAGCCCCGCAACTTCC-3′] and JA221 [(−3258) 5′-GCTCTGCTTCTGCTAATTCTGTCCA-3′] as described in Brandin et al. (2003). A second amplification was carried out using AmpliTaq Gold PCR Master Mix (Applied Biosystems) and the inner primers JA219 [(+1898) 5′-AGGGGCT(A/G)ACACCAACAGCAC-3′] and JA220MOD [(−3178) 5′-GTCTTTATICCTGGGTAGAI(T/G)TGTG-3′]. Cycle sequencing was accomplished using the Big Dye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems) according to the manufacturer’s recommendations and using four sequencing primers: JA219, JA220 [(−3178) 5′-GTCTTTAT(T/C)CCTGGGTAGATTTGTG-3′], JA222 [(+2525) 5′-ACCTCCAACTAATCCTTATAATACC-3′] and JA223 [(−2625) 5′-ACTGAATTTCTGTGAAATCTTGAGT-3′]. Purified products were run on an ABI Prism 3100 Genetic Analyzer (Applied Biosystems) according to the same protocol, but adjusted to HIV-2 specific settings. Nucleotide sequences were analysed with the SeqScape Software version 2.5 (Applied Biosystems).
GenBank accession numbers.
GenBank accession numbers, sampling dates and locations (either public or obtained following contact with relevant authors) are available in Supplementary Tables S1 and S2 (available in JGV Online). Newly determined nucleotide data were deposited in GenBank (Benson et al., 2005) under the accession numbers: JQ282087–JQ282154, JQ267759–JQ267771 and GU133364–GU133366.
Nucleotide sequence data compilation.
We retrieved all publicly available HIV-2A nucleotide sequences with known sampling location from the Los Alamos HIV database (www.hiv.lanl.gov/content/index). Multiple sequence alignments were conducted using MAFFT version 6.0 (Katoh et al., 2005) and sequences were edited manually with Se-Al v2.0 (Rambaut & Drummond, 2007). To improve the accuracy of the phylogenetic and geographical inference, we excluded sequences from the same patient and sequences from patients following antiretroviral treatment. In general, the nationality of the patients was unknown and, because we aimed to infer dispersal from the perspective of the virus, only the geographical locations where the virus was sampled were considered. The final datasets resulted in a total of 320 pol sequences (L = 283 nt, sampling interval: 1985–2009; positions 2651–2933 HIV-2 BEN) and 248 env sequences (L = 322 nt, sampling interval: 1985–2007; positions 7531–7852 HIV-2 BEN), spanning a total of 19 countries: Gambia (9 pol and 9 env), Senegal (0 and 13), Guinea-Bissau/Caió (16 and 48), Guinea-Bissau/Bissau (4 and 32), Burkina Faso (10 and 0), Nigeria (11 and 0), Côte d’Ivoire (31 and 4), Cape Verde (15 and 11), Ghana (1 and 0), Portugal (83 and 53), Spain (21 and 4), France (68 and 21), UK (13 and 0), Luxembourg (12 and 0), Belgium (6 and 0), Italy (3 and 0), Sweden (12 and 0), India (3 and 45), South Korea (0 and 8) and Brazil (2 and 0). Spatial locations are available in Supplementary Fig. S1.
Bayesian evolutionary analysis.
We estimated the parameters of a full probabilistic model, including timed sequence evolution, demographic history and spatial dynamics, using Bayesian statistical inference implemented in the beast software package v.1.6 (Drummond & Rambaut, 2007; Lemey et al., 2009) with beagle to improve run-time (Suchard & Rambaut, 2009). A Bayesian skyride demographic model with time-aware smoothing that does not require strong prior assumptions was used to estimate a posterior distribution of the effective population size through time (Minin et al., 2008). For each dataset, we used a standard general time-reversible substitution model with gamma distributed rate heterogeneity of four categories (GTR+4Γ) as selected by jModelTest (Posada, 2008).
To evaluate the ‘temporal signal’ in each alignment, the degree to which genetic divergence accumulates over the sampling time range was tested for the pol and env datasets independently using strict molecular clock models. Two evolutionary models were compared: H1 model (dated tips) and H0 model [contemporaneous tips, reflecting no accumulation of sequence divergence over the sampling period (Wertheim & Worobey, 2009)]. Model comparison was performed using BF estimates that compare the relative marginal likelihood of the models (Suchard et al., 2001). If model H1 did not outperform H0 [ln BF<3], data were considered to contain no significant temporal signal and hence insufficient to serve per se as evolutionary rate calibration (Table 1). This is analogous to a previously described likelihood ratio test that compares the dated-tip clock model against the standard clock model (Rambaut, 2000; Suchard et al., 2001), with the difference being in our ability to integrate over all possible phylogenies instead of conditioning a single topology. To investigate the degree to which geographical locations correlate with shared ancestry we used BaTS software (Parker et al., 2008). We first assigned each sequence to the country of sampling. We computed the AI and PS, which measure the degree of association between sampling location and the phylogeny and are inversely correlated with the degree of association (Parker et al., 2008). These statistics were computed for a null distribution with 1000 replicates and on a subset of a posterior sample of 500 trees generated from beast.
To reconstruct the spatial dynamics of HIV-2A we used a probabilistic model of discrete asymmetrical diffusion as implemented in the beast package (Drummond & Rambaut, 2007; Edwards et al., 2011). The original implementation models the unobserved diffusion process as a reversible CTMC process (Lemey et al., 2009), but recently a more realistic extension was developed that allows the transition rates in the CTMCs (and consequently the directionality of diffusion) to be asymmetrical (Edwards et al., 2011). Sequence evolution was modelled using an uncorrelated lognormal relaxed molecular clock model that accounts for rate variation among branches (Drummond et al., 2006). By incorporating uncertainty both in the phylogenetic estimates and location state substitution process, this approach permits the description of the diffusion process through estimating the posterior location-state probabilities along all branches of the phylogeny. In order to achieve statistical efficiency and identify significant epidemiological linkage among locations, we used a BSSVS procedure that allows rates to be zero with some probability (Lemey et al., 2009). Well-supported migratory events were summarized using the cross-platform SPREAD application (Bielejec et al., 2011) (discarding 20 % as burn-in) based on a BF cut-off above 15. Such a stringent cut-off value prevents us from drawing unwarranted conclusions on the epidemiological links since the distribution of the number of supported rates as a function of the BF cut-off was heavily tailed (not shown). The analyses were performed for the different prior setups both separately for each locus and by sharing the among-location exchange rate matrix between the two different genomic regions.
The standard implementation of the discrete asymmetrical phylogeographic approach assigns the same prior probability distribution to all of the transition rates in the CTMC. Since representative sampling was not feasible and both datasets were comprised by a higher number of non-West African to West African sequences, we specified a multivariate gamma distribution that considered location exchange rates from West African to non-West African localities to be 10, 100 and 1000 less likely than the complement rates (XMLs available from the authors upon request).
We estimated the joint posterior distribution of the full probabilistic model using Markov chain Monte Carlo (MCMC) techniques implemented in beast. Continuous parameters estimates are reported as posterior means and 95 % BCI. Each MCMC chain was run sufficiently along after convergence to the joint posterior distribution to ensure adequate mixing, typically between 50 and 150 million steps. Marginal posterior distributions of the continuous parameters and their corresponding effective sample sizes (ESS), a measure of the number of effectively independent samples from the marginal distributions, were inspected using Tracer v1.4.1 (Rambaut & Drummond, 2007). The log files for the location-exchange rate matrices obtained from the shared analyses for the prior configurations of 10 : 1, 100 : 1 and 1000 : 1 were combined using LogCombiner v.1.6.1 (Drummond & Rambaut, 2007).
MCC trees were summarized from the posterior distribution of trees (removing burn-in of 10 %) with TreeAnnotator and visualized with FigTree v1.3.1 (Rambaut & Drummond, 2007). The modal locations (most probable reconstructed locations at each node) were coloured along the phylogenies with branches width proportional to location posterior probability.
Acknowledgements
N. R. F. is supported by Fundação para a Ciência e Tecnologia (grant no. SFRH/BD/64530/2009). M. A. S. is supported by NIH R01 GM86887. We are grateful to Vítor Duque, Ana Treviño, José Miguel Pereira, Fausto Baldanti, Maurizio Zazzi, Carlo Perno, Catherine Brennan, Françoise Brun-Vezinet, Florence Damond, Diane Descamps, Gab Jung Kim, Sung Kim, Eric Arts, Carlo Torti, Robin Mukhopadhyaya, Anita Desai, Susmita Gurjan and Soo-Yon Rhee for valuable information concerning sampling dates and locations of the isolates, and Emmanuel Gilisseu for insight on the range of C. atys atys. The research leading to these results has received funding from the European Union Seventh Framework Programme [FP7/2007-2013] under grant agreement no. 278433 (PREDEMICS), grant agreement no. 223131 (CHAIN) and ERC grant agreement no. 260864. P. L. and M. A. S. acknowledge the support of the National Evolutionary Synthesis Center (NESCent) through a working group (Software for Bayesian Evolutionary Analysis).
Footnotes
Supplementary material is available with the online version of this paper.
References
- Ayanian J. Z., Maguire J. H., Marlink R. G., Essex M., Kanki P. J. (1989). HIV-2 infection in the United States. N Engl J Med 320, 1422–1423 10.1056/NEJM198905253202119 [DOI] [PubMed] [Google Scholar]
- Benson D. A., Karsch-Mizrachi I., Lipman D. J., Ostell J., Wheeler D. L. (2005). GenBank. Nucleic Acids Res 33 (Database issue), D34–D38 10.1093/nar/gki063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielejec F., Rambaut A., Suchard M. A., Lemey P. (2011). SPREAD: spatial phylogenetic reconstruction of evolutionary dynamics. Bioinformatics 27, 2910–2912 10.1093/bioinformatics/btr481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandin E., Lindborg L., Gyllensten K., Broström C., Hagberg L., Gisslen M., Tuvesson B., Blaxhult A., Albert J. (2003). pol gene sequence variation in Swedish HIV-2 patients failing antiretroviral therapy. AIDS Res Hum Retroviruses 19, 543–550 10.1089/088922203322230905 [DOI] [PubMed] [Google Scholar]
- Brennan C. A., Yamaguchi J., Vallari A. S., Hickman R. K., Devare S. G. (1997). Genetic variation in human immunodeficiency virus type 2: identification of a unique variant from human plasma. AIDS Res Hum Retroviruses 13, 401–404 10.1089/aid.1997.13.401 [DOI] [PubMed] [Google Scholar]
- Brücker G., Brun-Vezinet F., Rosenheim M., Rey M. A., Katlama C., Gentilini M. (1987). HIV-2 infection in two homosexual men in France. Lancet 329, 223 10.1016/S0140-6736(87)90043-2 [DOI] [PubMed] [Google Scholar]
- Chen Z., Telfier P., Gettie A., Reed P., Zhang L., Ho D. D., Marx P. A. (1996). Genetic characterization of new West African simian immunodeficiency virus SIVsm: geographic clustering of household-derived SIV strains with human immunodeficiency virus type 2 subtypes and genetically diverse viruses from a single feral sooty mangabey troop. J Virol 70, 3617–3627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Luckay A., Sodora D. L., Telfer P., Reed P., Gettie A., Kanu J. M., Sadek R. F., Yee J. & other authors (1997). Human immunodeficiency virus type 2 (HIV-2) seroprevalence and characterization of a distinct HIV-2 genetic subtype from the natural range of simian immunodeficiency virus-infected sooty mangabeys. J Virol 71, 3953–3960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costarelli S., Torti C., Rodella A., Baldanti F., Paolucci S., Lapadula G., Manca N., Quiros-Roldan E., Izzo I., Carosi G. (2008). Screening and management of HIV-2-infected individuals in Northern Italy. AIDS Patient Care STDS 22, 489–494 10.1089/apc.2007.0149 [DOI] [PubMed] [Google Scholar]
- da Silva Z. J., Oliveira I., Andersen A., Dias F., Rodrigues A., Holmgren B., Andersson S., Aaby P. (2008). Changes in prevalence and incidence of HIV-1, HIV-2 and dual infections in urban areas of Bissau, Guinea-Bissau: is HIV-2 disappearing? AIDS 22, 1195–1202 10.1097/QAD.0b013e328300a33d [DOI] [PubMed] [Google Scholar]
- Damond F., Apetrei C., Robertson D. L., Souquière S., Leprêtre A., Matheron S., Plantier J. C., Brun-Vézinet F., Simon F. (2001). Variability of human immunodeficiency virus type 2 (HIV-2) infecting patients living in France. Virology 280, 19–30 10.1006/viro.2000.0685 [DOI] [PubMed] [Google Scholar]
- Damond F., Worobey M., Campa P., Farfara I., Colin G., Matheron S., Brun-Vézinet F., Robertson D. L., Simon F. (2004). Identification of a highly divergent HIV type 2 and proposal for a change in HIV type 2 classification. AIDS Res Hum Retroviruses 20, 666–672 10.1089/0889222041217392 [DOI] [PubMed] [Google Scholar]
- De Cock K. M., Odehouri K., Colebunders R. L., Adjorlolo G., Lafontaine M. F., Porter A., Gnaore E., Diaby L., Moreau J. & other authors (1990). A comparison of HIV-1 and HIV-2 infections in hospitalized patients in Abidjan, Côte d’Ivoire. AIDS 4, 443–448 10.1097/00002030-199005000-00010 [DOI] [PubMed] [Google Scholar]
- de Sousa J. D., Müller V., Lemey P., Vandamme A. M. (2010). High GUD incidence in the early 20th century created a particularly permissive time window for the origin and initial spread of epidemic HIV strains. PLoS ONE 5, e9936 10.1371/journal.pone.0009936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djomand G., Greenberg A. E., Sassan-Morokro M., Tossou O., Diallo M. O., Ekpini E., Ghys P., Soro B., Brattegaard K. & other authors (1995). The epidemic of HIV/AIDS in Abidjan, Côte d’Ivoire: a review of data collected by Project RETRO-CI from 1987 to 1993. J Acquir Immune Defic Syndr Hum Retrovirol 10, 358–365 [PubMed] [Google Scholar]
- Drummond A. J., Rambaut A. (2007). beast: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7, 214 10.1186/1471-2148-7-214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A. J., Ho S. Y., Phillips M. J., Rambaut A. (2006). Relaxed phylogenetics and dating with confidence. PLoS Biol 4, e88 10.1371/journal.pbio.0040088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards C. J., Suchard M. A., Lemey P., Welch J. J., Barnes I., Fulton T. L., Barnett R., O’Connell T. C., Coxon P. & other authors (2011). Ancient hybridization and an Irish origin for the modern polar bear matriline. Curr Biol 21, 1251–1258 10.1016/j.cub.2011.05.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esu-Williams E., Mulanga-Kabeya C., Takena H., Zwandor A., Aminu K., Adamu I., Yetunde O., Akinsete I., Patrel D. & other authors (1997). Seroprevalence of HIV-1, HIV-2, and HIV-1 group O in Nigeria: evidence for a growing increase of HIV infection. J Acquir Immune Defic Syndr Hum Retrovirol 16, 204–210 10.1097/00042560-199711010-00010 [DOI] [PubMed] [Google Scholar]
- Gao F., Yue L., White A. T., Pappas P. G., Barchue J., Hanson A. P., Greene B. M., Sharp P. M., Shaw G. M., Hahn B. H. (1992). Human infection by genetically diverse SIVSM-related HIV-2 in West Africa. Nature 358, 495–499 10.1038/358495a0 [DOI] [PubMed] [Google Scholar]
- Garcia J. L., Jerónimo H. M., Rovisco M. L., da Silva P. A., Almeida C. M., Lopes J. C. (2000). Portugal Migrante – Emigrantes e Imigrados, dois estudos introdutórios Oeiras: CELTA [Google Scholar]
- Gomes P., Abecasis A., Almeida M., Camacho R., Mansinho K. (2003). Transmission of HIV-2. Lancet Infect Dis 3, 683–684 10.1016/S1473-3099(03)00797-7 [DOI] [PubMed] [Google Scholar]
- Grez M., Dietrich U., Balfe P., von Briesen H., Maniar J. K., Mahambre G., Delwart E. L., Mullins J. I., Rübsamen-Waigmann H. (1994). Genetic analysis of human immunodeficiency virus type 1 and 2 (HIV-1 and HIV-2) mixed infections in India reveals a recent spread of HIV-1 and HIV-2 from a single ancestor for each of these viruses. J Virol 68, 2161–2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn B. H., Shaw G. M., De Cock K. M., Sharp P. M. (2000). AIDS as a zoonosis: scientific and public health implications. Science 287, 607–614 10.1126/science.287.5453.607 [DOI] [PubMed] [Google Scholar]
- Holmes E. C. (2008). Evolutionary history and phylogeography of human viruses. Annu Rev Microbiol 62, 307–328 10.1146/annurev.micro.62.081307.162912 [DOI] [PubMed] [Google Scholar]
- Katoh K., Kuma K., Toh H., Miyata T. (2005). MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res 33, 511–518 10.1093/nar/gki198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura M., Yamazaki S., Ishikawa K., Kwofie T. B., Tsujimoto H., Hayami M. (1989). HIV-2 in West Africa in 1966. Lancet 333, 385 10.1016/S0140-6736(89)91760-1 [DOI] [PubMed] [Google Scholar]
- Keele B. F., Van Heuverswyn F., Li Y., Bailes E., Takehisa J., Santiago M. L., Bibollet-Ruche F., Chen Y., Wain L. V. & other authors (2006). Chimpanzee reservoirs of pandemic and nonpandemic HIV-1. Science 313, 523–526 10.1126/science.1126531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvinesdal B. B., Worm A. M., Lindhardt B. O., Jensen B. L., Nielsen C. M., Poulsen A. G. (1992). HIV-2 infection in Denmark. Scand J Infect Dis 24, 419–421 10.3109/00365549209052626 [DOI] [PubMed] [Google Scholar]
- Lakiss S., Kourouma K., Diallo M. P., Sabbatani S., Rezza G., Titti F., Rossi G. B., Verani P. HIV-1/2 seroprevalence in Guinea Conakry. In VII International Conference on AIDs 6, 16–21 Florence, Italy. [Google Scholar]
- Le Guenno B. (1989). HIV1 and HIV2: two ancient viruses for a new disease? Trans R Soc Trop Med Hyg 83, 847 10.1016/0035-9203(89)90350-7 [DOI] [PubMed] [Google Scholar]
- LeBreton M., Yang O., Tamoufe U., Mpoudi-Ngole E., Torimiro J. N., Djoko C. F., Carr J. K., Tassy Prosser A., Rimoin A. W. & other authors (2007). Exposure to wild primates among HIV-infected persons. Emerg Infect Dis 13, 1579–1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P., Pybus O. G., Wang B., Saksena N. K., Salemi M., Vandamme A. M. (2003). Tracing the origin and history of the HIV-2 epidemic. Proc Natl Acad Sci U S A 100, 6588–6592 10.1073/pnas.0936469100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P., Rambaut A., Drummond A. J., Suchard M. A. (2009). Bayesian phylogeography finds its roots. PLOS Comput Biol 5, e1000520 10.1371/journal.pcbi.1000520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabey D. C., Tedder R. S., Hughes A. S., Corrah P. T., Goodison S. J., O’Connor T., Shenton F. C., Lucas S. B., Whittle H. C., Greenwood B. M. (1988). Human retroviral infections in The Gambia: prevalence and clinical features. Br Med J (Clin Res Ed) 296, 83–86 10.1136/bmj.296.6615.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Månsson F., Biague A., da Silva Z. J., Dias F., Nilsson L. A., Andersson S., Fenyö E. M., Norrgren H. (2009). Prevalence and incidence of HIV-1 and HIV-2 before, during and after a civil war in an occupational cohort in Guinea-Bissau, West Africa. AIDS 23, 1575–1582 10.1097/QAD.0b013e32832cedfb [DOI] [PubMed] [Google Scholar]
- Marx P. A., Alcabes P. G., Drucker E. (2001). Serial human passage of simian immunodeficiency virus by unsterile injections and the emergence of epidemic human immunodeficiency virus in Africa. Philos Trans R Soc Lond B Biol Sci 356, 911–920 10.1098/rstb.2001.0867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minin V. N., Bloomquist E. W., Suchard M. A. (2008). Smooth skyride through a rough skyline: Bayesian coalescent-based inference of population dynamics. Mol Biol Evol 25, 1459–1471 10.1093/molbev/msn090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam J. G., Kim G. J., Baek J. Y., Suh S. D., Kee M. K., Lee J. S., Kim S. S. (2006). Molecular investigation of human immunodeficiency virus type 2 subtype a cases in South Korea. J Clin Microbiol 44, 1543–1546 10.1128/JCM.44.4.1543-1546.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann P. W., O’Shaughnessy M. V., Lepine D., D’Souza I., Major C., McLaughlin B. (1989). Laboratory diagnosis of the first cases of HIV-2 infection in Canada. CMAJ 140, 125–128 [PMC free article] [PubMed] [Google Scholar]
- Newmark P. (1988). HIV-2 detected in UK. Nature 332, 295 10.1038/332295d0 [DOI] [PubMed] [Google Scholar]
- Nielsen R., Beaumont M. A. (2009). Statistical inferences in phylogeography. Mol Ecol 18, 1034–1047 10.1111/j.1365-294X.2008.04059.x [DOI] [PubMed] [Google Scholar]
- OECD (2011). OECD.StatExtracts – http://stats.oecd.org/Index.aspx: Organisation for Economic Co-Operation and Development.
- Paraskevis D., Pybus O., Magiorkinis G., Hatzakis A., Wensing A. M., van de Vijver D. A., Albert J., Angarano G., Asjö B. & other authors (2009). Tracing the HIV-1 subtype B mobility in Europe: a phylogeographic approach. Retrovirology 6, 49 10.1186/1742-4690-6-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J., Rambaut A., Pybus O. G. (2008). Correlating viral phenotypes with phylogeny: accounting for phylogenetic uncertainty. Infect Genet Evol 8, 239–246 10.1016/j.meegid.2007.08.001 [DOI] [PubMed] [Google Scholar]
- Peeters M., Koumare B., Mulanga C., Brengues C., Mounirou B., Bougoudogo F., Ravel S., Bibollet-Ruche F., Delaporte E. (1998). Genetic subtypes of HIV type 1 and HIV type 2 strains in commercial sex workers from Bamako, Mali. AIDS Res Hum Retroviruses 14, 51–58 10.1089/aid.1998.14.51 [DOI] [PubMed] [Google Scholar]
- Pépin J., Plamondon M., Alves A. C., Beaudet M., Labbé A. C. (2006). Parenteral transmission during excision and treatment of tuberculosis and trypanosomiasis may be responsible for the HIV-2 epidemic in Guinea-Bissau. AIDS 20, 1303–1311 10.1097/01.aids.0000232239.05545.33 [DOI] [PubMed] [Google Scholar]
- Pieniazek D., Peralta J. M., Ferreira J. A., Krebs J. W., Owen S. M., Sion F. S., Filho C. F., Sereno A. B., de Sa C. A. & other authors (1991). Identification of mixed HIV-1/HIV-2 infections in Brazil by polymerase chain reaction. AIDS 5, 1293–1299 10.1097/00002030-199111000-00002 [DOI] [PubMed] [Google Scholar]
- Pieniazek D., Ellenberger D., Janini L. M., Ramos A. C., Nkengasong J., Sassan-Morokro M., Hu D. J., Coulibally I. M., Ekpini E. & other authors (1999). Predominance of human immunodeficiency virus type 2 subtype B in Abidjan, Ivory Coast. AIDS Res Hum Retroviruses 15, 603–608 10.1089/088922299311132 [DOI] [PubMed] [Google Scholar]
- Popovsky M. A., Carneski A., Hoff R., Fang C. T., Krieger M. (1990). Infection with HIV-2 in a resident of the United States. N Engl J Med 322, 1887 10.1056/NEJM199006283222614 [DOI] [PubMed] [Google Scholar]
- Popper S. J., Sarr A. D., Travers K. U., Guèye-Ndiaye A., Mboup S., Essex M. E., Kanki P. J. (1999). Lower human immunodeficiency virus (HIV) type 2 viral load reflects the difference in pathogenicity of HIV-1 and HIV-2. J Infect Dis 180, 1116–1121 10.1086/315010 [DOI] [PubMed] [Google Scholar]
- Posada D. (2008). jModelTest: phylogenetic model averaging. Mol Biol Evol 25, 1253–1256 10.1093/molbev/msn083 [DOI] [PubMed] [Google Scholar]
- Poulsen A.-G., Aaby P., Frederiksen K., Kvinesdal B., Mlbak K., Dias F., Lauritzen E. (1989). Prevalence of and mortality from human immunodeficiency virus type 2 in Bissau, West Africa. Lancet 333, 827–831 10.1016/S0140-6736(89)92281-2 [DOI] [PubMed] [Google Scholar]
- Poulsen A. G., Aaby P., Gottschau A., Kvinesdal B. B., Dias F., Mølbak K., Lauritzen E. (1993). HIV-2 infection in Bissau, West Africa, 1987-1989: incidence, prevalences, and routes of transmission. J Acquir Immune Defic Syndr 6, 941–948 [PubMed] [Google Scholar]
- Poulsen A. G., Aaby P., Larsen O., Jensen H., Nauclér A., Lisse I. M., Christiansen C. B., Dias F., Melbye M. (1997). 9-year HIV-2-associated mortality in an urban community in Bissau, West Africa. Lancet 349, 911–914 10.1016/S0140-6736(96)04402-9 [DOI] [PubMed] [Google Scholar]
- Pybus O. G., Rambaut A. (2009). Evolutionary analysis of the dynamics of viral infectious disease. Nat Rev Genet 10, 540–550 10.1038/nrg2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. (2000). Estimating the rate of molecular evolution: incorporating non-contemporaneous sequences into maximum likelihood phylogenies. Bioinformatics 16, 395–399 10.1093/bioinformatics/16.4.395 [DOI] [PubMed] [Google Scholar]
- Rambaut A., Drummond A. (2007). Molecular evolution, phylogenetics and epidemiology – http://tree.bio.ed.ac.uk/software/
- Ricard D., Wilkins A., N’Gum P. T., Hayes R., Morgan G., Da Silva A. P., Whittle H. (1994). The effects of HIV-2 infection in a rural area of Guinea-Bissau. AIDS 8, 977–982 10.1097/00002030-199407000-00016 [DOI] [PubMed] [Google Scholar]
- Ruelle J., Sanou M., Liu H. F., Vandenbroucke A. T., Duquenne A., Goubau P. (2007). Genetic polymorphisms and resistance mutations of HIV type 2 in antiretroviral-naive patients in Burkina Faso. AIDS Res Hum Retroviruses 23, 955–964 10.1089/aid.2007.0034 [DOI] [PubMed] [Google Scholar]
- Santiago M. L., Range F., Keele B. F., Li Y., Bailes E., Bibollet-Ruche F., Fruteau C., Noë R., Peeters M. & other authors (2005). Simian immunodeficiency virus infection in free-ranging sooty mangabeys (Cercocebus atys atys) from the Taï Forest, Côte d’Ivoire: implications for the origin of epidemic human immunodeficiency virus type 2. J Virol 79, 12515–12527 10.1128/JVI.79.19.12515-12527.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallman-Raynor M., Cliff A. (1991). The spread of human immunodeficiency virus type 2 into Europe: a geographical analysis. Int J Epidemiol 20, 480–489 10.1093/ije/20.2.480 [DOI] [PubMed] [Google Scholar]
- Smith S. M., Christian D., de Lame V., Shah U., Austin L., Gautam R., Gautam A., Apetrei C., Marx P. A. (2008). Isolation of a new HIV-2 group in the US. Retrovirology 5, 103 10.1186/1742-4690-5-103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares R. S., Tendeiro R., Foxall R. B., Baptista A. P., Cavaleiro R., Gomes P., Camacho R., Valadas E., Doroana M. & other authors (2011). Cell-associated viral burden provides evidence of ongoing viral replication in aviremic HIV-2-infected patients. J Virol 85, 2429–2438 10.1128/JVI.01921-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano V., Gomes P., Heneine W., Holguín A., Doruana M., Antunes R., Mansinho K., Switzer W. M., Araujo C. & other authors (2000). Human immunodeficiency virus type 2 (HIV-2) in Portugal: clinical spectrum, circulating subtypes, virus isolation, and plasma viral load. J Med Virol 61, 111–116 [DOI] [PubMed] [Google Scholar]
- Suchard M. A., Rambaut A. (2009). Many-core algorithms for statistical phylogenetics. Bioinformatics 25, 1370–1376 10.1093/bioinformatics/btp244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchard M. A., Weiss R. E., Sinsheimer J. S. (2001). Bayesian selection of continuous-time Markov chain evolutionary models. Mol Biol Evol 18, 1001–1013 [DOI] [PubMed] [Google Scholar]
- Takehisa J., Osei-Kwasi M., Ayisi N. K., Hishida O., Miura T., Igarashi T., Brandful J., Ampofo W., Netty V. B. & other authors (1997). Phylogenetic analysis of HIV type 2 in Ghana and intrasubtype recombination in HIV type 2. AIDS Res Hum Retroviruses 13, 621–623 10.1089/aid.1997.13.621 [DOI] [PubMed] [Google Scholar]
- Treviño A., de Mendoza C., Caballero E., Rodríguez C., Parra P., Benito R., Cabezas T., Roc L., Aguilera A. & other authors (2011). Drug resistance mutations in patients infected with HIV-2 living in Spain. J Antimicrob Chemother 66, 1484–1488 10.1093/jac/dkr164 [DOI] [PubMed] [Google Scholar]
- UNAIDS (2006). AIDS epidemic update 2006. Geneva, Switzerland: Joint United Nations Programme on HIV/AIDS (UNAIDS) and World Heatlth Organization (WHO) 2004, pp. 1–94 [Google Scholar]
- United Nations (2010). Population Division, Population Estimates and Projections Section – http://stats.oecd.org/Index.aspx
- Valadas E., França L., Sousa S., Antunes F. (2009). 20 years of HIV-2 infection in Portugal: trends and changes in epidemiology. Clin Infect Dis 48, 1166–1167 10.1086/597504 [DOI] [PubMed] [Google Scholar]
- van Tienen C., van der Loeff M. S., Zaman S. M., Vincent T., Sarge-Njie R., Peterson I., Leligdowicz A., Jaye A., Rowland-Jones S. & other authors (2009). Two distinct epidemics: the rise of HIV-1 and decline of HIV-2 infection between 1990 and 2007 in rural Guinea-Bissau. J Acquir Immune Defic Syndr 53, 640–647 [DOI] [PubMed] [Google Scholar]
- Wertheim J. O., Worobey M. (2009). Dating the age of the SIV lineages that gave rise to HIV-1 and HIV-2. PLOS Comput Biol 5, e1000377 10.1371/journal.pcbi.1000377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins A., Oelman B., Pepin J., Cham K., Corrah T., Hughes A., Manneh K., Njai R., Rolfe M. & other authors (1991). Trends in HIV-1 and HIV-2 infection in The Gambia. AIDS 5, 1529–1530 10.1097/00002030-199112000-00018 [DOI] [PubMed] [Google Scholar]
- Wolfe N. D., Daszak P., Kilpatrick A. M., Burke D. S. (2005). Bushmeat hunting, deforestation, and prediction of zoonoses emergence. Emerg Infect Dis 11, 1822–1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeh C., Pieniazek D., Agwale S. M., Robbins K. E., Odama L., Sani-Gwarzo N., Gboun M. S., Inyang U. S., Folks T. M. & other authors (2005). Nigerian HIV type 2 subtype A and B from heterotypic HIV type 1 and HIV type 2 or monotypic HIV type 2 infections. AIDS Res Hum Retroviruses 21, 17–27 10.1089/aid.2005.21.17 [DOI] [PubMed] [Google Scholar]