

Abstract
LL-37, the only human cathelicidin, is a cationic antimicrobial peptide with antibacterial and antifungal activity. LL-37 is released from neutrophil granules and produced by epithelial cells. It has been implicated in host defence against influenza A virus (IAV) in recent studies. We now demonstrate dose-related neutralizing activity of LL-37 against several seasonal and mouse-adapted IAV strains. The ability of LL-37 to inhibit these IAV strains resulted mainly from direct effects on the virus, since pre-incubation of virus with LL-37 was needed for optimal inhibition. LL-37 bound high-density lipoprotein (HDL), and pre-incubation of LL-37 with human serum or HDL reduced its antiviral activity. LL-37 did not inhibit viral association with epithelial cells as assessed by quantitative RT-PCR or confocal microscopy. This finding contrasted with results obtained with surfactant protein D (SP-D). Unlike collectins or human neutrophil defensins (HNPs), LL-37 did not induce viral aggregation under electron microscopy. In the electron microscopy studies, LL-37 appeared to cause disruption of viral membranes. LL-37 had additive antiviral activity when combined with other innate inhibitors like SP-D, surfactant protein A and HNPs. Unlike HNPs, LL-37 did not bind SP-D significantly. These findings indicate that LL-37 contributes to host defence against IAV through a mechanism distinct from that of SP-D and HNPs.
Introduction
A cathelicidin was first identified as a lipopolysaccharide (LPS)-binding protein in rabbit granulocytes (Larrick et al., 1991). Like the defensins, the cathelicidins are a large family of cationic antimicrobial peptides expressed in many species and have broad-spectrum antimicrobial activity; however, the only human cathelicidin is hCAP18/LL-37 (Burton & Steel, 2009; Lai & Gallo, 2009). The cathelicidins contain a signal peptide, a cathelin-like domain and an antimicrobial domain. hCAP18/LL-37 is produced by neutrophils, macrophages and various epithelial cells. The predominant antimicrobial product, composed of 37 amino acids, is released by cleavage by proteases, and this domain is termed LL-37. There are other cleavage products with antimicrobial activity. LL-37 is released from specific granules of neutrophils upon cell activation (Sørensen et al., 2001). LL-37 can activate respiratory epithelial cells by interacting with epidermal growth factor receptors (EGFRs) (Tjabringa et al., 2003). Respiratory epithelial cells also produce LL-37 in response to inflammatory stimuli. LL-37 expression by the respiratory epithelium is upregulated by active metabolites of vitamin D (Hansdottir et al., 2008; Liu et al., 2006).
The murine homologue of LL-37 is cathelicidin-related antimicrobial peptide (CRAMP). Mice lacking CRAMP have impaired defence against urinary, gastro-intestinal, respiratory, pulmonary and skin infections (Chromek et al., 2006; Doss et al., 2010). Mouse models have been useful to study upregulation of β-defensins and CRAMP during respiratory bacterial infections (Braff et al., 2007; Caverly et al., 2003). Recently, human research also indicates that LL-37 plays important roles in respiratory infection (e.g. tuberculosis; Liu et al., 2007).
Although the defensins have well-established antiviral activities, relatively less attention has been paid to antiviral activity of LL-37. LL-37 has been shown to have antiviral activity against human immunodeficiency virus (HIV) in vitro (Bergman et al., 2007) and to improve the outcome of influenza A virus (IAV) infection in mice through inhibition of viral replication and reduction of virus-induced pro-inflammatory cytokine generation (Barlow et al., 2011). Upregulation of LL-37 expression by stimulation with leukotriene B4 (LTB4) correlated with improved outcome of influenza infection in mice (Gaudreault & Gosselin, 2008).
In this paper, we determine the activity of LL-37 and related peptides against several strains of IAV, compare the mechanism of antiviral activity of LL-37 with that of other soluble innate inhibitors and determine its interactions with other innate antiviral proteins.
Results
LL-37 binds seasonal H3N2 IAV but does not inhibit haemagglutination (HA) activity
Binding of LL-37 to IAV was tested by ELISA. Binding was compared to binding to Staphylococcus aureus. As shown in Fig. 1(a), LL-37 bound the Phil82 strain of IAV to an extent similar to binding of S. aureus. For comparison, we tested binding of LL-37 to a known high-density lipoprotein (HDL) (Lau et al., 2006).
Fig. 1.

Binding of LL-37 to IAV or S. aureus and neutralization of IAV by LL-37. (a) Binding assessed by solid-phase ELISA. Results are means±sem of four separate experiments. Binding of LL-37 to IAV (Phil82 strain), S. aureus and HDL was significantly greater than binding to BSA at all concentrations tested. (b–d) Neutralization assessed using the fluorescent focus assay for detection of viral nucleoprotein. All results are means±sem of four or more separate experiments. The Phil82 IAV strain was used. (b) Effect of pre-incubation of Phil82 IAV with LL-37 versus scrambled LL-37 (sLL-37). (c) Effects of varying the time of addition of LL-37. Using all methods, LL-37 reduced viral infectivity significantly compared with the control, but inhibition was significantly less when cells were incubated with LL-37 either before or after infection than when the virus was pre-incubated with LL-37. (d) Effects of HDL, apolipoprotein A1 or the EGFR-signalling inhibitor AG1478 on antiviral activity of LL-37. *, P<0.05 compared with binding to BSA in (a) and compared with infectivity of untreated virus in (b) and (c). **, P<0.05 compared with the control, sLL-37 (b), pre-incubation of cells with LL-37 or delayed addition of LL-37 (c) as assessed by ANOVA. #, HDL reduced antiviral activity of LL-37 significantly as assessed by ANOVA.
Like the defensins (Doss et al., 2009), LL-37 did not inhibit HA activity of the Phil82 or PR-8 strains of IAV as assessed using the HA inhibition assay. LL-37 was tested up to a concentration of 32 µg ml−1 without any evidence of HA inhibition (data not shown; n = 5).
Viral neutralization of seasonal H3N2 IAV and mouse-adapted IAV by LL-37
As a measure of viral neutralization, we assessed the ability of LL-37 to reduce the number of Madin–Darby canine kidney (MDCK) cells expressing viral nucleoprotein after infection. Fig. 1(b) depicts neutralizing activity of LL-37 against the Phil82 H3N2 strain of IAV. The m.o.i. for these experiments was 0.1. We pre-incubated LL-37 with the virus and then infected MDCK cells. There was clear dose-dependent inhibition, with a 50 % neutralizing concentration of LL-37 of ~2 µg ml−1. For comparison, we tested the effect of a scrambled version of LL-37 (sLL-37). sLL-37 had no inhibitory effect on viral replication up to 8 µg ml−1, although it did have a slight inhibitory effect at 16 µg ml−1 (Fig. 1b). LL-37 has been shown to cause death of respiratory epithelial cells (Lau et al., 2006). Release of lactate dehydrogenase (LDH) into the cell supernatant is a sensitive assay for cell death; hence, we measured LDH release under the conditions of the experiments in Fig. 1. There was no increase in LDH release from the cells in the presence of IAV and/or LL-37 (or sLL-37) up to concentrations of 32 µg ml−1 under the conditions of this assay (e.g. LDH release was not significantly greater than in uninfected control cells for any sample after 7 h of incubation; data not shown).
LL-37 inhibits seasonal H3N2 mainly by direct effects on the virus
As a first step in determining whether the antiviral activity of LL-37 depends on interaction of the peptide with the virus or with epithelial cells, we tested the effects of adding LL-37 to the cells after viral internalization (e.g. 45 min after incubation of virus with cells). In these experiments, the residual extracellular virus was washed off prior to addition of LL-37. As shown in Fig. 1(c) (see curve labelled ‘Delayed addition of LL-37’), LL-37 caused some inhibition in this setting, but the effect was reduced substantially compared with pre-incubating the virus with LL-37 prior to infection. In this case, the 50 % neutralizing concentration of LL-37 was ~20 µg ml−1, and complete inhibition was not achieved even at a concentration of 64 µg ml−1. It is worth noting that we did not test for degradation of LL-37 over time in culture, so the results of these experiments could have been affected by degradation of LL-37. Very similar results were obtained when the MDCK cells were pre-incubated with LL-37, followed by washing of cells and addition of virus (Fig. 1c). Hence, although LL-37 has some direct effects on MDCK cells to inhibit viral replication, the most potent activity involves direct interactions of the virus with LL-37.
As noted above, effects of LL-37 in some epithelial cells are mediated by signalling through EGFR. Pre-incubation of the MDCK cells overnight with either 2 or 4 µM of the EGFR-signalling inhibitor AG1478 did not alter the activity of LL-37 (Fig. 1d shows results with 4 µM). AG1478 did not alter viral replication on its own in these experiments (data not shown). Hence, the antiviral activity of LL-37 in this setting is not mediated by EGFR signalling.
Pre-incubation with HDL blocks the antiviral activity of LL-37
LL-37 binds serum components, and its antibacterial activity is inhibited by serum. We first tested the effects of pre-incubation of LL-37 with human serum followed by addition to virus and measurement of viral infectivity. Although the serum did reduce the apparent neutralizing activity of LL-37, serum alone also strongly inhibited viral infectivity, making the results hard to interpret (data not shown). It has been reported that the key element in serum binding to LL-37 is HDL. As shown in Fig. 1(a), LL-37 bound avidly to HDL. Concentrations of up to 200 µg HDL ml−1 did not inhibit viral infectivity on their own (although dose-related inhibition was noted above this concentration). Pre-incubation of LL-37 with 50 µg HDL ml−1 inhibited its antiviral activity (Fig. 1d). Interestingly, apolipoprotein A1 did not alter the antiviral activity of LL-37 in parallel experiments (Fig. 1d). Similar results were reported by Lau et al. (2006).
Effects of LL-37 on replication of IAV in human primary respiratory epithelial cells and effects of LL-37 or its mouse homologue on replication of mouse-adapted IAV strains
To evaluate the effects of LL-37 in a more physiological cellular model, we tested neutralization of Phil82 IAV in primary human tracheobronchial epithelial (HTBE) cells (Fig. 2a). LL-37 showed similar inhibition in these cells as in MDCK cells. The effects of LL-37 were significantly greater than those of sLL-37, although sLL-37 again caused inhibition of viral replication at higher concentrations.
Fig. 2.

Inhibition of infectivity of IAV strains by LL-37 or CRAMP in HBTE cells or MDCK cells. These experiments were performed as in Fig. 1(b). (a) Effects of LL-37 or sLL-37 on replication of Phil82 in HTBE cells. sLL-37 caused increased viral replication at the lowest concentration tested, but reduced replication at the highest concentrations tested. LL-37 inhibited viral replication at all concentrations tested, and its effects were significantly greater than those of sLL-37 as compared by ANOVA. (b) Inhibition of three mouse-adapted IAV strains by LL-37 in MDCK cells. In this set of experiments, sLL-37 did not inhibit replication of the WSN strain significantly. (c) CRAMP inhibits replication of Phil82 and PR-8 in MDCK cells. Results are means±sem of four or more experiments. *, P<0.05 compared with controls; **, P<0.005 compared with controls [in (b) and (c), * and ** apply to all viruses tested].
We also tested effects of LL-37 on mouse-adapted strains of IAV, PR-8, WSN and WSNHAnc-Asp225Gly, using MDCK cells. These viral strains were all inhibited by LL-37 (Fig. 2b) at a 50 % neutralizing concentration of ~4 µg ml−1. Since these are all mouse-adapted strains, they could be useful in murine models aimed at evaluating the antiviral activity of LL-37 (Barlow et al., 2011; Crouch et al., 2011; Smee et al., 2008). sLL-37 did not inhibit replication of WSN significantly in these experiments. Overall, the neutralizing activity of LL-37 for all these viral strains and Phil82 was similar to that of human neutrophil defensins (HNPs) (Doss et al., 2009). The murine homologue of LL-37 (CRAMP) also inhibited the PR-8 and Phil82 strains (Fig. 2c).
LL-37 does not cause viral aggregation or inhibit viral uptake by respiratory epithelial cells
We next studied the effects of LL-37 on viral infection of A549 cells using fluorescently labelled IAV and confocal microscopy. As shown in Fig. 3, LL-37 did not appear to alter significantly the pattern of IAV binding or initial uptake by cells. This was in striking contrast to findings obtained with surfactant protein D (SP-D), which caused marked viral aggregation and effective reduction in particle numbers. The confocal microscopy results were obtained using an approximately 200-fold higher m.o.i. (i.e. m.o.i. of 200) than was used in the infectious focus assay. This was necessary because it was difficult to visualize virus consistently using lower concentrations. We next used quantitative RT-PCR (qPCR) to detect whether LL-37 or SP-D alter viral uptake by cells, since this was more sensitive and could be used under the same conditions as the infectious focus assay. As shown in Fig. 4(a), LL-37 did not reduce the amount of virus associated with cells after 45 min of infection. In contrast, SP-D reduced cell-associated virus significantly at this time (Fig. 4c). Using the same technique, however, we confirmed that both LL-37 and SP-D reduced the amount of virus present in cells or in cell supernatant significantly 24 h after infection (Fig. 4b, c).
Fig. 3.
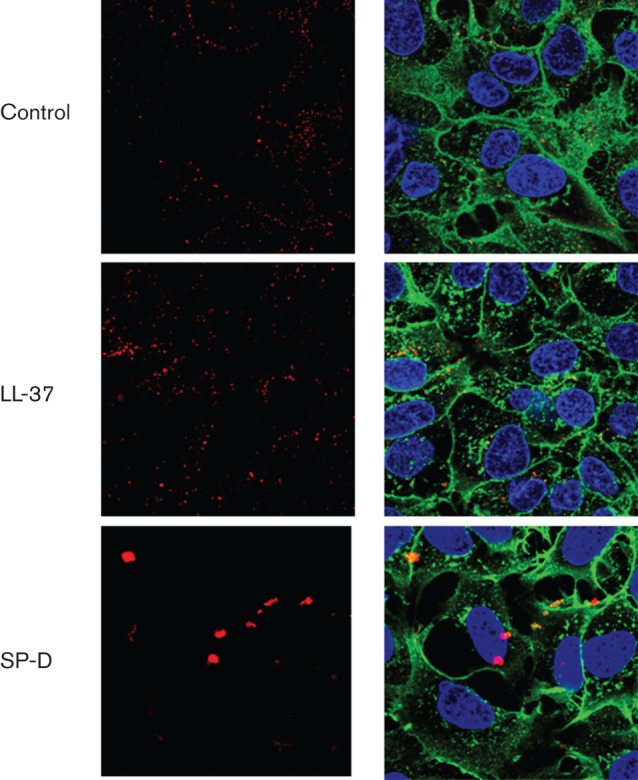
Effects of LL-37 on viral interactions with A549 cells under confocal microscopy. Alexa Fluor 594-labelled Phil82 IAV was incubated with control buffer, LL-37 or SP-D followed by incubation of A549 cell monolayers with the virus. The virus appears red; cell nuclei were stained with DAPI 350 and appear blue and cell membranes were labelled with WGA–Oregon green 488. Results are representative of four experiments. Virus alone is shown in the upper panels. The left column of images was taken at the wavelength of IAV alone (red); the right panel of images combines wavelengths of virus, cell membranes and nuclei. No consistent alteration in the pattern of virus binding to cells was seen with LL-37; however, SP-D caused formation of large viral aggregates. The concentration of LL-37 used was 8 µg ml−1, but similar results were obtained with 4 and 16 µg ml−1. The concentration of SP-D used was 2 µg ml−1.
Fig. 4.
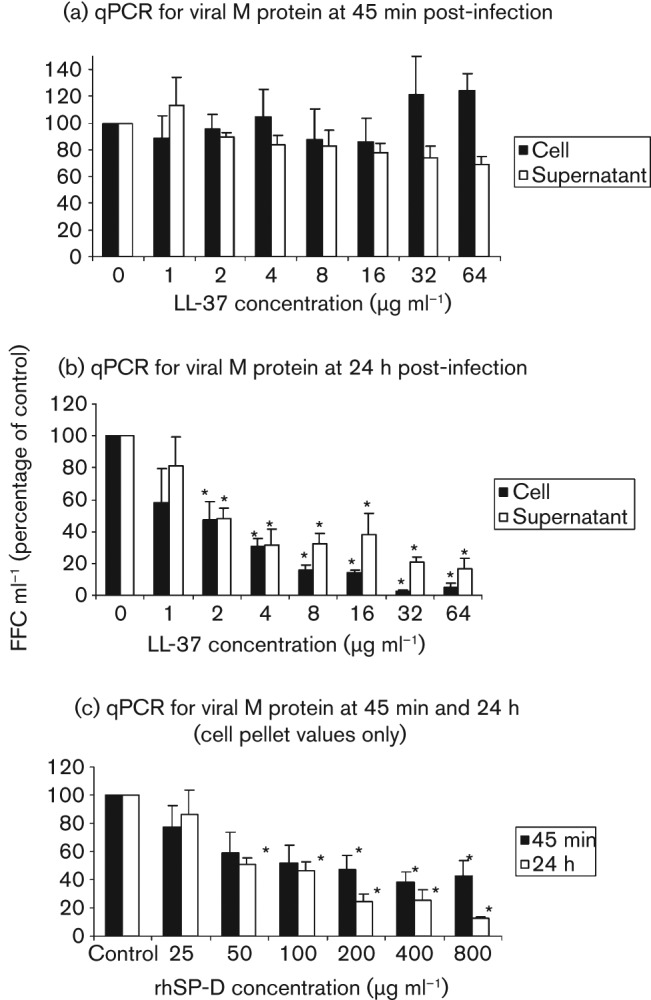
Effects of LL-37 on viral replication as assessed by qPCR. IAV (strain Phil82) was pre-incubated with LL-37 as in Fig. 1(b) and the virus was then incubated with A549 cells for 45 min. The cell supernatant was then harvested and the cells were washed several times prior to preparing cell homogenates. In (a), cell supernatants and cell homogenates were obtained after the 45 min period of initial infection with IAV. RNA was isolated and qPCR was performed. Results are expressed as mean no. of fluorescent foci (FFC) ml−1. The mean FFC ml−1 in control samples for (a) was 190 846 for supernatant and 6216 for cell-associated virus. Results shown are percentages of control copy numbers for samples treated with LL-37. There were no significant differences in viral RNA quantities between control and LL-37-treated samples in (a). (b) RNA was isolated from cells and supernatants after 24 h of infection. In this case, the control FFC ml−1 was 2645 and 158 584, respectively, for supernatant and cells (i.e. cell-associated virus increased markedly in cells compared with 45 min samples). LL-37 at concentrations ≥2 µg ml−1 decreased viral RNA significantly in supernatants and cells in (b). (c) SP-D caused significant reductions in cell-associated viral RNA at both 45 min and 24 h after infection. The quantities of RNA at both time points were reduced significantly at SP-D concentrations of 50 ng ml−1 or higher. All results are means±sem of four or more separate experiments. *, P<0.05 compared with controls.
We have shown that collectins, HNPs and retrocyclins induce viral aggregation, which may contribute importantly to their antiviral activity (Doss et al., 2009; Hartshorn et al., 1994). No viral aggregation was apparent in LL-37-treated samples in the confocal microscopy experiments (see Fig. 3). We evaluated virus-aggregating activity directly using two additional assays. As shown in Fig. 5(a), LL-37 did not cause any detectable viral aggregation as assessed by optical density assays. Results obtained with SP-D are shown for comparison. LL-37 also did not alter significantly the ability of SP-D to induce viral aggregation. In our earlier experiments, viral aggregation induced by HNPs was relatively subtle compared with that induced by SP-D in light-scattering assays; however, HNPs clearly induced aggregation as assessed using electron microscopy (EM) (Tecle et al., 2007). LL-37 also caused minimal viral aggregation when assessed by EM, in contrast to findings with HNP-1 or retrocyclin 2 (RC2) (Fig. 5b; upper row of images). Of note, in higher-magnification images, LL-37 was found to cause disruption of viral membranes (Fig. 5b; lower row). In our previous experiments, HNPs did not cause any evident alterations in viral membrane structure (data not shown).
Fig. 5.

LL-37 does not cause viral aggregation. (a) Viral aggregation assessed by measuring increases in light scattering through the viral suspension induced by either LL-37 or SP-D. SP-D induced viral aggregation strongly, as reported previously, but LL-37 did not. LL-37 also did not alter the aggregating effects of SP-D significantly (SP-D+LL-37). All results are means±sem of seven separate experiments. (b) Effects of LL-37 or defensins on appearance of virus under EM. Phil82 IAV (Phil) was pre-incubated with LL-37 (64 µg ml−1), HNP-1 (40 µg ml−1) or RC2 (40 µg ml−1) for 30 min and evaluated by EM. Images in the upper panel were taken at lower magnification and used the Phil82 IAV strain. This panel shows the lack of viral aggregation induced by LL-37 compared with controls. Aggregation was also not evident using lower concentrations of LL-37 (not shown). The effects of HNP-1 and RC2 are shown for comparison. The experiment shown is representative of six experiments using various concentrations of LL-37 (2–64 µg ml−1). The lower panels show higher-power views of either Phil82 or PR-8 virus particles treated with control buffer or LL-37. Note the intact hair-like haemagglutinin spikes on the surface of the control viruses. The Phil82 sample was treated with LL-37 (32 µg ml−1) and shows viral membrane breaks and blebbing. The PR-8 sample treated with LL-37 (16 µg ml−1) shows marked disruption of particle morphology. These results are representative of three or more experiments.
Interaction with other inhibitors
We have reported previously that HNPs bind SP-D and interfere with the antiviral activity of SP-D against viral strain Phil82 (Hartshorn et al., 2006). Fig. 6(a) shows that binding of SP-D to LL-37 was minimal and much less than binding of SP-D to HNP-2. LL-37 had additive neutralizing activity when combined with SP-D (Fig. 6b). Similar results were obtained when combining LL-37 with HNPs and surfactant protein A (SP-A) (Table 1).
Fig. 6.

Interactions of LL-37 with SP-D. In (a), LL-37 or HNP-2 were coated on ELISA plates and binding of SP-D was tested. SP-D showed significant dose-related binding to HNP-2 but not to LL-37. In (b), viral neutralization was assessed in the presence of LL-37 alone or LL-37 combined with 50 ng SP-D ml−1. The combination of LL-37 with 4 µg SP-D ml−1 caused significantly greater reduction in infectivity than either LL-37 or SP-D alone. Similar results were obtained using combinations of LL-37 with 100 ng SP-D ml−1 (not shown). Results are means±sem of four experiments. *, P<0.05 compared with controls; **, P<0.005 compared with controls.
Table 1. Viral neutralization by LL-37 in combination with SP-A, HNP-1 or HNP-2.
Results are expressed as percentages of control infectious foci. HNP results are means±sem from four experiments; SP-A results are means±sem from seven experiments. nt, Not tested.
| Experiment | Control buffer | LL-37 | |
| 2 µg ml−1 | 4 µg ml−1 | ||
| Control buffer | 100 | 73±5 | 49±5 |
| SP-A 1 µg ml−1 | 80±10 | 43±4* | 22±3.5† |
| SP-A 2 µg ml−1 | 70±11 | 34±4* | 19±3† |
| SP-A 4 µg ml−1 | 45±10 | 31±6* | 16±3* |
| HNP-1 5 µg ml−1 | 75±9 | nt | 30±6‡ |
| HNP-1 10 µg ml−1 | 51±7 | nt | 22±4‡ |
| HNP-1 20 µg ml−1 | 33±5 | nt | 11±3‡ |
| HNP-2 5 µg ml−1 | 43±4 | nt | 20±7‡ |
| HNP-2 10 µg ml−1 | 30±4 | nt | 12±‡ |
| HNP-2 20 µg ml−1 | 20±3 | nt | 5±2‡ |
P<0.05 compared with LL-37 alone.
P<0.05 compared with LL-37 or SP-A alone.
P<0.05 compared with LL-37 or HNP alone.
Discussion
Our findings extend recent observations that LL-37 inhibits the PR-8 strain of IAV in vitro and in vivo (Barlow et al., 2011) by evaluating the activity of LL-37 against other IAV strains and by studying the mechanism of antiviral activity of LL-37 compared with other soluble innate inhibitors. The potency of LL-37 for viral inhibition was similar to that of HNPs and greater than that of human β-defensins, although less than that of retrocyclins (Doss et al., 2009). We first characterized the antiviral activity of LL-37 using the Phil82 strain, which is representative of recent seasonal H3N2 strains in terms of its envelope proteins. LL-37 inhibited IAV replication to similar extents in MDCK cells, A549 cells (not shown) and HTBE cells. The effects of LL-37 were largely specific, since the scrambled peptide had much less activity; however, higher concentrations of sLL-37 did begin to inhibit viral growth in some instances. This did not appear to be due to an effect of sLL-37 on MDCK cell viability, based on LDH assays. LL-37 bound HDL, and HDL inhibited its antiviral activity. A major part of the anti-influenza activity of LL-37 resulted from direct interaction with the virus, since addition of LL-37 directly to MDCK cells at times before or after viral infection was less effective at inhibiting viral infectivity than when the virus and LL-37 were pre-incubated prior to infection of cells. In addition, inhibitors of EGFR signalling did not alter the activity of LL-37.
Since the infectious focus assay assesses viral nucleoprotein production at 6 h after infection, the action of LL-37 occurs in an early phase of the viral life cycle. LL-37 also reduced viral M protein RNA generation in cells and release of viral RNA at 24 h. LL-37 did not reduce cell-associated virus, as assessed by qPCR for viral RNA after 45 min of incubation. There was also no apparent alteration in viral binding using confocal microscopy. Interestingly, we found that higher concentrations of LL-37 damaged viral membranes, as visualized by EM. It is possible that disruption of viral membranes by LL-37 affects other early events in the viral life cycle (e.g. pH-mediated release of viral nucleoprotein from endosomes), although further experiments will be needed to clarify this. The PR-8 strain of IAV lacks glycosylation on the head of the haemagglutinin molecule and was inhibited by LL-37; hence, glycosylation of haemagglutinin is not important for the antiviral activity of LL-37. The lack of HA-inhibitory activity of LL-37, and the failure of LL-37 to inhibit viral binding or uptake, also suggest that LL-37 does not mediate its effects by inhibiting binding of the haemagglutinin molecule.
We conclude that the mechanism of antiviral activity of LL-37 differs from that of HNPs or retrocyclins in that it does not cause viral aggregation and appears to cause viral membrane degradation. Salvatore et al. (2007) have demonstrated that HNPs inhibit IAV replication through a direct interaction of the HNPs with epithelial cells. Based on our results, LL-37 exerts its effects predominantly through direct interactions with the virus. The mechanism of action of LL-37 also differs from that of SP-D in several ways. To reach this conclusion, we further clarified the mechanism of action of SP-D in this paper. As demonstrated previously, SP-D causes viral aggregation and inhibits viral HA activity, and its activity is dependent on binding and inhibiting the actions of haemagglutinin (Hartshorn, 2010; Hartshorn et al., 2000b). SP-D requires the presence of N-linked glycans on the haemagglutinin head domain for binding (e.g. SP-D does not inhibit PR-8 or WSN strains) (Crouch et al., 2011). We now confirm through the use of confocal microscopy and qPCR assays that SP-D reduces the initial attachment or uptake of IAV by epithelial cells. In all of these respects, LL-37 differs in its activity from SP-D.
Like HNPs, LL-37 is packaged in large quantities in neutrophil secondary granules and can be released through various stimuli. Neutrophils have been shown in a variety of studies (Hashimoto et al., 2007; Tate et al., 2009; White et al., 2008) to contribute to host defence against IAV, and neutrophil-derived LL-37 could contribute to this activity. LL-37 is distinguished from HNPs, but is similar to β-defensins, in being produced by epithelial cells in response to various activating stimuli. LL-37 and β-defensins serve as part of the initial barrier against infection, play a role in repair of damaged epithelium and also act as ‘alarmins’ to trigger recruitment of inflammatory cells (Oppenheim & Yang, 2005). LL-37 expression is regulated by distinct mechanisms from the β-defensins, and its receptors on immune cells are distinct from those of the defensins. As noted, there is evidence that release of LL-37 and β-defensin in response to LTB4 contributes to host defence against IAV (Gaudreault & Gosselin, 2008). Regulation of LL-37 production by vitamin D could also contribute to host defence against IAV.
Unlike HNPs, LL-37 did not bind SP-D significantly and did not inhibit SP-D’s antiviral activity. In fact, LL-37 had additive antiviral activity when combined with SP-D. Binding of HNPs to SP-D appears to be mediated by charge interactions, since it is increased at low ionic strength and pH (Hartshorn et al., 2006). LL-37 is strongly positively charged (net positive charge 6), comparable to or greater than many defensins. Hence, charge alone is not responsible for binding to SP-D. Note also that β-defensins do not bind SP-D, and have additive antiviral activity when combined with SP-D (Doss et al., 2009). Thus, antimicrobial peptides produced by respiratory epithelial cells have cooperative activities with the surfactant collectins.
In conclusion, LL-37 inhibits a variety of IAV strains through a mechanism that predominantly involves direct interactions with the virus but that does not involve viral aggregation or inhibition of binding or uptake of virus by cells. LL-37 may be an important contributor to the initial innate defence against IAV.
Methods
Virus preparations.
IAV strain A/Philippines/82/H3N2 (Phil82) was kindly provided by Dr E. Margot Anders (University of Melbourne, Melbourne, Australia). Strain A/PR-8/34/H1N1 (PR-8) was graciously provided by Jon Abramson (Wake Forest University, Winston-Salem, NC, USA). These IAV strains were grown in chorioallantoic fluid of 10-day-old chicken eggs and purified on a discontinuous sucrose gradient, as described previously (Hartshorn et al., 1988). The virus was dialysed against PBS to remove sucrose, aliquotted and stored at −80 °C until needed. After thawing, the viral stocks contained ~5×108 infectious f.f.u. ml−1. The mouse-adapted strain A/WSN/33/H1N1 (WSN) was purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA). Strain A/WSN/33 HAnc-Asp225Gly/H1N1 (WSN mutant) was kindly provided by Dr Donald Smee (Utah State University, Logan, UT, USA) (Smee et al., 2008) and grown in MDCK cells.
LL-37, collectin and HNP preparations.
LL-37 was purchased from Phoenix Pharmaceuticals. The scrambled LL-37 preparation was purchased from Abgent Inc. Recombinant human SP-D dodecamers were produced in CHO cells as described previously (Hartshorn et al., 1996). The endotoxin level of all SP-D preparations was 0.1–0.5 EU ml−1 (Limulus lysate assay; Cambrex). SP-A was prepared from bronchoalveolar lavage fluids of patients with alveolar proteinosis as described by Gaynor et al. (1995) and was kindly provided by Dr Frank McCormack (University of Cincinnati School of Medicine, Cincinnati, OH, USA). HNP-1 and HNP-2 were purchased from Bachem. HDL (catalogue no. L-1567) and apolipoprotein A1 (A-0744) were obtained from Sigma-Aldrich.
HA inhibition assay.
HA inhibition was measured by serially diluting LL-37 in round-bottomed 96-well plates (Serocluster U-vinyl plates; Costar) using PBS as a diluent and human type-O red blood cells as described previously (Hartshorn et al., 1994).
Binding of LL-37 to IAV, S. aureus, HDL or SP-D.
Binding of LL-37 was assessed by solid-phase ELISA. Plates were coated with IAV, S. aureus or HDL (10 µg ml−1; 3×106 fluorescent foci per well) in coating buffer (15 mM Na2CO3, 35 mM NaHCO3, pH 9.6) overnight at 4 °C. PBS containing 2.5 % BSA (fraction V, fatty-acid-free and low endotoxin; Sigma-Aldrich) was coated onto plates as a background control. Following three washings with PBS, the plates were blocked with 2.5 % BSA for 3 h. The plates were then incubated with LL-37 for 30 min at 37 °C and then washed with PBS with 0.02 % Tween 20, followed by addition of rabbit polyclonal antibodies against LL-37 (Phoenix Pharmaceuticals). Bound anti-LL-37 antibody was detected with HRP-labelled goat anti-rabbit antibodies using tetramethylbenzidine as a substrate (Bio-Rad). The reaction was stopped using 0.5 M sulfuric acid. The A450 was measured on an ELISA plate-reader. Each individual data point was determined in duplicate. Background non-specific binding was assessed by coating plates with fatty-acid-free BSA but no IAV or bacteria as outlined earlier. To test binding of SP-D to LL-37 or HNP-2, plates were coated with LL-37 or HNP-2 overnight as described above and various concentrations of SP-D were added the next day followed by detection with murine mAb directed against SP-D.
Fluorescent focus assay of IAV infectivity.
MDCK cell monolayers were prepared in 96-well plates and grown to confluence. These layers were then infected with diluted IAV preparations for 45 min at 37 °C in PBS. MDCK cells were tested for the presence of IAV-infected cells after 7 h of virus addition using a mAb directed against the IAV nucleoprotein (provided by Dr Nancy Cox, CDC, Atlanta, GA, USA) as described previously (Hartshorn et al., 2000a). The number of fluorescent foci (FFC) per ml of inoculum was calculated from this. IAV was pre-incubated for 30 min at 37 °C with various concentrations of LL-37 or control buffer, followed by addition of these viral samples to the MDCK cells. In some experiments, IAV was added to MDCK cells directly (without pre-incubation with LL-37) followed by incubation for 45 min at 37 °C. In some assays, primary human tracheobronchial epithelial (HTBE) cells were used. These cells were purchased from the ATCC and propagated in the undifferentiated state in standard tissue culture flasks. After 45 min, the plate was washed and various concentrations of LL-37 were then added, followed by incubation for 6.15 h at 37 °C. LDH release from cells was tested using a kit purchased from Roche.
Measurement of viral aggregation by LL-37.
Viral aggregation caused by LL-37 alone and in combination with SP-D was measured by assessing light scattering by suspensions of IAV. This was done by measuring optical density using a Perkin Elmer Lambda 35 UV/Vis spectrophotometer. In addition, viral aggregation was assessed using EM as described previously (Tecle et al., 2007).
Measurement of viral RNA.
RNA for the viral M protein was measured using real-time PCR. A549 cells were infected with Phil82 virus strain incubated for 30 min at 37 °C with or without various doses of LL-37. RNA extraction was done at 45 min and 24 h post-infection using a Magmax viral RNA isolation kit (Applied Biosystems) according to the manufacturer’s instructions. Both lysed cells and cell supernatant were used for extraction. Viral RNA was also extracted from different concentrations of virus with known FFC ml−1, which was used as a standard series. RNA was reverse-transcribed using TaqMan reverse transcription reagents (Applied Biosystems). The reaction mixture and cycle conditions were chosen according to the manufacturer’s instructions. For real-time PCR, primers specific for the IAV M protein (forward, 5′-AGACCAATCCTGTCACCTCTGA; reverse, 5′-CTGCAGTCCTCGCTCACT) were used. The primers and TaqMan-labelled probes with non-fluorescent minor groove binder (MGB) moieties were designed manually using the Primer Express software version 3.0 (Applied Biosystems) and were also synthesized by Applied Biosystems. The assay sequences were examined for specificity by nucleotide blast. The experiment was performed in a 7500 Real-time PCR system (Applied Biosystems) in a total volume of 20 µl containing 2 µl template cDNA, 0.9 µM primer, 0.25 µM 6-FAM-labelled TaqMan MGB probe (6-FAM–5′-ATTTGTGTTCACGCTCACCGTG–MGB) and 1× TaqMan Universal PCR master mix (Applied Biosystems). Thermal cycling proceeded at 50 °C for 2 min and 95 °C for 10 min followed by 40 cycles of 95 °C for 15 s, 60 °C for 1 min and 72 °C for 30 s. For calculation of FFC ml−1 from the cycle threshold (Ct) values, we first plotted a standard curve using known values of log10(FFC ml−1) and corresponding Ct values. The Ct values of samples were converted to values of log10(FFC ml−1) (x) using the formula y = mx+c, where y is the Ct value, m is the slope and c is the intercept. Slope and intercept were calculated from the standard curve using Microsoft Excel. Values of log10(FFC ml−1) were converted to FFC ml−1 by taking the antilogarithm (10^log10).
Confocal microscopy.
For these experiments, the Phil82 strain of IAV was labelled with Alexa Fluor 594. The Alexa Fluor 594 carboxylic acid, succinimidyl ester labelling kit was purchased from Molecular Probes and labelling was carried out using the manufacturer’s recommendations with some modifications. In brief, concentrated virus stock was incubated with Alexa Fluor in sodium bicarbonate buffer (pH 8.3) for 1 h at room temperature. The preparation was then dialysed overnight against PBS at 4 °C. After this procedure, there was no reduction in viral HA titre. MDCK cells were pre-incubated with the labelled virus for 45 min, followed by washing and fixation using 1 % paraformaldehyde. Before this, the IAV was pre-incubated with either control buffer, LL-37 or SP-D for 30 min at 37 °C in the same manner as in the infectious focus assay. Wheat-germ agglutinin (WGA)–Oregon green 488 (4 µg ml−1) and DAPI 350 were used to stain the cell membrane and nucleus, respectively. Confocal pictures were taken with a Zeiss LSM510 microscope (LSEB) at 100× resolution.
Statistics.
Statistical comparisons were made using Student’s paired, two-tailed t test or ANOVA with post hoc test (Tukey’s). ANOVA was used for multiple comparisons to a single control.
Acknowledgements
This work was supported by NIH grants HL06931 (K. L. H.) and AI083222 (K. L. H.).
References
- Barlow P. G., Svoboda P., Mackellar A., Nash A. A., York I. A., Pohl J., Davidson D. J., Donis R. O. (2011). Antiviral activity and increased host defense against influenza infection elicited by the human cathelicidin LL-37. PLoS ONE 6, e25333. 10.1371/journal.pone.0025333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman P., Walter-Jallow L., Broliden K., Agerberth B., Söderlund J. (2007). The antimicrobial peptide LL-37 inhibits HIV-1 replication. Curr HIV Res 5, 410–415 10.2174/157016207781023947 [DOI] [PubMed] [Google Scholar]
- Braff M. H., Jones A. L., Skerrett S. J., Rubens C. E. (2007). Staphylococcus aureus exploits cathelicidin antimicrobial peptides produced during early pneumonia to promote staphylokinase-dependent fibrinolysis. J Infect Dis 195, 1365–1372 10.1086/513277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton M. F., Steel P. G. (2009). The chemistry and biology of LL-37. Nat Prod Rep 26, 1572–1584 10.1039/b912533g [DOI] [PubMed] [Google Scholar]
- Caverly J. M., Diamond G., Gallup J. M., Brogden K. A., Dixon R. A., Ackermann M. R. (2003). Coordinated expression of tracheal antimicrobial peptide and inflammatory-response elements in the lungs of neonatal calves with acute bacterial pneumonia. Infect Immun 71, 2950–2955 10.1128/IAI.71.5.2950-2955.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chromek M., Slamová Z., Bergman P., Kovács L., Podracká L., Ehrén I., Hökfelt T., Gudmundsson G. H., Gallo R. L. & other authors (2006). The antimicrobial peptide cathelicidin protects the urinary tract against invasive bacterial infection. Nat Med 12, 636–641 10.1038/nm1407 [DOI] [PubMed] [Google Scholar]
- Crouch E., Nikolaidis N., McCormack F. X., McDonald B., Allen K., Rynkiewicz M. J., Cafarella T. M., White M., Lewnard K. & other authors (2011). Mutagenesis of surfactant protein D informed by evolution and x-ray crystallography enhances defenses against influenza A virus in vivo. J Biol Chem 286, 40681–40692 10.1074/jbc.M111.300673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doss M., White M. R., Tecle T., Gantz D., Crouch E. C., Jung G., Ruchala P., Waring A. J., Lehrer R. I., Hartshorn K. L. (2009). Interactions of alpha-, beta-, and theta-defensins with influenza A virus and surfactant protein D. J Immunol 182, 7878–7887 10.4049/jimmunol.0804049 [DOI] [PubMed] [Google Scholar]
- Doss M., White M. R., Tecle T., Hartshorn K. L. (2010). Human defensins and LL-37 in mucosal immunity. J Leukoc Biol 87, 79–92 10.1189/jlb.0609382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudreault E., Gosselin J. (2008). Leukotriene B4 induces release of antimicrobial peptides in lungs of virally infected mice. J Immunol 180, 6211–6221 [DOI] [PubMed] [Google Scholar]
- Gaynor C. D., McCormack F. X., Voelker D. R., McGowan S. E., Schlesinger L. S. (1995). Pulmonary surfactant protein A mediates enhanced phagocytosis of Mycobacterium tuberculosis by a direct interaction with human macrophages. J Immunol 155, 5343–5351 [PubMed] [Google Scholar]
- Hansdottir S., Monick M. M., Hinde S. L., Lovan N., Look D. C., Hunninghake G. W. (2008). Respiratory epithelial cells convert inactive vitamin D to its active form: potential effects on host defense. J Immunol 181, 7090–7099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartshorn K. L. (2010). Role of surfactant protein A and D (SP-A and SP-D) in human antiviral host defense. Front Biosci (Schol Ed) 2, 527–546 10.2741/s83 [DOI] [PubMed] [Google Scholar]
- Hartshorn K. L., Collamer M., Auerbach M., Myers J. B., Pavlotsky N., Tauber A. I. (1988). Effects of influenza A virus on human neutrophil calcium metabolism. J Immunol 141, 1295–1301 [PubMed] [Google Scholar]
- Hartshorn K. L., Crouch E. C., White M. R., Eggleton P., Tauber A. I., Chang D., Sastry K. (1994). Evidence for a protective role of pulmonary surfactant protein D (SP-D) against influenza A viruses. J Clin Invest 94, 311–319 10.1172/JCI117323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartshorn K., Chang D., Rust K., White M., Heuser J., Crouch E. (1996). Interactions of recombinant human pulmonary surfactant protein D and SP-D multimers with influenza A. Am J Physiol 271, L753–L762 [DOI] [PubMed] [Google Scholar]
- Hartshorn K. L., Sastry K. N., Chang D., White M. R., Crouch E. C. (2000a). Enhanced anti-influenza activity of a surfactant protein D and serum conglutinin fusion protein. Am J Physiol Lung Cell Mol Physiol 278, L90–L98 [DOI] [PubMed] [Google Scholar]
- Hartshorn K. L., White M. R., Voelker D. R., Coburn J., Zaner K., Crouch E. C. (2000b). Mechanism of binding of surfactant protein D to influenza A viruses: importance of binding to haemagglutinin to antiviral activity. Biochem J 351, 449–458 10.1042/0264-6021:3510449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartshorn K. L., White M. R., Tecle T., Holmskov U., Crouch E. C. (2006). Innate defense against influenza A virus: activity of human neutrophil defensins and interactions of defensins with surfactant protein D. J Immunol 176, 6962–6972 [DOI] [PubMed] [Google Scholar]
- Hashimoto Y., Moki T., Takizawa T., Shiratsuchi A., Nakanishi Y. (2007). Evidence for phagocytosis of influenza virus-infected, apoptotic cells by neutrophils and macrophages in mice. J Immunol 178, 2448–2457 [DOI] [PubMed] [Google Scholar]
- Lai Y., Gallo R. L. (2009). AMPed up immunity: how antimicrobial peptides have multiple roles in immune defense. Trends Immunol 30, 131–141 10.1016/j.it.2008.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrick J. W., Morgan J. G., Palings I., Hirata M., Yen M. H. (1991). Complementary DNA sequence of rabbit CAP18 – a unique lipopolysaccharide binding protein. Biochem Biophys Res Commun 179, 170–175 10.1016/0006-291X(91)91350-L [DOI] [PubMed] [Google Scholar]
- Lau Y. E., Bowdish D. M., Cosseau C., Hancock R. E., Davidson D. J. (2006). Apoptosis of airway epithelial cells: human serum sensitive induction by the cathelicidin LL-37. Am J Respir Cell Mol Biol 34, 399–409 10.1165/rcmb.2005-0170OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P. T., Stenger S., Li H., Wenzel L., Tan B. H., Krutzik S. R., Ochoa M. T., Schauber J., Wu K. & other authors (2006). Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 311, 1770–1773 10.1126/science.1123933 [DOI] [PubMed] [Google Scholar]
- Liu P. T., Stenger S., Tang D. H., Modlin R. L. (2007). Cutting edge: vitamin D-mediated human antimicrobial activity against Mycobacterium tuberculosis is dependent on the induction of cathelicidin. J Immunol 179, 2060–2063 [DOI] [PubMed] [Google Scholar]
- Oppenheim J. J., Yang D. (2005). Alarmins: chemotactic activators of immune responses. Curr Opin Immunol 17, 359–365 10.1016/j.coi.2005.06.002 [DOI] [PubMed] [Google Scholar]
- Salvatore M., Garcia-Sastre A., Ruchala P., Lehrer R. I., Chang T., Klotman M. E. (2007). α-Defensin inhibits influenza virus replication by cell-mediated mechanism(s). J Infect Dis 196, 835–843 10.1086/521027 [DOI] [PubMed] [Google Scholar]
- Smee D. F., Bailey K. W., Wong M. H., O’Keefe B. R., Gustafson K. R., Mishin V. P., Gubareva L. V. (2008). Treatment of influenza A (H1N1) virus infections in mice and ferrets with cyanovirin-N. Antiviral Res 80, 266–271 10.1016/j.antiviral.2008.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen O. E., Follin P., Johnsen A. H., Calafat J., Tjabringa G. S., Hiemstra P. S., Borregaard N. (2001). Human cathelicidin, hCAP-18, is processed to the antimicrobial peptide LL-37 by extracellular cleavage with proteinase 3. Blood 97, 3951–3959 10.1182/blood.V97.12.3951 [DOI] [PubMed] [Google Scholar]
- Tate M. D., Deng Y. M., Jones J. E., Anderson G. P., Brooks A. G., Reading P. C. (2009). Neutrophils ameliorate lung injury and the development of severe disease during influenza infection. J Immunol 183, 7441–7450 10.4049/jimmunol.0902497 [DOI] [PubMed] [Google Scholar]
- Tecle T., White M. R., Gantz D., Crouch E. C., Hartshorn K. L. (2007). Human neutrophil defensins increase neutrophil uptake of influenza A virus and bacteria and modify virus-induced respiratory burst responses. J Immunol 178, 8046–8052 [DOI] [PubMed] [Google Scholar]
- Tjabringa G. S., Aarbiou J., Ninaber D. K., Drijfhout J. W., Sørensen O. E., Borregaard N., Rabe K. F., Hiemstra P. S. (2003). The antimicrobial peptide LL-37 activates innate immunity at the airway epithelial surface by transactivation of the epidermal growth factor receptor. J Immunol 171, 6690–6696 [DOI] [PubMed] [Google Scholar]
- White M. R., Doss M., Boland P., Tecle T., Hartshorn K. L. (2008). Innate immunity to influenza virus: implications for future therapy. Expert Rev Clin Immunol 4, 497–514 10.1586/1744666X.4.4.497 [DOI] [PMC free article] [PubMed] [Google Scholar]