

Abstract
The human immunodeficiency virus (HIV) and acquired immunodeficiency syndrome (AIDS) pandemic is amongst the most important current worldwide public health threats. While much research has been focused on AIDS vaccines that target the surface viral envelope (Env) protein, including gp120 and the gp41 ectodomain, the C-terminal tail (CTT) of gp41 has received relatively little attention. Despite early studies highlighting the immunogenicity of a particular CTT sequence, the CTT has been classically portrayed as a type I membrane protein limited to functioning in Env trafficking and virion incorporation. Recent studies demonstrate, however, that the Env CTT has other important functions. The CTT has been shown to additionally modulate Env ectodomain structure on the cell and virion surface, affect Env reactivity and viral sensitivity to conformation-dependent neutralizing antibodies, and alter cell–cell and virus–cell fusogenicity of Env. This review provides an overview of the Env structure and function with a particular emphasis on the CTT and recent studies that highlight its functionally rich nature.
Introduction
Human immunodeficiency virus (HIV) is the aetiological agent of acquired immunodeficiency syndrome (AIDS). Despite a significant number of breakthroughs since its discovery in 1983 (Barré-Sinoussi et al., 1983; Gallo et al., 1984), the infection remains pandemic. According to the WHO, 2.7 million people were infected worldwide in 2010 (the last date for which data are available) bringing the total infected population to 34 million individuals, with sub-Saharan Africa being the most affected region with 22.9 million people infected. For nearly three decades, scientists and physicians have been working together to fight the epidemic. Different approaches have been assessed, and some of them have shown significant advances to slow the progression of the disease, such as highly active anti-retroviral therapy. But these strategies, despite successfully increasing the lifespan for treated patients, do not indefinitely halt the progression of the infection. Thus, many efforts have also been made towards the development of an HIV vaccine through the testing of numerous candidates in human clinical trials (for review see McElrath & Haynes, 2010). To date, no or only modest efficacy has been observed. Most of these trials have focused on the HIV-1 envelope as the primary target.
HIV is a member of the family Retroviridae, and belongs to the genus Lentiviridae. As a retrovirus, RNA is the genetic material for HIV, and each virion contains two positive-strand copies of the viral genome containing a 5′ cap and a 3′ poly(A) tail, and as such, the viral genome is not infectious (Luciw et al., 2002). Morphologically, HIV is an irregular, roughly spherical virus with a characteristic electron-dense conical core surrounded by a lipid envelope that is derived from the host cell during the budding process (Fig. 1) (Luciw et al., 2002). The virus commonly infects CD4+ T-cells, macrophages or dendritic cells. The envelope protein (Env) of the virus is key to the viral infection process as it functions to bind to the receptor and co-receptors at the surface of target cells. Env is the only viral protein exposed on the virion surface, and it is the main target of the host humoral immune system (McElrath & Haynes, 2010). The targeting of Env, through direction of the immune response by vaccination or by traditional small-molecule pharmaceuticals provides arguably the best hope for controlling the epidemic as the goal of these therapies is to prevent the initial infection event (McElrath & Haynes, 2010). As such, the study of Env structure and function has yielded important insights into the properties and features of these viral proteins.
Fig. 1.
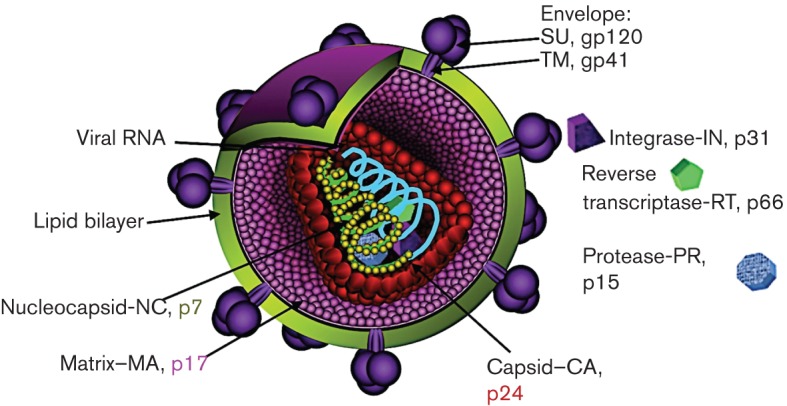
HIV virion structure. Schematic representation of a mature HIV virion detailing the localization of viral proteins and the approximate virion structure. The representation is not to scale.
Functional Env is a trimer of heterodimers derived from extensive processing of the env gene product, as presented schematically in Fig. 2. Env is translated and co-translationally N-glycosylated as a 160 kDa polyprotein (gp160) in the endoplasmic reticulum (ER). Env gp160 is thought to multimerize in the ER into predominantly trimers before trafficking to the Golgi (Earl et al., 1990, 1991; Pinter et al., 1989). In the Golgi, Env gp160 is cleaved by a cellular furin-like protease at a conserved K/R(X)K/RR motif into gp120 [or surface unit (SU)] and gp41 (or transmembrane) (Freed et al., 1989; McCune et al., 1988). The cleaved gp120 and gp41 products non-covalently associate to form the active trimeric Env unit.
Fig. 2.
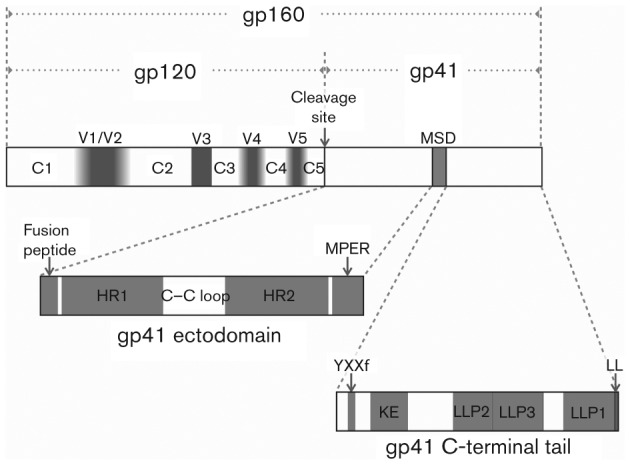
HIV Env primary structure. The Env primary structure is presented schematically, with the immature polyprotein labelled gp160 and the mature, cleaved protein labelled gp120 and gp41. Protein domains are labelled within each mature protein. In gp120 the constant regions are labelled C1–C5 and the variable domains are labelled V1–V5. The shading in the variable regions indicates the relative plasticity of variable region length, with V1/V2 ranging from 50 to 90 aa, V4 from 19 to 44 and V5 from 14 to 36 aa, while V3 does not vary appreciably in size (reviewed by Checkley et al., 2011). In the gp41 ectodomain, the heptad repeat regions (also called the N- and C-heptad repeats) are labelled HR1 and HR2, respectively, and the membrane proximal external region is labelled MPER. In the gp41 CTT, the Kennedy epitope is labelled KE and the lentivirus lytic peptide sequences are labelled LLP1, LLP2 and LLP3. Functional endocytic motifs are labelled YXXΦ (near the N terminus) and LL (at the C terminus).
gp120 structure and function
The primary function of gp120 is to initiate and modulate virus–cell interactions through binding to the primary receptor, CD4, and the co-receptor, predominantly CCR5 or CXCR4. gp120 initiates virus–cell contact by binding to CD4 and subsequently undergoes structural rearrangements that result in the formation of the co-receptor-binding site. These conformational changes allow for high affinity binding of gp120 to co-receptor that in turn leads to conformational changes in gp41 to begin the process of virus–cell membrane fusion (Myszka et al., 2000). Because of its receptor binding functionality and the identification of antibodies that exhibit cross-clade neutralizing activity (broadly neutralizing antibodies), gp120 is a major focus for experimental vaccines (McElrath & Haynes, 2010). In addition, one commercially licensed small molecule therapeutic, maraviroc, targets the gp120–co-receptor-binding interaction (Dragic et al., 2000). However, maraviroc acts by binding CCR-5, and does not specifically interact with gp120 and is inactive against viruses that utilize CXCR4 as co-receptor (Dragic et al., 2000).
gp120 is a structurally complex molecule. Sequence analyses of gp120 from diverse HIV-1 isolates identified a high degree of variation, resulting from the high error rate of reverse transcriptase and biological selection (Preston et al., 1988; Roberts et al., 1988). Currently, variation in gp120 is understood to reflect an evolutionary struggle between the host immune system and the virus, with circulating sequences reflecting viruses not yet recognized by the host immune response. gp120 is characterized as containing five variable regions (V1–V5) sequentially occurring in the primary sequence with five relatively conserved regions (C1–C5) (Starcich et al., 1986; Willey et al., 1986). Sequence analyses also identified 18 highly conserved cysteine residues that form nine disulfide bonds that play an important role in the formation of gp120 tertiary structure (Leonard et al., 1990). The variable regions determined by sequence analyses coincided with loop structures that were predicted to be formed by the disulfide bonds (Leonard et al., 1990). The presence of these loops has been confirmed by X-ray crystal structures of gp120 proteins (Huang et al., 2005; Kwong et al., 1998). In addition, gp120 contains 20–30 canonical N-linked glycosylation sites and is extensively glycosylated with oligosaccharides accounting for approximately half of its molecular mass. The location and number of glycosylation sites also varies by strain and change over time during the course of infection (Allan et al., 1985; Montefiori et al., 1988). The variations in sequence and the uniquely dense glycosylation are thought to be important mechanisms by which the virus evades the humoral immune response.
HIV-1 gp120 has been studied extensively, and a number of atomic resolution structures have been elucidated, both in the unliganded form (Kwon et al., 2012) and in conjunction with one or more binding partners, including CD4 and co-receptor-binding site mAbs 17B (Kwong et al., 1998) and X5 (Huang et al., 2005), or CD4-binding site mAbs b12 (Zhou et al., 2007), VRC01 and VRC03 (Wu et al., 2010). Due to what is likely extensive flexibility, the V1/V2 loops were deleted from gp120 proteins used for structure determination (Kwong et al., 1998). The relative location of the V1/V2 loops on the virion-associated Env trimer has been recently determined, however, by fitting (V1/V2 deleted) gp120-CD4 crystal structures into cryo-electron microscopy (cryo-EM) density maps of CD4-bound Env (Liu et al., 2008). The inclusion of CD4 in the cryo-EM Env structure determination allowed for unambiguous orientation and fitting of pre-determined CD4/gp120 crystal structures that led to the identification of V1/V2 density in the cryo-EM Env structure (Fig. 3) (Liu et al., 2008; White et al., 2010).
Fig. 3.
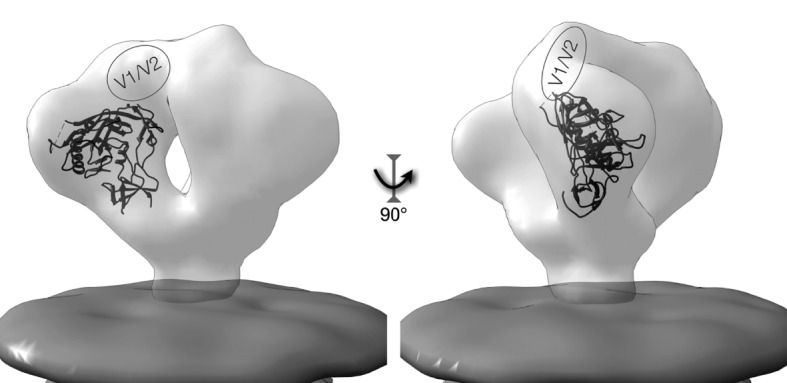
Structure of virion-associated Env. The structure of HIV Env is presented with the relative localization of a gp120 subunit within the virion-associated trimeric structure. The Env volume is displayed in light grey, while the membrane is darker grey at the bottom of the figure. The figure was constructed using the EM Database files EMD-5019 (Env) and EMD-5022 (membrane) (Liu et al., 2008) and the gp120 co-ordinates from PDB file 2NY7 (Zhou et al., 2007), with gp120 positioned into the EM density map using the ‘Fit in Map’ tool in UCSF Chimera (Pettersen et al., 2004).
gp41 structure and function
gp41 can be structurally sub-divided into three major domains (see Fig. 2): the extracellular domain (or ectodomain); the membrane-spanning domain (MSD); and the C-terminal tail (CTT), also referred to classically as the intracytoplasmic tail. Of these, experimental protein structures for only the gp41 ectodomain have been determined at atomic detail, and then only for the six-helix bundle of what is generally considered the fusion-competent state (Caffrey et al., 1998; Chan et al., 1997; Shi et al., 2010). Little to no structural information exists on the native structure of gp41. Even recent cryo-EM structures of Env on the virion surface provide little insight into the structure of gp41 beyond that it appears to present as a thin stalk supporting the gp120 domains (Liu et al., 2008; White et al., 2010).
gp41 ectodomain
The location and function of the gp41 ectodomain is well-studied and generally unquestioned. gp41 is also N-glycosylated, although to a much lesser extent than gp120, with three to five potential sites (Montefiori et al., 1988). Initial modelling studies of the gp41 ectodomain from the primary amino acid sequence revealed a remarkable similarity of the predicted structure to the crystal structure of the influenza HA protein, despite the lack of significant amino acid sequence homology (Gallaher et al., 1989). Subsequent structural studies of gp41 ectodomain sequences, including X-ray crystallography, confirmed the predicted structure of the gp41 ectodomain. The ectodomain contains the major determinants of membrane fusion (Fig. 2): a hydrophobic N-terminal domain termed the fusion peptide (FP) (Bosch et al., 1989; Freed et al., 1990, 1992); two heptad repeat regions, termed HR1 and HR2 (also referred to as the N- and C-helix, respectively) that form α-helical coiled-coil structures and are linked by a disulfide-bridged loop (Caffrey et al., 1998; Chan et al., 1997; Dubay et al., 1992; Lu et al., 1995; Weissenhorn et al., 1997); and a tryptophan-rich region referred to as the membrane-proximal external region (MPER) (Muñoz-Barroso et al., 1999; Salzwedel et al., 1999). Additionally, the MPER is an important target for broadly neutralizing mAbs (Montero et al., 2008).
The primary function of the gp41 ectodomain is to drive virus–cell membrane fusion. The gp41 FP is normally buried in the gp120/gp41 complex, but following conformational rearrangements induced by receptor and co-receptor binding, FP is exposed and inserts into the target cell membrane. The three HR1 domains then fold into a stable six-helix bundle in an anti-parallel fashion with the three HR2 domains (Fig. 4) to bring the virus and cell into close proximity to allow fusion to occur (Chan et al., 1997; Melikyan, 2011; Weissenhorn et al., 1997). The mechanism by which the actual fusion step occurs is not well elucidated. However, peptide analogues of both the FP and the MPER have been demonstrated to both disorder and reduce the bending modulus for lipid bilayers, and thus may lower the free energy barrier to membrane fusion, a decidedly non-spontaneous process (Greenwood et al., 2008; Tristram-Nagle et al., 2010; Tristram-Nagle & Nagle, 2007).
Fig. 4.

Schematic representation of the virus and cell fusion. (a) Virus binds to the target cell through the interaction of gp120 SU with CD4 cellular receptor. (b) This interaction activates a conformational change of the viral envelope leading to unmasking of the FP from gp41, which inserts in the cellular plasma membrane. The segment portion of HR2 interacts with HR1 in an anti-parallel way in the trimer, forming the six helix bundle. (c) This conformational change of the envelope brings the viral and cellular plasma membrane in close proximity, allowing their fusion. Only the external leaflets of both membranes are fused such that this step is called the hemi-fusion intermediate. (d) Both external and inner leaflets of the membranes are fused, leading to the complete fusion.
gp41 membrane-spanning domain
In contrast to the gp41 ectodomain, the gp41 MSD remains, despite much study, structurally relatively undefined. Early studies identified a hydrophobic 25 aa sequence that acted as a ribosomal stop-transfer signal, leading to the classical identification of the gp41 ectodomain, MSD, and intracytoplasmic tail (Haffar et al., 1988). The MSD amino acid sequence is highly conserved among HIV isolates, and contains a conserved GXXXG helix–helix interaction motif and a highly conserved midspan arginine residue. The classical structural view of the MSD is that of a single membrane-spanning α-helix. The presence of the midspan arginine, as well as results from both previous and more recent studies, however, lead to conflicting views of the sequence and structural characteristics of the MSD in contrast to the classical definition. These studies are briefly discussed below.
Prior studies attempting to generate antibodies (in rabbits) to peptides spanning the full Env protein (gp120 and gp41) identified a strongly reactive peptide derived from positions 728 to 745 that is referred to as the Kennedy epitope (KE) (Chanh et al., 1986; Kennedy et al., 1986) located in the CTT. Intriguingly, the sera directed to the KE strongly neutralized virus (Chanh et al., 1986), suggesting an external localization of the epitope on the virion, as antibodies cannot cross intact lipid membranes. These results were largely dismissed in light of the identification of the stop-transfer signal and the division of gp41 into the classical external, membrane-spanning and intracytoplasmic domains (Haffar et al., 1988). More recently, however, accumulating evidence also points to possibly different MSD sequence and/or structure. Studies examining the exposure of the KE on the surface of Env-expressing cells suggest that under some circumstances the KE is extracellularly exposed, indicating a need for either additional MSD sequences, or an alternative structure for the MSD other than the traditionally held α-helix (Cheung et al., 2005; Cleveland et al., 2003; Heap et al., 2005; Reading et al., 2003). The two proposed MSD models are shown in Fig. 5.
Fig. 5.

Proposed models for the membrane-spanning sequences of HIV gp41. The two models for the HIV gp41 MSD are shown schematically using the sequence from HIV-1 89.6. (Left) The traditional model, with the MSD as a single α-helix. (Right) The alternative model proposed by Hollier & Dimmock (2005) with three membrane-spanning β-sheets. Residues are numbered according to the standard HXB2 sequence, and residues 706–746 are left out for simplicity.
One of the most successful techniques for examining the structure of the MSD has been the use of molecular dynamics (MD) simulations. Two recent studies use MD simulations to examine the stability of monomeric and trimeric forms of the MSD in lipid membranes (Kim et al., 2009) and conformational variation in the MSD (Gangupomu & Abrams, 2010). A general caveat of these techniques, however, is that the starting assumptions can have a major impact on the outcome. As such, both studies begin with the assumption that the MSD sequence adopts a helical conformation in the membrane. In general, this is a reasonable assumption given that the most energetically favourable conformation of a peptide in a membrane is an α-helix due to the internal stabilization of all potential hydrogen-bonding partners (White & Wimley, 1999). It does not, however, account for the experimental data that suggests alternative gp41 topologies, and thus alternative sequences or structures for the MSD. With that understanding, both studies provide interesting insights into the potential structure of the MSD. Engelman and colleagues demonstrated a remarkable stability of the HIV MSD sequence (using FIMIVGGLVGLRIVFAVLSI) as a monomeric helix in the lipid membrane, but rapid unfolding of the helix in water (Kim et al., 2009). They also demonstrated conformational stability of a right-handed helical bundle that increased further when the central arginine residues were deprotonated (Kim et al., 2009). These results suggest that a right-handed helical bundle with deprotonated arginine residues is the most stable trimeric conformation for the gp41 MSD sequence. In contrast, Gangupomu and Abrams examined the conformational flexibility of a monomeric MSD sequence (using KLFIMIVGGLVGLRIVFAVLSIVNRVR). Their findings indicate that the MSD monomer forms a stable-tilted helix in the membrane, but that metastable states are formed when the midspan arginine ‘snorkels’ to either side of the membrane (Gangupomu & Abrams, 2010). When the arginine snorkels towards the outer leaflet, the N-terminal end of the MSD is unfolded. However, when the midspan arginine snorkels towards the inner leaflet the MSD kinks at phenylalanine 697, and the remaining 10 aa form a helix along the inner leaflet at the water–membrane interface (Gangupomu & Abrams, 2010). Collectively, and importantly, these MD simulation results suggest multiple (meta)stable conformations of the MSD, even in an α-helical state.
Functionally, the primary role of the MSD is to anchor the Env protein to the cellular and viral membrane. Additional studies have identified a role for the MSD in overall Env function. In particular, mutations in the MSD have been shown to have an effect on Env-mediated fusion such that mutations of conserved residues in the MSD, including the midspan arginine, led to decreased cell–cell fusion and decreased viral infection (Kondo et al., 2010; Shang & Hunter, 2010; Shang et al., 2008; Yue et al., 2009). The mechanisms by which these mutations act to decrease fusion are not well understood.
gp41 C-terminal tail
Structure
Of the three gp41 domains, the structure of the gp41 CTT is the least understood. Results from early topogenesis studies led to the view of gp41 (and thus Env as a whole) as a type I membrane protein, with an extracellular N terminus, a single MSD, and an approximately 150 aa long cytoplasmic CTT (Haffar et al., 1988). Comparisons of this model to other retroviral Env proteins supported this view, with a major difference in the length of the proposed cytoplasmic CTT. For most retroviral Env proteins, the cytoplasmic CTT sequences are generally between 20 and 40 aa long (Checkley et al., 2011). The presence of a very long CTT is not unique to HIV, as other lentiviruses such as the simian immunodeficiency virus (SIV) and the equine infectious anemia virus have similarly long CTTs, at 150 and 200 aa, respectively (Rushlow et al., 1986). The presence of a long CTT in most lentiviruses suggests an important functional role, as viruses do not generally replicate non-functional sequences. This functional importance is supported by the finding that truncation of the CTT leads to in vivo suppression of viral replication in animal models (Shacklett et al., 2000). A decade earlier, however, studies demonstrated that the CTT was dispensable for in vitro viral replication (Chakrabarti et al., 1989; Hirsch et al., 1989; Kodama et al., 1989), leading to a long-held view that the CTT was not functionally important. This view was subsequently popularized by the finding that truncation of the CTT led to increased Env incorporation into the virion, which was an important consideration at the time as a means by which to boost the anti-Env immune response in vaccine studies. It is now generally accepted that the CTT plays multiple important functional roles in the virus life cycle. Structurally, very little is still known and no atomic level structures exist for full-length CTT sequences.
Structural information has been determined for some CTT sequences. The most well-studied domains, the lentivirus lytic peptides (LLPs), are sequences that were initially identified by sequence scanning as having extraordinarily high hydrophobic moments (Eisenberg & Wesson, 1990; Miller et al., 1991). Subsequent studies on peptide analogues of these domains demonstrated high levels of structural similarity with naturally occurring cytolytic amphipathic cationic anti-microbial peptides (Miller et al., 1991) as well as the ability of the peptides to alter cell membrane permeability (Miller et al., 1993a). Peptide analogues of these domains have been demonstrated to be generally unstructured in aqueous buffer, but to rapidly adopt amphipathic α-helical structure in membrane or membrane-mimetic environments as determined by circular dichroism spectroscopy (Chernomordik et al., 1994; Kliger & Shai, 1997; Srinivas et al., 1992; Steckbeck et al., 2011). There are, however, no atomic level structures of the peptides interacting with membranes or membrane mimetics. Recently, the physico-chemical and structural characteristics of the LLPs were determined to be highly conserved among sequences from numerous clades and groups, despite substantial variations in primary amino acid sequences (Steckbeck et al., 2011). In particular, conservation of the membrane-associating amphipathic α-helical structures suggests that the LLP regions may function in anchoring the CTT to the cellular and viral membranes. In addition, the higher and unusual degree of conservation of arginines in the LLP sequences might be of importance in regulating the interaction of the CTT with the membrane (Steckbeck et al., 2011).
Topology and function
As the intracytoplasmic localization of the CTT has been predominantly reinforced implicitly with functional data, the topology and function of the CTT will be presented together. As discussed above, the topology of the CTT has long been considered to have an entirely cytoplasmic localization. Accumulating recent evidence suggests that this may not be the case, or is at least not always the case, and will be developed further in the next section. Early topogenesis studies suggested the organization of Env as a type I membrane protein (Haffar et al., 1988), a view that is predominant and has been implicitly reinforced by a number of experimental functional studies. This is largely due to genetic evidence demonstrating functional interactions of the CTT with known intracellular or intravirion partners. These functional interactions have been found to regulate, or be regulated by: (i) Env incorporation into viral particles (Freed & Martin, 1995; 1996; Jiang & Aiken, 2007; Murakami, 2008; Murakami & Freed, 2000a, b); (ii) virion maturation (Jiang & Aiken, 2007; Joyner et al., 2011; Kol et al., 2007; Wyma et al., 2004); and (iii) endocytosis of Env from the cell surface to late endosomes (Byland et al., 2007; Ohno et al., 1997).
Virion Env incorporation
In the mid-1990s, studies were published that demonstrated a functionally important role for the CTT. These studies are crucial to the current CTT field in that they established conclusively that the CTT was of functional importance, in contrast to the view that arose from studies published in the late 1980s, indicating that the CTT was dispensable for virus replication (Chakrabarti et al., 1989; Hirsch et al., 1989; Kodama et al., 1989). In addition to the effect of CTT-deletion on replication, these studies demonstrated increased Env content in virions containing a CTT-deleted Env and proposed that deletion of the CTT would be a means to boost the anti-Env immune response through the increase in Env per virion. This was the prevailing attitude until seminal studies by Freed and Martin demonstrated that the CTT was implicated in the incorporation of Env into virions through interactions with the MA domain of Gag. Their first study used site-directed mutagenesis to demonstrate that mutations in specific amino acids in Gag MA resulted in deficiencies in incorporation of Env with a full-length CTT (Freed & Martin, 1995). The MA mutations did not, however, lead to reduced incorporation of heterologous retroviral Env proteins with naturally short CTT sequences. Further, specific truncation of CTT sequences to seven or 47 residues (from the original 150) reversed the Env-incorporation block imposed by the MA mutations. This paper has demonstrated for the first time conclusive evidence of a functional role for the CTT through direct or indirect interactions with an intracellular partner, Gag MA.
The role and localization of sequences important in the MA–CTT interaction has been further elucidated. A subsequent paper by the same authors demonstrated that truncating up to 56 residues from the CTT (leaving a 94 residue CTT) eliminated Env incorporation in viruses containing MA mutations leading to deficiencies in Env incorporation, but that truncations of 93 aa or greater (leaving only 57 aa CTT) relieved the block caused by MA mutations, resulting in efficient Env incorporation (Freed & Martin, 1996). Later papers from this group localized the MA–CTT interaction to LLP2 in the CTT (Murakami & Freed, 2000a) and further demonstrated that the interaction was crucial for Env virion incorporation in a cell-type-specific manner (Murakami & Freed, 2000b). It remains unclear whether the interaction between MA and the CTT occurs through a direct or indirect mechanism. In 1996 Cosson published the first, and to date only, in vitro demonstration of a direct interaction between these two proteins by GST pulldown (Cosson, 1996). However, in 2006 another group showed that a cellular protein, TIP47 (tail-interacting protein of 47 kDa) was necessary for a functional interaction between Env and Gag (Lopez-Vergès et al., 2006). The role of TIP47 in the Env–Gag interaction is currently unclear, and further studies will be necessary to determine whether the Gag–Env interaction occurs through a direct or indirect mechanism (Checkley et al., 2011).
The Env–Gag interaction is also of importance for the correct localization of Env in the plasma membrane and consequently for the efficient incorporation of Env in the virions. Indeed, the processed Env protein is transported to the plasma membrane and more specifically to the lipid rafts where it interacts with Gag, participating in viral assembly and virus budding (Nguyen & Hildreth, 2000). The palmitoylation of two cysteines in the cytoplasmic domain of gp41 has been indicated as necessary for the correct targeting of Env to these regions (Bhattacharya et al., 2004; Rousso et al., 2000). Later, it was shown that Gag was responsible for the recruitment of Env to the lipid rafts (Bhattacharya et al., 2006; Patil et al., 2010) and that palmitoylation of cysteines was not involved (Chan et al., 2005). Indeed, deletion or mutation of Gag renders Env unable to localize to lipid rafts, limiting its incorporation into virions (Bhattacharya et al., 2006; Patil et al., 2010). Interaction with Gag and a membrane-proximal tyrosine-based motif overlapping the endocytosis motif in the CTT have also been shown to be important to polarized assembly and release of the virus (Deschambeault et al., 1999; Lodge et al., 1994, 1997). Whether the interaction between Gag and Env occurs at the plasma membrane or during their transport to the plasma membrane has not yet been determined.
Some published data suggest a role for the interaction between Env and Gag in HIV infectivity during virion maturation, as described below.
Virion maturation
Aiken and colleagues examined virion organization by characterizing the effect of pelleting immature HIV virus particles (in which the Pr55Gag has not been cleaved) through detergent (Wyma et al., 2000). Pelleting particles through detergent strips away the viral lipid membrane in which the CTT is embedded, leaving viral protein cores. Their reasoning was if the CTT and Pr55Gag interact, then gp41 should be associated with the pelleted viral cores. They found that the pelleted cores retained the major fraction of the gp41 found in untreated virions (Wyma et al., 2000). Virions treated similarly but with a truncated CTT did not retain the gp41 with the pelleted cores (Wyma et al., 2000). This study does not preclude, however, the presence of a mediating protein that facilitates the interaction between gp41 CTT and PR55Gag. Interestingly, a previous study using mature viral particles for a similar analysis demonstrated no association of the CTT with MA (Kotov et al., 1999). Thus, taken together, these results suggested that the association of the CTT with Pr55Gag is dependent on the maturation state of the virion.
This maturation-dependent association of the CTT with Gag has been extended to examine the role of the CTT in viral fusion. It is well established that HIV particles are not infectious until proteolytic cleavage of the Pr55Gag into its constituent domains (MA, CA and NC, predominantly). Using a reporter assay to measure virus–cell fusion, Wyma et al. demonstrated that immature virions were less fusogenic than mature virions in a manner that was dependent on the CTT, as truncations of the CTT resulted in identical fusogenicity of immature and mature viral particles (Wyma et al., 2004). More specifically, the interaction of gp41 CTT with an unprocessed Gag inhibits fusion, which is restored by Gag processing or CTT deletion (Murakami et al., 2004). More recent results suggest that the extreme C terminus of the CTT, LLP1, modulates the maturation dependence of infectivity, while deletion of this region does not affect the incorporation of Env into immature viral particles (Jiang & Aiken, 2007). As such, the interaction between the CTT and Gag is regulated by the maturation state of the viral particles, thereby modulating fusion and thus the efficiency of infectivity. The CTT has also been shown to modulate the mechanical stability of immature virus particles relative to mature virus particles, presumably through its interactions with Pr55Gag (Kol et al., 2007). These results collectively demonstrate that the interaction of the CTT and Gag have specific effects on the infectivity and structural stability of HIV particles.
Env endocytosis
Another functional role of the CTT has implicitly reinforced the traditional topological model. The gp41 CTT of both SIV and HIV have long been known to contain endocytic signals. This was first demonstrated in HIV-1 in a study of the Env internalization pathway that allows processing for antigen presentation by the class II MHC (Rowell et al., 1995). In this study the authors observed a high rate of Env internalization that was dependent on a tyrosine residue located six positions from the MSD. In parallel, similar observations were reported for SIV where the CTT had been shown to contain an endocytic signal (Sauter et al., 1996). The transfer of the SIV CTT to a chimeric CD4 fusion protein (external CD4 domain associated to the MSD and CTT of SIV) resulted in the internalization of the chimeric protein in a manner dependent on 703tyrosine (Sauter et al., 1996). A consensus YXXΦ motif in the CTT of HIV-1 was then shown to interact with members of the adaptor protein medium chain family (Ohno et al., 1997). It has also been demonstrated that CTT sequences interact specifically with the AP-2 clathrin adaptor (Boge et al., 1998). Most recently, Byland et al. demonstrated that the CTT contains two functional endocytic signals: a 711GYXXΦ motif located near the N terminus of the CTT, and a dileucine motif at the extreme C terminus (Byland et al., 2007). Their results indicate that in order to completely abolish endocytosis of Env from the cell surface to late endosomes, both motifs must be mutated, suggesting that both the N- and C-termini of the CTT are cytoplasmically localized.
Additional tyrosine and dileucine motifs have been identified in the CTT and studied in a recent publication by Hunter and colleagues, demonstrating that these motifs do not affect cell surface expression of Env, but rather its incorporation into virions, with cell–cell fusion and virus infectivity also being affected (Bhakta et al., 2011), suggesting that these additional motifs are not involved in Env endocytosis. As noted by the authors, a number of these motifs overlap the LLP regions, and the observed phenotypic impairment could be due to alterations of LLP functions. However, the 802YW803 motif plays a role in internalization through an interaction with TIP47, allowing the Env retrograde transport (Blot et al., 2003). As mentioned previously, TIP47 is thought to bridge Gag and Env and is necessary for Env incorporation into virions (Lopez-Vergès et al., 2006). Thus, the addition of Gag might allow a redirection of Env from an internalization process to incorporation into virions (Lopez-Vergès et al., 2006).
Overall, known CTT functions in Env virion incorporation and maturation as well as endocytosis of Env from the cell surface provide support for the traditional intracytoplasmic (intravirion) model for the CTT. Fig. 6 presents an overview of the known functionalities associated with the CTT sequences.
Fig. 6.
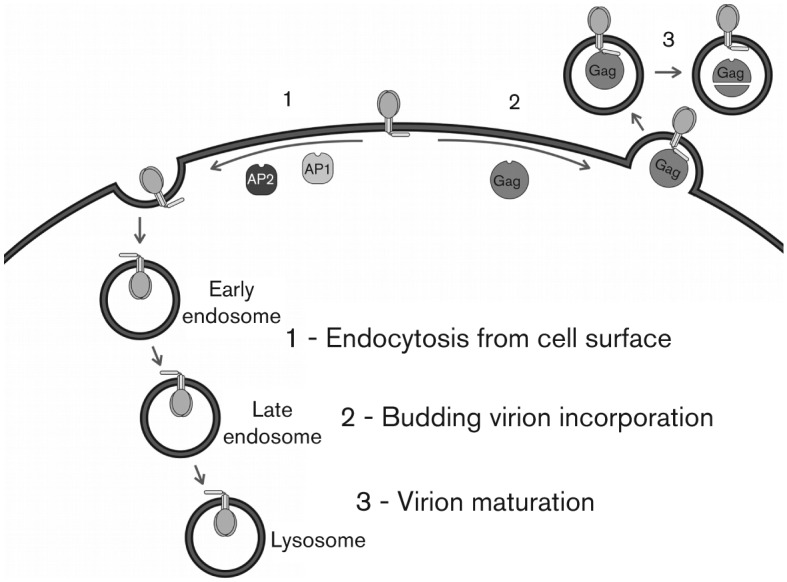
CTT function. Known functional roles of the CTT are presented schematically. (1) The CTT has been demonstrated to influence endocytosis of Env from the cell surface through interactions with AP1 and AP2. (2) Through interactions with Gag, the CTT plays a role in incorporation of Env into budding virions. (3) The interaction between Gag and the CTT may play a role in virion maturation.
CTT cell functions
Little is known about the functions of the CTT beyond its role in trafficking and Env incorporation. Indeed, in addition to the interactions detailed above, the CTT may also have a role during virus entry and in circumventing some cellular functions.
One of the first identified functions of the CTT concerned the LLP domains having a cytolytic and cell–cell fusion inhibition activity as peptides based on functional interactions with lipid membranes (Srinivas et al., 1992). The gp41 subunit also physically interacts with cellular proteins. Indeed, the calmodulin cellular protein binds to the CTT through the LLP1 domain, leading to the inhibition of the calmodulin activity (Miller et al., 1993b; Tencza et al., 1995). Point mutations of arginines in the LLP1 domain are associated with a decreased binding of LLP1 peptides to calmodulin and lead to an impairment of the cytolytic activity (Miller et al., 1993b; Tencza et al., 1995). Interestingly, the LLP peptides also inhibit T-cell activation, one of the calmodulin cellular functions (Srinivas et al., 1993), and Env is associated with an increased sensitivity of T-cells to FAS-mediated apoptosis in an LLP1-calmodulin-binding-dependent manner (Micoli et al., 2000). Thus, these functions of the LLP domains have been proposed to contribute to the cytopathic effect of HIV-1 on T-cells.
Recent studies have demonstrated that the CTT could interact with or affect other cellular pathways. For instance, the HIV-1 CTT has been shown to activate the canonical NF-κB pathway through a direct or indirect interaction with TAK1 cellular protein (Postler & Desrosiers, 2012). This activation appears to be of critical importance in suboptimally activated T-cells. Additionally, the CTT, and more specifically the three LLP domains, has been reported to directly interact with the cellular protein p115-RhoGEF, which is involved in cell proliferation (Zhang et al., 1999). Such an interaction prevented the activation of serum response factor transcription factor, but did not prevent its activation by other factors. In addition, the impairment of this interaction led to a defect in HIV-1 replication in T-cells.
Finally, Luman, a cellular protein belonging to the CREB/ATF family of bZIP transcription factors, also interacts with the CTT (Blot et al., 2006). Luman is anchored in the ER, and following proteolytic cleavage, the truncated, active form of Luman is translocated in the nucleus where it inhibits HIV-1 LTR transactivation by interacting with the viral Tat protein. However, the CTT of gp41 interacts with the ER-embedded inactive form of Luman and decreases the stability of the cellular protein. As such, by acting on the precursor of Luman, gp41 may decrease the amount of active translocated Luman available to inhibit the LTR transactivation by Tat (Blot et al., 2006).
These functions of the CTT are likely of importance for the virus life cycle and disease progression, but significant progress remains to be made to better understand their implications.
Evidence for alternative CTT topologies
Surface exposure of the KE
The derivation of an alternative topological model for CTT comes from a combination of indirect (inferred from neutralization of HIV infection by anti-CTT antibodies) and direct biochemical evidence. Initial studies suggestive of a non-cytoplasmic localization for the CTT were not explicitly performed to determine the CTT topology, but provided the earliest indirect evidence for potential exposure. These studies found that serum from a rabbit immunized with a gp41 peptide (residues 728–745, the KE) bound to HIV Env, and that serum from HIV-1-infected patients bound the synthetic peptide (Kennedy et al., 1986). Subsequent studies demonstrated that anti-KE serum could specifically neutralize HIV in vitro (Chanh et al., 1986). As antibodies cannot cross intact lipid membranes, these results implied the localization of the KE on the outer surface of the virion, in direct contrast to the presumed intravirion localization.
More recently, the exposure of CTT sequences has been explored predominantly by Nigel Dimmock’s lab (for review see Dimmock, 2005). Over 3 years and five manuscripts, his group provided evidence for the extravirion and/or extracellular localization of CTT sequences, and it is his group that proposed the alternative topological model that is contrasted with the traditional model in Fig. 7.
Fig. 7.

Proposed topology models for HIV gp41 CTT. The two predominant CTT topology models are presented. (Left) Traditional CTT topology model, with completely intracytoplasmic CTT. (Right) model proposed by Hollier & Dimmock (2005), with the KE (yellow) externally localized. Transmembrane domains are as presented in detail in Fig. 5. Red boxes indicate functional endocytic sequences.
The initial evidence for external localization of the KE sequences were the result of antibody neutralization and binding studies of anti-CTT antibodies to viral particles (Cleveland et al., 2003). This study used epitope-purified antiserum (against the epitope 746ERDRD750 and termed EPES) to demonstrate high levels of neutralization [concentration necessary to achieve 50 % neutralization (NC50) = 0.3 µg ml−1] of HIV in vitro. Importantly, EPES did not have any neutralizing activity against viruses with a CTT-deleted Env. They further demonstrated that EPES could bind to intact viral particles (where intact particles were demonstrated by a lack of binding by anti-MA and anti-CA antibodies), and that this binding was abolished by protease treatment of the particles (Cleveland et al., 2003). This report provided the first direct evidence for exposure of CTT sequences on viral particles.
A second study from Dimmock’s group described a mAb, termed SAR1, that is directed against the KE, as well as its neutralization activity (Reading et al., 2003). SAR1 was found to bind to both soluble peptides and proteins containing a GERDRDR sequence. More interesting, however, was that SAR1 bound to cells infected with all different HIV strains tested, but only to some virions representing the same strains. Moreover, SAR1 did not exhibit neutralization activity in a standard neutralization assay, but was able to neutralize effectively in a post-attachment neutralization (PAN) assay. In a standard neutralization assay, antibody and virus are incubated together prior to adding to the cells to allow the interaction to reach equilibrium before addition to target cells; neutralizing antibodies reduce viral infection in a concentration-dependent manner. In a PAN assay, however, antibody, virus and target cells are incubated together, but at temperatures that do not support virus–cell fusion (usually ≤31 °C) for a defined period of time before washing free virus from the cells and incubating at 37 °C to allow infection to occur (or not, if the antibody neutralizes) (Reading et al., 2003). The inability of SAR1 to neutralize in a traditional assay, but exhibit PAN activity, suggested a transient exposure of the KE during the fusion process. The mechanism of the PAN activity of SAR1 was confirmed in a subsequent paper to act through the blocking of virus–cell fusion (Heap et al., 2005). Reading et al. also demonstrated that SAR1 acted to block the production of infectious progeny virus from infected cells (Reading et al., 2003). These results provided increased evidence for an alternative topology for the CTT, where the KE is exposed to antibody binding, although not universally as SAR1 did not bind to virions from all HIV strains tested. Indeed, it is noteworthy that the KE seems to be exposed differently at the surface of virions and of the cells. Recent studies have demonstrated that two different mAbs directed against the KE (Chessie 8 and SAR1) bind exclusively at the surface of the cells and not to virions, suggesting different conformations for the CTT that are dependent on context (Cheung et al., 2005; Steckbeck et al., 2010). However, the cell surface exposure of the CTT in SIV has recently been proposed to be a consequence of Env shed from transfected cells binding non-specifically to other cells in culture thus exposing the CTT to antibody binding (Postler et al., 2012). A conclusive model for functional CTT topology remains to be elucidated.
Finally, in an effort to provide a condensation of results from these studies and to provide a working topological model explaining their results, Hollier and Dimmock used sequence analyses to provide theoretical support to their observed exposure of CTT sequences. Using a combination of secondary structure predictions with hydrophobicity analyses, Hollier and Dimmock proposed that the CTT might form a tail loop structure supported by three membrane-spanning β-sheets that result in the extracellular localization of the KE (Hollier & Dimmock, 2005). This is the alternative topological model presented in Fig. 7. Hollier and Dimmock acknowledge that their proposed topological model positions the N-terminal GYXXΦ endocytic signal in an extracellular position, implying that it cannot be functional. As discussed above, this model is in direct contrast with subsequent results, demonstrating the GYXXΦ signal to be active in AP-2-mediated endocytosis (Byland et al., 2007). Hollier and Dimmock propose that the majority of cell surface Env is in a state similar to the traditional topological model, with a single MSD, but that a minor population exists in their newly proposed topological state, and that it is this KE-exposed Env that is preferentially incorporated into progeny virions (Hollier & Dimmock, 2005). A major argument against this model is the improbable existence of an MSD composed entirely of β-sheet with an odd number of membrane-interacting sequences. One of the ‘construction rules’ of β-sheet membrane proteins is that they must have an even number of MSDs, as β-sheets hydrogen bond most efficiently in an anti-parallel fashion (Schulz, 2000). An odd number of β-sheet MSD sequences would leave a lone β-strand and its hydrogen-bonding backbone amide and carboxyl groups exposed to the hydrocarbon interior of the membrane, an energetically unfavourable situation (Schulz, 2000). This unfavourable arrangement would be further compounded upon Env trimerization, where there would now be three naked β-sheets in the interior of the membrane, all arranged in a parallel fashion and unable to relieve the unfavourable interactions.
In contrast to the Hollier and Dimmock model, other recent studies support the incorporation of a single MSD gp41 in virions. It has been demonstrated that HIV-1 develops a resistance to antibiotic amphotericin B methyl ester through mutations in gp41 that create proteolytic cleavage sites in the cytoplasmic tail that allow truncation of the CTT by the viral protease in the virions, but not in cells (Waheed et al., 2007; Waheed et al., 2010). This observation is consistent with the existence of different Env conformations at the virus and cell surfaces which could be responsible for different accessibility of the cleavage sites to the protease (Waheed et al., 2007). Interestingly, 10 % of gp41 molecules are not cleaved in virions, suggesting the possible co-existence of alternative CTT structures as a minority population (Waheed et al., 2007). By contrast, these CTT cleavage sites are inaccessible in cells, suggesting that either interaction with viral or cellular proteins or an alternative CTT conformation prevents protease access and cleavage.
Dynamic exposure of LLP2 sequences
In addition to the evidence for possible KE exposure, recent studies present data supporting the exposure of additional CTT sequences, in particular the LLP2 domain. A study by Lu et al. determined the exposure of CTT sequences in Env-expressing cells under native conditions and during the fusion process (Lu et al., 2008). Using antibodies directed at the C-terminal 90 aa of the CTT and to the LLP2 region only, Lu et al. determined that no exposure of the C-terminal 90 aa was detected under normal conditions, nor did they observe exposure during cell–cell fusion carried out at 37 °C as measured by flow cytometry. They did, however, observe LLP2 exposure during cell–cell fusion by slowing the fusion reaction by incubation at 31.5 °C (Lu et al., 2008). These same antibodies were also demonstrated to inhibit cell–cell fusion in a concentration-dependent manner at 31.5 °C, but not at 37 °C. Their results suggest a transient exposure of LLP2 sequences during membrane fusion, and are consistent with results demonstrating membrane association of LLP2 both pre- and post-fusion (Viard et al., 2008).
CTT influences overall Env structure
The final known function of the CTT is its role in modulating overall Env structure and functional properties. One of the earliest observations that the CTT could influence overall Env structure was done in SIV and showed that truncation of the CTT increased the accessibility of the reactive amino acids from the envelope in a cell surface biotinylation assay (Spies et al., 1994). Further evidence of this phenomenon was provided by the insight that HIV-1 with CTT-deleted Env proteins could infect target cells in a CD4-independent manner (Edinger et al., 1997). Until that time, the paradigm for HIV infection of target cells was that gp120 binding to CD4 was necessary to induce conformational changes that allowed binding to co-receptor. Edinger et al. demonstrated that viruses with a CTT-deleted Env were able to infect CD4 negative/co-receptor-positive cells, suggesting the existence of different conformations of Env that are dependent on the presence or absence of the CTT.
Initially, direct evidence for CTT-dependent alterations in Env structure was provided by differential reactivity between CTT-deleted and wild-type Env with conformationally dependent antibodies. Edwards et al. demonstrated that truncation of the CTT to 27 aa resulted in increased binding by mAbs directed to both the CD4-binding site and the CD4-induced (CD4i) co-receptor-binding site (Edwards et al., 2002). In Env with a full-length CTT, mAbs directed to the co-receptor-binding site only bound if the Env was preincubated with soluble CD4. The study also demonstrated differential reactivity of conformational mAbs directed to the ectodomain of gp41 between CTT-truncated and full-length CTT (Edwards et al., 2002). This study was the first to demonstrate that the CTT modulates the conformation of overall Env structure, even in the non-covalently attached gp120 that is presumably on the opposite side of the membrane. These results were further confirmed by a study from Wyss et al. showing that the CTT truncation not only exposed the CD4i epitope, but also increased fusion efficiency (Wyss et al., 2005). Subramaniam and colleagues recently published cryo-EM structures of CTT-deleted Env on the surface of SIV viral particles (CP-mac variant) (White et al., 2010). In comparison to wild-type virus containing the full-length CTT, CTT-deleted Env existed in a naturally ‘open’ state, displaying large differences in the localization of electron density that are consistent with the conformational changes normally associated with CD4 binding. The CP-mac variant also contains mutations in the gp120 and gp41 ectodomains that may contribute to the open conformation (LaBranche et al., 1994), as other studies have demonstrated that deletion of the CTT alone does not confer a CD4-independent phenotype in HIV-1 (Edwards et al., 2001). Indeed, the combination of CTT truncation and gp120 mutations allows the CD4 independence, indicating that the CTT deletion is necessary but not sufficient to confer a CD4-independent phenotype (Edwards et al., 2001). Other studies, discussed below, provide additional evidence for the influence of the CTT on overall Env structure.
Kalia et al. also demonstrated that alterations in the CTT could modulate the antigenic conformation of both the gp120 and gp41 ectodomains. Instead of utilizing large deletions in the CTT, however, Kalia demonstrated that mutation of two conserved arginine residues in LLP2 to glutamate was sufficient to alter the conformation of both gp120 and the gp41 ectodomain on the surface of Env-expressing cells (Kalia et al., 2005). The mutations were shown previously to have no effect on the levels of virion Env incorporation or viral replication, but did decrease the efficiency of cell–cell fusion (Kalia et al., 2003). In addition to demonstrating antigenic distinctions between wild-type and mutant Env on the cell surface, differences were seen in the viral sensitivity to antibody-mediated neutralization by antibodies directed at the CD4-binding site, with the mutant virus demonstrating an approximately 40-fold decrease in neutralization sensitivity (Kalia et al., 2005). This study importantly demonstrated that point mutations in the CTT were sufficient to alter overall Env conformation and antigenicity, and additionally provided the first data regarding the critical nature of the conserved arginine residues in the LLP regions. In addition, this study revealed the potential role of minor CTT variation in Env antigenic variation and immune escape.
A recent paper has extended the study of CTT-dependent alterations in overall Env antigenicity from the cell surface to the surface of viral particles (Joyner et al., 2011). While investigating maturation-induced cloaking of neutralization epitopes, Joyner et al. demonstrated that Env on immature virions (protease deleted) reacted differently with a number of conformationally dependent mAbs in a manner that was dependent on the presence or absence of the CTT (Joyner et al., 2011). This study provided the first direct evidence that the CTT plays a major role in modulating the conformation of the CTT in the virion in addition to being on the cell surface.
The potential exposure of the CTT and its modulation of Env conformation could also have some implications in the virus life cycle. Indeed, it has been shown in T-cells that the virus neutralizing EPES antibody binding to the KE was able to inhibit the cell–cell fusion between an HIV-1-infected cell and its target uninfected cell, suggesting a role for the CTT exposure at the cell surface in the virus propagation (Cheung et al., 2005). These results suggest that the presence of the CTT may be important to a dynamic change of conformation of the envelope during the cell–cell fusion. The binding of this antibody to KE epitope could prevent the conformational modification of the envelope at the cell surface and thus inhibit cell–cell fusion. A summary of the different antibodies binding to the CTT and their characteristics is presented in Table 1.
Table 1. List of the published antibodies binding to the gp41 CTT.
na, Not available.
| Name of the antibody | Targeted region | Epitope sequence | Epitope characteristics | Species/clonality | Antibody characteristics | References |
| Chessie 8 | KE | PDRPEG | Conformational | Murine IgG | Non-neutralizing binds to HIV-infected cells | Abacioglu et al. (1994) |
| SAR1 | KE | GERDRDR | Conformational | Murine IgG | PAN-neutralizing binds to HIV-infected cells and most virions, no binding to intact NL4.3 virions | Reading et al. (2003), Heap et al. (2005), Steckbeck et al. (2010) |
| EPES (epitope-purified ERDRD specific) | KE | ERDRD | Conformational | Murine polyclonal | Neutralizing binds to HIV-infected cells and virions | Buratti et al. (1998), Cleveland et al. (2000b, 2003) |
| anti-LLP2 | LLP2 | na | na | Rabbit polyclonal IgG | na | Lu et al. (2008) |
| 1575 | KE | IEEE | Linear immunodominant | Murine monoclonal | Non-neutralizing binds to HIV-infected cells and virions | Buratti et al. (1996, 1997, 1998), Cleveland et al. (2000a), Evans et al. (1989), Heap et al. (2005), Vella et al. (1993) |
| 1577 | KE | ERDRD | Conformational | Murine monoclonal IgG | Non-neutralizing binding to HIV-infected cells and virions (Dimmock), no binding to intact virions (Steckbeck), epitope binding inhibited by mAb1575 | Beaumont et al. (2000), Buratti et al. (1996, 1998), Chomont et al. (2008), Cleveland et al. (2000a, 2003), Evans et al. (1989), Holl et al. (2006), Steckbeck et al. (2010), Vella et al. (1993) |
| 1583 | KE | ERDRD | Conformational | Murine monoclonal IgG | Non-neutralizing binds to virions | Evans et al. (1989), Buratti et al. (1996, 1998), Vella et al. (1993), Cleveland et al. (2003), Heap et al. (2005) |
Conclusion and perspectives
Most studies have focused on the gp120 subunit toward developing an HIV-1 vaccine, removing all or much of gp41 (and always the CTT), to increase protein expression and produce a soluble immunogen. However, as has been summarized above, the CTT plays an important role in Env structure and consequently in the virus life cycle, underscoring the importance of using the entirety of Env, including the CTT, in further immunization studies. It may, therefore, be useful to theorize on the means by which the CTT can exert influence on the structure of the Env ectodomain in an effort to provide a framework for understanding the mechanism(s) by which the influence occurs.
The conservation of the physico-chemical properties is particularly interesting for the LLP regions in light of their proposed membrane associating characteristics (Chernomordik et al., 1994; Fujii et al., 1992; Kliger & Shai, 1997; Srinivas et al., 1992), where hydrophobic moment and charge may complement one another. The hydrophobic moment has been proposed as a measure of the tendency of a sequence to prefer the chemically complex interfacial boundary between the hydrocarbon membrane interior and the aqueous phase (Eisenberg et al., 1984). The preference for the chemically complex membrane–water interface is suggestive of a role for the LLP sequences as membrane anchors. The (partial) insertion of the LLP sequences into the interfacial region would have a physical influence on the local membrane environment through introduction of local curvature stress as well as by affecting the lateral pressure profile in the membrane interior (Booth et al., 2001; Cantor, 1997b, 1999; Tillman & Cascio, 2003; van den Brink-van der Laan et al., 2004). These physical alterations of the membrane environment by interfacially localized LLP sequences may provide an explanation for the modulation of Env ectodomain conformation induced by point mutations in the LLP (Kalia et al., 2005). While the mutations introduced by Kalia et al. maintain the amphipathic potential of the LLP2 region, they alter the net charge of the region from +3 to −1 (Kalia et al., 2005). The resulting overall negative charge of the region may impact its association with the negatively charged inner membrane leaflet by introducing charge–charge incompatibilities with the negatively charged phosphatidylserine headgroups. Altered association (i.e. reduced/abrogated association) of LLP2 with the inner leaflet of the membrane would lead to a change in the local lateral pressure profile of the membrane, which in turn could lead to conformational changes in the protein by altering the arrangement of the transmembrane helices (Cantor, 1997a, 1999, 2002; Marsh, 2007; Tillman & Cascio, 2003; van den Brink-van der Laan et al., 2004).
Arginine conservation relative to lysine in the CTT is another area that invites further consideration. Studies demonstrating a transient exposure of LLP2 sequences during the cell–cell fusion process are suggestive of a mechanism to prefer arginine conservation relative to replacement with lysine. Arginine-rich peptides have been demonstrated to cross cellular membranes, while peptides with lysine instead of arginine do not (Futaki, 2005; Mitchell et al., 2000; Tung & Weissleder, 2003). In addition, arginine-rich peptides have been demonstrated to deliver soluble proteins into the cytoplasm of live cells (Inomata et al., 2009). The apparent traversing of the membrane by LLP2 during the fusion process would seem, then, to require the presence of arginine.
Implications for the overall observed conservation of arginine in the CTT become more interesting in the context of a potentially dynamic CTT topology. Published studies suggest that the topology of the CTT apparently can be distinct between the cellular and virion surface (‘can be’ as a function of the fact that published cellular data does not preclude the existence of virion-like CTT in the cell). This discovery of a cellular topology that is distinct from the virion topology is suggestive of either an alternative, stable topology that is apparently functionally irrelevant to the virion, or a dynamic rearrangement of the topology from the extracellular (lumenal or exposed) state to the internally localized (intracytoplasmic) form.
For the sake of argument, if CTT-exposed Env undergoes dynamic rearrangement by ‘flipping’ sides of the membrane (crossing the hydrocarbon interior in the process) to reach a different topology as was demonstrated for LLP2 (Lu et al., 2008), the presence of arginine relative to lysine would be energetically preferred according to the biological hydrophobicity scale (Hessa et al., 2005a). A movement of arginine-rich sequences across a membrane when captive as part of a larger protein complex (as opposed to soluble peptide fragments) is perhaps best demonstrated in the voltage-sensor paddle of the potassium-gated ion channel KvAP, where the arginine-rich S4 helix (with four arginine residues) is proposed to cross the membrane hydrocarbon interior in response to changes in the membrane electrical field (Jiang et al., 2003). Interestingly, this same helical segment has demonstrated potential to insert into the membrane as a transmembrane helix in spite of the four arginine residues (Hessa et al., 2005b).
Overall, the data summarized in this review demonstrate an intriguing evolution of the prevailing perspective on HIV CTT sequences from a general consensus of being unnecessary and unimportant to the virus, to the identification of numerous diverse functional motifs and roles that influence overall Env structural and critical functional properties. It is intriguing to consider that these CTT-mediated functions may be further investigated to reveal new cellular co-factors that participate in Env trafficking and incorporation into virions and that may serve as novel targets for antiviral drugs. In addition, the data indicating a dynamic topology for the CTT may upon further study reveal heretofore undefined functions associated with the selectively arginine-rich, amphipathic helical sequences that are characteristic of the CTT domains of all HIV Env species, and of all animal lentiviral species.
Acknowledgements
This work was supported in part by the National Institutes of Health (grant 5R01 AI087533).
References
- Abacioglu Y. H., Fouts T. R., Laman J. D., Claassen E., Pincus S. H., Moore J. P., Roby C. A., Kamin-Lewis R., Lewis G. K. (1994). Epitope mapping and topology of baculovirus-expressed HIV-1 gp160 determined with a panel of murine monoclonal antibodies. AIDS Res Hum Retroviruses 10, 371–381 10.1089/aid.1994.10.371 [DOI] [PubMed] [Google Scholar]
- Allan J. S., Coligan J. E., Barin F., McLane M. F., Sodroski J. G., Rosen C. A., Haseltine W. A., Lee T. H., Essex M. (1985). Major glycoprotein antigens that induce antibodies in AIDS patients are encoded by HTLV-III. Science 228, 1091–1094 10.1126/science.2986290 [DOI] [PubMed] [Google Scholar]
- Barré-Sinoussi F., Chermann J. C., Rey F., Nugeyre M. T., Chamaret S., Gruest J., Dauguet C., Axler-Blin C., Vézinet-Brun F. & other authors (1983). Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 220, 868–871 10.1126/science.6189183 [DOI] [PubMed] [Google Scholar]
- Beaumont T., Broersen S., van Nuenen A., Huisman H. G., de Roda Husman A. M., Heeney J. L., Schuitemaker H. (2000). Increased neutralization sensitivity and reduced replicative capacity of human immunodeficiency virus type 1 after short-term in vivo or in vitro passage through chimpanzees. J Virol 74, 7699–7707 10.1128/JVI.74.17.7699-7707.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakta S. J., Shang L., Prince J. L., Claiborne D. T., Hunter E. (2011). Mutagenesis of tyrosine and di-leucine motifs in the HIV-1 envelope cytoplasmic domain results in a loss of Env-mediated fusion and infectivity. Retrovirology 8, 37 10.1186/1742-4690-8-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya J., Peters P. J., Clapham P. R. (2004). Human immunodeficiency virus type 1 envelope glycoproteins that lack cytoplasmic domain cysteines: impact on association with membrane lipid rafts and incorporation onto budding virus particles. J Virol 78, 5500–5506 10.1128/JVI.78.10.5500-5506.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya J., Repik A., Clapham P. R. (2006). Gag regulates association of human immunodeficiency virus type 1 envelope with detergent-resistant membranes. J Virol 80, 5292–5300 10.1128/JVI.01469-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blot G., Janvier K., Le Panse S., Benarous R., Berlioz-Torrent C. (2003). Targeting of the human immunodeficiency virus type 1 envelope to the trans-Golgi network through binding to TIP47 is required for env incorporation into virions and infectivity. J Virol 77, 6931–6945 10.1128/JVI.77.12.6931-6945.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blot G., Lopez-Vergès S., Treand C., Kubat N. J., Delcroix-Genête D., Emiliani S., Benarous R., Berlioz-Torrent C. (2006). Luman, a new partner of HIV-1 TMgp41, interferes with Tat-mediated transcription of the HIV-1 LTR. J Mol Biol 364, 1034–1047 10.1016/j.jmb.2006.09.080 [DOI] [PubMed] [Google Scholar]
- Boge M., Wyss S., Bonifacino J. S., Thali M. (1998). A membrane-proximal tyrosine-based signal mediates internalization of the HIV-1 envelope glycoprotein via interaction with the AP-2 clathrin adaptor. J Biol Chem 273, 15773–15778 10.1074/jbc.273.25.15773 [DOI] [PubMed] [Google Scholar]
- Booth P. J., Templer R. H., Meijberg W., Allen S. J., Curran A. R., Lorch M. (2001). In vitro studies of membrane protein folding. Crit Rev Biochem Mol Biol 36, 501–603 10.1080/20014091074246 [DOI] [PubMed] [Google Scholar]
- Bosch M. L., Earl P. L., Fargnoli K., Picciafuoco S., Giombini F., Wong-Staal F., Franchini G. (1989). Identification of the fusion peptide of primate immunodeficiency viruses. Science 244, 694–697 10.1126/science.2541505 [DOI] [PubMed] [Google Scholar]
- Buratti E., Tisminetzky S. G., Scodeller E. S., Baralle F. E. (1996). Conformational display of two neutralizing epitopes of HIV-1 gp41 on the Flock House virus capsid protein. J Immunol Methods 197, 7–18 10.1016/0022-1759(96)00097-X [DOI] [PubMed] [Google Scholar]
- Buratti E., Tisminetzky S. G., D’Agaro P., Baralle F. E. (1997). A neutralizing monoclonal antibody previously mapped exclusively on human immunodeficiency virus type 1 gp41 recognizes an epitope in p17 sharing the core sequence IEEE. J Virol 71, 2457–2462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E., McLain L., Tisminetzky S., Cleveland S. M., Dimmock N. J., Baralle F. E. (1998). The neutralizing antibody response against a conserved region of human immunodeficiency virus type 1 gp41 (amino acid residues 731–752) is uniquely directed against a conformational epitope. J Gen Virol 79, 2709–2716 [DOI] [PubMed] [Google Scholar]
- Byland R., Vance P. J., Hoxie J. A., Marsh M. (2007). A conserved dileucine motif mediates clathrin and AP-2-dependent endocytosis of the HIV-1 envelope protein. Mol Biol Cell 18, 414–425 10.1091/mbc.E06-06-0535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caffrey M., Cai M., Kaufman J., Stahl S. J., Wingfield P. T., Covell D. G., Gronenborn A. M., Clore G. M. (1998). Three-dimensional solution structure of the 44 kDa ectodomain of SIV gp41. EMBO J 17, 4572–4584 10.1093/emboj/17.16.4572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor R. S. (1997a). The lateral pressure profile in membranes: a physical mechanism of general anesthesia. Biochemistry 36, 2339–2344 10.1021/bi9627323 [DOI] [PubMed] [Google Scholar]
- Cantor R. S. (1997b). Lateral pressures in cell membranes: a mechanism for modulation of protein function. J Phys Chem B 101, 1723–1725 10.1021/jp963911x [DOI] [Google Scholar]
- Cantor R. S. (1999). The influence of membrane lateral pressures on simple geometric models of protein conformational equilibria. Chem Phys Lipids 101, 45–56 10.1016/S0009-3084(99)00054-7 [DOI] [PubMed] [Google Scholar]
- Cantor R. S. (2002). Size distribution of barrel-stave aggregates of membrane peptides: influence of the bilayer lateral pressure profile. Biophys J 82, 2520–2525 10.1016/S0006-3495(02)75595-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti L., Emerman M., Tiollais P., Sonigo P. (1989). The cytoplasmic domain of simian immunodeficiency virus transmembrane protein modulates infectivity. J Virol 63, 4395–4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan D. C., Fass D., Berger J. M., Kim P. S. (1997). Core structure of gp41 from the HIV envelope glycoprotein. Cell 89, 263–273 10.1016/S0092-8674(00)80205-6 [DOI] [PubMed] [Google Scholar]
- Chan W.-E., Lin H.-H., Chen S. S.-L. (2005). Wild-type-like viral replication potential of human immunodeficiency virus type 1 envelope mutants lacking palmitoylation signals. J Virol 79, 8374–8387 10.1128/JVI.79.13.8374-8387.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanh T. C., Dreesman G. R., Kanda P., Linette G. P., Sparrow J. T., Ho D. D., Kennedy R. C. (1986). Induction of anti-HIV neutralizing antibodies by synthetic peptides. EMBO J 5, 3065–3071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Checkley M. A., Luttge B. G., Freed E. O. (2011). HIV-1 envelope glycoprotein biosynthesis, trafficking, and incorporation. J Mol Biol 410, 582–608 10.1016/j.jmb.2011.04.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomordik L., Chanturiya A. N., Suss-Toby E., Nora E., Zimmerberg J. (1994). An amphipathic peptide from the C-terminal region of the human immunodeficiency virus envelope glycoprotein causes pore formation in membranes. J Virol 68, 7115–7123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung L., McLain L., Hollier M. J., Reading S. A., Dimmock N. J. (2005). Part of the C-terminal tail of the envelope gp41 transmembrane glycoprotein of human immunodeficiency virus type 1 is exposed on the surface of infected cells and is involved in virus-mediated cell fusion. J Gen Virol 86, 131–138 10.1099/vir.0.80439-0 [DOI] [PubMed] [Google Scholar]
- Chomont N., Hocini H., Gody J. C., Bouhlal H., Becquart P., Krief-Bouillet C., Kazatchkine M., Bélec L. (2008). Neutralizing monoclonal antibodies to human immunodeficiency virus type 1 do not inhibit viral transcytosis through mucosal epithelial cells. Virology 370, 246–254 10.1016/j.virol.2007.09.006 [DOI] [PubMed] [Google Scholar]
- Cleveland S. M., Buratti E., Jones T. D., North P., Baralle F., McLain L., McInerney T., Durrani Z., Dimmock N. J. (2000a). Immunogenic and antigenic dominance of a nonneutralizing epitope over a highly conserved neutralizing epitope in the gp41 envelope glycoprotein of human immunodeficiency virus type 1: its deletion leads to a strong neutralizing response. Virology 266, 66–78 10.1006/viro.1999.0041 [DOI] [PubMed] [Google Scholar]
- Cleveland S. M., Jones T. D., Dimmock N. J. (2000b). Properties of a neutralizing antibody that recognizes a conformational form of epitope ERDRD in the gp41 C-terminal tail of human immunodeficiency virus type 1. J Gen Virol 81, 1251–1260 [DOI] [PubMed] [Google Scholar]
- Cleveland S. M., McLain L., Cheung L., Jones T. D., Hollier M., Dimmock N. J. (2003). A region of the C-terminal tail of the gp41 envelope glycoprotein of human immunodeficiency virus type 1 contains a neutralizing epitope: evidence for its exposure on the surface of the virion. J Gen Virol 84, 591–602 10.1099/vir.0.18630-0 [DOI] [PubMed] [Google Scholar]
- Cosson P. (1996). Direct interaction between the envelope and matrix proteins of HIV-1. EMBO J 15, 5783–5788 [PMC free article] [PubMed] [Google Scholar]
- Deschambeault J., Lalonde J. P., Cervantes-Acosta G., Lodge R., Cohen E. A., Lemay G. (1999). Polarized human immunodeficiency virus budding in lymphocytes involves a tyrosine-based signal and favors cell-to-cell viral transmission. J Virol 73, 5010–5017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmock N. J. (2005). The complex antigenicity of a small external region of the C-terminal tail of the HIV-1 gp41 envelope protein: a lesson in epitope analysis. Rev Med Virol 15, 365–381 10.1002/rmv.476 [DOI] [PubMed] [Google Scholar]
- Dragic T., Trkola A., Thompson D. A., Cormier E. G., Kajumo F. A., Maxwell E., Lin S. W., Ying W., Smith S. O. & other authors (2000). A binding pocket for a small molecule inhibitor of HIV-1 entry within the transmembrane helices of CCR5. Proc Natl Acad Sci U S A 97, 5639–5644 10.1073/pnas.090576697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubay J. W., Roberts S. J., Brody B., Hunter E. (1992). Mutations in the leucine zipper of the human immunodeficiency virus type 1 transmembrane glycoprotein affect fusion and infectivity. J Virol 66, 4748–4756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earl P. L., Doms R. W., Moss B. (1990). Oligomeric structure of the human immunodeficiency virus type 1 envelope glycoprotein. Proc Natl Acad Sci U S A 87, 648–652 10.1073/pnas.87.2.648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earl P. L., Koenig S., Moss B. (1991). Biological and immunological properties of human immunodeficiency virus type 1 envelope glycoprotein: analysis of proteins with truncations and deletions expressed by recombinant vaccinia viruses. J Virol 65, 31–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edinger A. L., Mankowski J. L., Doranz B. J., Margulies B. J., Lee B., Rucker J., Sharron M., Hoffman T. L., Berson J. F. & other authors (1997). CD4-independent, CCR5-dependent infection of brain capillary endothelial cells by a neurovirulent simian immunodeficiency virus strain. Proc Natl Acad Sci U S A 94, 14742–14747 10.1073/pnas.94.26.14742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards T. G., Hoffman T. L., Baribaud F., Wyss S., LaBranche C. C., Romano J., Adkinson J., Sharron M., Hoxie J. A., Doms R. W. (2001). Relationships between CD4 independence, neutralization sensitivity, and exposure of a CD4-induced epitope in a human immunodeficiency virus type 1 envelope protein. J Virol 75, 5230–5239 10.1128/JVI.75.11.5230-5239.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards T. G., Wyss S., Reeves J. D., Zolla-Pazner S., Hoxie J. A., Doms R. W., Baribaud F. (2002). Truncation of the cytoplasmic domain induces exposure of conserved regions in the ectodomain of human immunodeficiency virus type 1 envelope protein. J Virol 76, 2683–2691 10.1128/JVI.76.6.2683-2691.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg D., Wesson M. (1990). The most highly amphiphilic α-helices include two amino acid segments in human immunodeficiency virus glycoprotein 41. Biopolymers 29, 171–177 10.1002/bip.360290122 [DOI] [PubMed] [Google Scholar]
- Eisenberg D., Weiss R. M., Terwilliger T. C. (1984). The hydrophobic moment detects periodicity in protein hydrophobicity. Proc Natl Acad Sci U S A 81, 140–144 10.1073/pnas.81.1.140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans D. J., McKeating J., Meredith J. M., Burke K. L., Katrak K., John A., Ferguson M., Minor P. D., Weiss R. A., Almond J. W. (1989). An engineered poliovirus chimaera elicits broadly reactive HIV-1 neutralizing antibodies. Nature 339, 385–388, 340 10.1038/339385a0 [DOI] [PubMed] [Google Scholar]
- Freed E. O., Martin M. A. (1995). Virion incorporation of envelope glycoproteins with long but not short cytoplasmic tails is blocked by specific, single amino acid substitutions in the human immunodeficiency virus type 1 matrix. J Virol 69, 1984–1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed E. O., Martin M. A. (1996). Domains of the human immunodeficiency virus type 1 matrix and gp41 cytoplasmic tail required for envelope incorporation into virions. J Virol 70, 341–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed E. O., Myers D. J., Risser R. (1989). Mutational analysis of the cleavage sequence of the human immunodeficiency virus type 1 envelope glycoprotein precursor gp160. J Virol 63, 4670–4675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed E. O., Myers D. J., Risser R. (1990). Characterization of the fusion domain of the human immunodeficiency virus type 1 envelope glycoprotein gp41. Proc Natl Acad Sci U S A 87, 4650–4654 10.1073/pnas.87.12.4650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed E. O., Delwart E. L., Buchschacher G. L., Jr, Panganiban A. T. (1992). A mutation in the human immunodeficiency virus type 1 transmembrane glycoprotein gp41 dominantly interferes with fusion and infectivity. Proc Natl Acad Sci U S A 89, 70–74 10.1073/pnas.89.1.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii G., Horvath S., Woodward S., Eiserling F., Eisenberg D. (1992). A molecular model for membrane fusion based on solution studies of an amphiphilic peptide from HIV gp41. Protein Sci 1, 1454–1464 10.1002/pro.5560011107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futaki S. (2005). Membrane-permeable arginine-rich peptides and the translocation mechanisms. Adv Drug Deliv Rev 57, 547–558 10.1016/j.addr.2004.10.009 [DOI] [PubMed] [Google Scholar]
- Gallaher W. R., Ball J. M., Garry R. F., Griffin M. C., Montelaro R. C. (1989). A general model for the transmembrane proteins of HIV and other retroviruses. AIDS Res Hum Retroviruses 5, 431–440 10.1089/aid.1989.5.431 [DOI] [PubMed] [Google Scholar]
- Gallo R. C., Salahuddin S. Z., Popovic M., Shearer G. M., Kaplan M., Haynes B. F., Palker T. J., Redfield R., Oleske J. & other authors (1984). Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS. Science 224, 500–503 10.1126/science.6200936 [DOI] [PubMed] [Google Scholar]
- Gangupomu V. K., Abrams C. F. (2010). All-atom models of the membrane-spanning domain of HIV-1 gp41 from metadynamics. Biophys J 99, 3438–3444 10.1016/j.bpj.2010.09.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood A. I., Pan J., Mills T. T., Nagle J. F., Epand R. M., Tristram-Nagle S. (2008). CRAC motif peptide of the HIV-1 gp41 protein thins SOPC membranes and interacts with cholesterol. Biochim Biophys Acta 1778, 1120–1130 10.1016/j.bbamem.2008.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffar O. K., Dowbenko D. J., Berman P. W. (1988). Topogenic analysis of the human immunodeficiency virus type 1 envelope glycoprotein, gp160, in microsomal membranes. J Cell Biol 107, 1677–1687 10.1083/jcb.107.5.1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heap C. J., Reading S. A., Dimmock N. J. (2005). An antibody specific for the C-terminal tail of the gp41 transmembrane protein of human immunodeficiency virus type 1 mediates post-attachment neutralization, probably through inhibition of virus-cell fusion. J Gen Virol 86, 1499–1507 10.1099/vir.0.80414-0 [DOI] [PubMed] [Google Scholar]
- Hessa T., Kim H., Bihlmaier K., Lundin C., Boekel J., Andersson H., Nilsson I., White S. H., von Heijne G. (2005a). Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature 433, 377–381 10.1038/nature03216 [DOI] [PubMed] [Google Scholar]
- Hessa T., White S. H., von Heijne G. (2005b). Membrane insertion of a potassium-channel voltage sensor. Science 307, 1427 10.1126/science.1109176 [DOI] [PubMed] [Google Scholar]
- Hirsch V. M., Edmondson P., Murphey-Corb M., Arbeille B., Johnson P. R., Mullins J. I. (1989). SIV adaptation to human cells. Nature 341, 573–574 10.1038/341573a0 [DOI] [PubMed] [Google Scholar]
- Holl V., Peressin M., Decoville T., Schmidt S., Zolla-Pazner S., Aubertin A. M., Moog C. (2006). Nonneutralizing antibodies are able to inhibit human immunodeficiency virus type 1 replication in macrophages and immature dendritic cells. J Virol 80, 6177–6181 10.1128/JVI.02625-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollier M. J., Dimmock N. J. (2005). The C-terminal tail of the gp41 transmembrane envelope glycoprotein of HIV-1 clades A, B, C, and D may exist in two conformations: an analysis of sequence, structure, and function. Virology 337, 284–296 10.1016/j.virol.2005.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C. C., Tang M., Zhang M.-Y., Majeed S., Montabana E., Stanfield R. L., Dimitrov D. S., Korber B., Sodroski J. & other authors (2005). Structure of a V3-containing HIV-1 gp120 core. Science 310, 1025–1028 10.1126/science.1118398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inomata K., Ohno A., Tochio H., Isogai S., Tenno T., Nakase I., Takeuchi T., Futaki S., Ito Y. & other authors (2009). High-resolution multi-dimensional NMR spectroscopy of proteins in human cells. Nature 458, 106–109 10.1038/nature07839 [DOI] [PubMed] [Google Scholar]
- Jiang J, Aiken C. (2007). Maturation-dependent HIV-1 particle fusion requires a carboxyl-terminal region of the gp41 cytoplasmic tail. J Virol 81, 9999-10008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y., Ruta V., Chen J., Lee A., MacKinnon R. (2003). The principle of gating charge movement in a voltage-dependent K+ channel. Nature 423, 42–48 10.1038/nature01581 [DOI] [PubMed] [Google Scholar]
- Joyner A. S., Willis J. R., Crowe J. E., Jr, Aiken C. (2011). Maturation-induced cloaking of neutralization epitopes on HIV-1 particles. PLoS Pathog 7, e1002234 10.1371/journal.ppat.1002234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia V., Sarkar S., Gupta P., Montelaro R. C. (2003). Rational site-directed mutations of the LLP-1 and LLP-2 lentivirus lytic peptide domains in the intracytoplasmic tail of human immunodeficiency virus type 1 gp41 indicate common functions in cell-cell fusion but distinct roles in virion envelope incorporation. J Virol 77, 3634–3646 10.1128/JVI.77.6.3634-3646.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia V., Sarkar S., Gupta P., Montelaro R. C. (2005). Antibody neutralization escape mediated by point mutations in the intracytoplasmic tail of human immunodeficiency virus type 1 gp41. J Virol 79, 2097–2107 10.1128/JVI.79.4.2097-2107.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy R. C., Henkel R. D., Pauletti D., Allan J. S., Lee T. H., Essex M., Dreesman G. R. (1986). Antiserum to a synthetic peptide recognizes the HTLV-III envelope glycoprotein. Science 231, 1556–1559 10.1126/science.3006246 [DOI] [PubMed] [Google Scholar]
- Kim J. H., Hartley T. L., Curran A. R., Engelman D. M. (2009). Molecular dynamics studies of the transmembrane domain of gp41 from HIV-1. Biochim Biophys Acta 1788, 1804–1812 10.1016/j.bbamem.2009.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliger Y., Shai Y. (1997). A leucine zipper-like sequence from the cytoplasmic tail of the HIV-1 envelope glycoprotein binds and perturbs lipid bilayers. Biochemistry 36, 5157–5169 10.1021/bi962935r [DOI] [PubMed] [Google Scholar]
- Kodama T., Wooley D. P., Naidu Y. M., Kestler H. W., III, Daniel M. D., Li Y., Desrosiers R. C. (1989). Significance of premature stop codons in env of simian immunodeficiency virus. J Virol 63, 4709–4714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kol N., Shi Y., Tsvitov M., Barlam D., Shneck R. Z., Kay M. S., Rousso I. (2007). A stiffness switch in human immunodeficiency virus. Biophys J 92, 1777–1783 10.1529/biophysj.106.093914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo N., Miyauchi K., Meng F., Iwamoto A., Matsuda Z. (2010). Conformational changes of the HIV-1 envelope protein during membrane fusion are inhibited by the replacement of its membrane-spanning domain. J Biol Chem 285, 14681–14688 10.1074/jbc.M109.067090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotov A., Zhou J., Flicker P., Aiken C. (1999). Association of Nef with the human immunodeficiency virus type 1 core. J Virol 73, 8824–8830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y. D., Finzi A., Wu X., Dogo-Isonagie C., Lee L. K., Moore L. R., Schmidt S. D., Stuckey J., Yang Y. & other authors (2012). Unliganded HIV-1 gp120 core structures assume the CD4-bound conformation with regulation by quaternary interactions and variable loops. Proc Natl Acad Sci U S A 109, 5663–5668 10.1073/pnas.1112391109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong P. D., Wyatt R., Robinson J., Sweet R. W., Sodroski J., Hendrickson W. A. (1998). Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393, 648–659 10.1038/31405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBranche C. C., Sauter M. M., Haggarty B. S., Vance P. J., Romano J., Hart T. K., Bugelski P. J., Hoxie J. A. (1994). Biological, molecular, and structural analysis of a cytopathic variant from a molecularly cloned simian immunodeficiency virus. J Virol 68, 7665–7667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard C. K., Spellman M. W., Riddle L., Harris R. J., Thomas J. N., Gregory T. J. (1990). Assignment of intrachain disulfide bonds and characterization of potential glycosylation sites of the type 1 recombinant human immunodeficiency virus envelope glycoprotein (gp120) expressed in Chinese hamster ovary cells. J Biol Chem 265, 10373–10382 [PubMed] [Google Scholar]
- Liu J., Bartesaghi A., Borgnia M. J., Sapiro G., Subramaniam S. (2008). Molecular architecture of native HIV-1 gp120 trimers. Nature 455, 109–113 10.1038/nature07159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge R., Göttlinger H., Gabuzda D., Cohen E. A., Lemay G. (1994). The intracytoplasmic domain of gp41 mediates polarized budding of human immunodeficiency virus type 1 in MDCK cells. J Virol 68, 4857–4861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge R., Lalonde J. P., Lemay G., Cohen E. A. (1997). The membrane-proximal intracytoplasmic tyrosine residue of HIV-1 envelope glycoprotein is critical for basolateral targeting of viral budding in MDCK cells. EMBO J 16, 695–705 10.1093/emboj/16.4.695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Vergès S., Camus G., Blot G., Beauvoir R., Benarous R., Berlioz-Torrent C. (2006). Tail-interacting protein TIP47 is a connector between Gag and Env and is required for Env incorporation into HIV-1 virions. Proc Natl Acad Sci U S A 103, 14947–14952 10.1073/pnas.0602941103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M., Blacklow S. C., Kim P. S. (1995). A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat Struct Biol 2, 1075–1082 10.1038/nsb1295-1075 [DOI] [PubMed] [Google Scholar]
- Lu L., Zhu Y., Huang J., Chen X., Yang H., Jiang S., Chen Y.-H. (2008). Surface exposure of the HIV-1 env cytoplasmic tail LLP2 domain during the membrane fusion process: interaction with gp41 fusion core. J Biol Chem 283, 16723–16731 10.1074/jbc.M801083200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciw P. A., Fields B. N., Knipe D. M., Howley P. M. (editors) (2002). Fields' Virology. Philadelphia: Lippincott-Raven [Google Scholar]
- Marsh D. (2007). Lateral pressure profile, spontaneous curvature frustration, and the incorporation and conformation of proteins in membranes. Biophys J 93, 3884–3899 10.1529/biophysj.107.107938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCune J. M., Rabin L. B., Feinberg M. B., Lieberman M., Kosek J. C., Reyes G. R., Weissman I. L. (1988). Endoproteolytic cleavage of gp160 is required for the activation of human immunodeficiency virus. Cell 53, 55–67 10.1016/0092-8674(88)90487-4 [DOI] [PubMed] [Google Scholar]
- McElrath M. J., Haynes B. F. (2010). Induction of immunity to human immunodeficiency virus type-1 by vaccination. Immunity 33, 542–554 10.1016/j.immuni.2010.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan G. B. (2011). Membrane fusion mediated by human immunodeficiency virus envelope glycoprotein. Curr Top Membr 68, 81–106 10.1016/B978-0-12-385891-7.00004-0 [DOI] [PubMed] [Google Scholar]
- Micoli K. J., Pan G., Wu Y., Williams J. P., Cook W. J., McDonald J. M. (2000). Requirement of calmodulin binding by HIV-1 gp160 for enhanced FAS-mediated apoptosis. J Biol Chem 275, 1233–1240 10.1074/jbc.275.2.1233 [DOI] [PubMed] [Google Scholar]
- Miller M. A., Garry R. F., Jaynes J. M., Montelaro R. C. (1991). A structural correlation between lentivirus transmembrane proteins and natural cytolytic peptides. AIDS Res Hum Retroviruses 7, 511–519 10.1089/aid.1991.7.511 [DOI] [PubMed] [Google Scholar]
- Miller M. A., Cloyd M. W., Liebmann J., Rinaldo C. R., Jr, Islam K. R., Wang S. Z., Mietzner T. A., Montelaro R. C. (1993a). Alterations in cell membrane permeability by the lentivirus lytic peptide (LLP-1) of HIV-1 transmembrane protein. Virology 196, 89–100 10.1006/viro.1993.1457 [DOI] [PubMed] [Google Scholar]
- Miller M. A., Mietzner T. A., Cloyd M. W., Robey W. G., Montelaro R. C. (1993b). Identification of a calmodulin-binding and inhibitory peptide domain in the HIV-1 transmembrane glycoprotein. AIDS Res Hum Retroviruses 9, 1057–1066 10.1089/aid.1993.9.1057 [DOI] [PubMed] [Google Scholar]
- Mitchell D. J., Steinman L., Kim D. T., Fathman C. G., Rothbard J. B. (2000). Polyarginine enters cells more efficiently than other polycationic homopolymers. J Pept Res 56, 318–325 10.1034/j.1399-3011.2000.00723.x [DOI] [PubMed] [Google Scholar]
- Montefiori D. C., Robinson W. E., Jr, Mitchell W. M. (1988). Role of protein N-glycosylation in pathogenesis of human immunodeficiency virus type 1. Proc Natl Acad Sci U S A 85, 9248–9252 10.1073/pnas.85.23.9248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero M., van Houten N. E., Wang X., Scott J. K. (2008). The membrane-proximal external region of the human immunodeficiency virus type 1 envelope: dominant site of antibody neutralization and target for vaccine design. Microbiol Mol Biol Rev 72, 54–84 10.1128/MMBR.00020-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Barroso I., Salzwedel K., Hunter E., Blumenthal R. (1999). Role of the membrane-proximal domain in the initial stages of human immunodeficiency virus type 1 envelope glycoprotein-mediated membrane fusion. J Virol 73, 6089–6092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T. (2008). Roles of the interactions between Env and Gag proteins in the HIV-1 replication cycle. Microbiol Immunol 52, 287–295 10.1111/j.1348-0421.2008.00008.x [DOI] [PubMed] [Google Scholar]
- Murakami T., Freed E. O. (2000a). Genetic evidence for an interaction between human immunodeficiency virus type 1 matrix and alpha-helix 2 of the gp41 cytoplasmic tail. J Virol 74, 3548–3554 10.1128/JVI.74.8.3548-3554.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T., Freed E. O. (2000b). The long cytoplasmic tail of gp41 is required in a cell type-dependent manner for HIV-1 envelope glycoprotein incorporation into virions. Proc Natl Acad Sci U S A 97, 343–348 10.1073/pnas.97.1.343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T., Ablan S., Freed E. O., Tanaka Y. (2004). Regulation of human immunodeficiency virus type 1 Env-mediated membrane fusion by viral protease activity. J Virol 78, 1026–1031 10.1128/JVI.78.2.1026-1031.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myszka D. G., Sweet R. W., Hensley P., Brigham-Burke M., Kwong P. D., Hendrickson W. A., Wyatt R., Sodroski J., Doyle M. L. (2000). Energetics of the HIV gp120-CD4 binding reaction. Proc Natl Acad Sci U S A 97, 9026–9031 10.1073/pnas.97.16.9026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D. H., Hildreth J. E. (2000). Evidence for budding of human immunodeficiency virus type 1 selectively from glycolipid-enriched membrane lipid rafts. J Virol 74, 3264–3272 10.1128/JVI.74.7.3264-3272.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno H., Aguilar R. C., Fournier M. C., Hennecke S., Cosson P., Bonifacino J. S. (1997). Interaction of endocytic signals from the HIV-1 envelope glycoprotein complex with members of the adaptor medium chain family. Virology 238, 305–315 10.1006/viro.1997.8839 [DOI] [PubMed] [Google Scholar]
- Patil A., Gautam A., Bhattacharya J. (2010). Evidence that Gag facilitates HIV-1 envelope association both in GPI-enriched plasma membrane and detergent resistant membranes and facilitates envelope incorporation onto virions in primary CD4+ T cells. Virol J 7, 3 10.1186/1743-422X-7-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004). UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- Pinter A., Honnen W. J., Tilley S. A., Bona C., Zaghouani H., Gorny M. K., Zolla-Pazner S. (1989). Oligomeric structure of gp41, the transmembrane protein of human immunodeficiency virus type 1. J Virol 63, 2674–2679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postler T. S., Desrosiers R. C. (2012). The cytoplasmic domain of the HIV-1 glycoprotein gp41 induces NF-κB activation through TGF-β-activated kinase 1. Cell Host Microbe 11, 181–193 10.1016/j.chom.2011.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postler T. S., Martinez-Navio J. M., Yuste E., Desrosiers R. C. (2012). Evidence against extracellular exposure of a highly immunogenic region in the C-terminal domain of the simian immunodeficiency virus gp41 transmembrane protein. J Virol 86, 1145–1157 10.1128/JVI.06463-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston B. D., Poiesz B. J., Loeb L. A. (1988). Fidelity of HIV-1 reverse transcriptase. Science 242, 1168–1171 10.1126/science.2460924 [DOI] [PubMed] [Google Scholar]
- Reading S. A., Heap C. J., Dimmock N. J. (2003). A novel monoclonal antibody specific to the C-terminal tail of the gp41 envelope transmembrane protein of human immunodeficiency virus type 1 that preferentially neutralizes virus after it has attached to the target cell and inhibits the production of infectious progeny. Virology 315, 362–372 10.1016/S0042-6822(03)00533-6 [DOI] [PubMed] [Google Scholar]
- Roberts J. D., Bebenek K., Kunkel T. A. (1988). The accuracy of reverse transcriptase from HIV-1. Science 242, 1171–1173 10.1126/science.2460925 [DOI] [PubMed] [Google Scholar]
- Rousso I., Mixon M. B., Chen B. K., Kim P. S. (2000). Palmitoylation of the HIV-1 envelope glycoprotein is critical for viral infectivity. Proc Natl Acad Sci U S A 97, 13523–13525 10.1073/pnas.240459697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowell J. F., Stanhope P. E., Siliciano R. F. (1995). Endocytosis of endogenously synthesized HIV-1 envelope protein. Mechanism and role in processing for association with class II MHC. J Immunol 155, 473–488 [PubMed] [Google Scholar]
- Rushlow K., Olsen K., Stiegler G., Payne S. L., Montelaro R. C., Issel C. J. (1986). Lentivirus genomic organization: the complete nucleotide sequence of the Env gene region of equine infectious anemia virus. Virology 155, 309–321 10.1016/0042-6822(86)90195-9 [DOI] [PubMed] [Google Scholar]
- Salzwedel K., West J. T., Hunter E. (1999). A conserved tryptophan-rich motif in the membrane-proximal region of the human immunodeficiency virus type 1 gp41 ectodomain is important for Env-mediated fusion and virus infectivity. J Virol 73, 2469–2480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauter M. M., Pelchen-Matthews A., Bron R., Marsh M., LaBranche C. C., Vance P. J., Romano J., Haggarty B. S., Hart T. K. & other authors (1996). An internalization signal in the simian immunodeficiency virus transmembrane protein cytoplasmic domain modulates expression of envelope glycoproteins on the cell surface. J Cell Biol 132, 795–811 10.1083/jcb.132.5.795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz G. E. (2000). beta-Barrel membrane proteins. Curr Opin Struct Biol 10, 443–447 10.1016/S0959-440X(00)00120-2 [DOI] [PubMed] [Google Scholar]
- Shacklett B. L., Weber C. J., Shaw K. E., Keddie E. M., Gardner M. B., Sonigo P., Luciw P. A. (2000). The intracytoplasmic domain of the Env transmembrane protein is a locus for attenuation of simian immunodeficiency virus SIVmac in rhesus macaques. J Virol 74, 5836–5844 10.1128/JVI.74.13.5836-5844.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang L., Hunter E. (2010). Residues in the membrane-spanning domain core modulate conformation and fusogenicity of the HIV-1 envelope glycoprotein. Virology 404, 158–167 10.1016/j.virol.2010.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang L., Yue L., Hunter E. (2008). Role of the membrane-spanning domain of human immunodeficiency virus type 1 envelope glycoprotein in cell-cell fusion and virus infection. J Virol 82, 5417–5428 10.1128/JVI.02666-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W., Bohon J., Han D. P., Habte H., Qin Y., Cho M. W., Chance M. R. (2010). Structural characterization of HIV gp41 with the membrane-proximal external region. J Biol Chem 285, 24290–24298 10.1074/jbc.M110.111351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spies C. P., Ritter G. D., Jr, Mulligan M. J., Compans R. W. (1994). Truncation of the cytoplasmic domain of the simian immunodeficiency virus envelope glycoprotein alters the conformation of the external domain. J Virol 68, 585–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas S. K., Srinivas R. V., Anantharamaiah G. M., Segrest J. P., Compans R. W. (1992). Membrane interactions of synthetic peptides corresponding to amphipathic helical segments of the human immunodeficiency virus type-1 envelope glycoprotein. J Biol Chem 267, 7121–7127 [PubMed] [Google Scholar]
- Srinivas S. K., Srinivas R. V., Anantharamaiah G. M., Compans R. W., Segrest J. P. (1993). Cytosolic domain of the human immunodeficiency virus envelope glycoproteins binds to calmodulin and inhibits calmodulin-regulated proteins. J Biol Chem 268, 22895–22899 [PubMed] [Google Scholar]
- Starcich B. R., Hahn B. H., Shaw G. M., McNeely P. D., Modrow S., Wolf H., Parks E. S., Parks W. P., Josephs S. F. & other authors (1986). Identification and characterization of conserved and variable regions in the envelope gene of HTLV-III/LAV, the retrovirus of AIDS. Cell 45, 637–648 10.1016/0092-8674(86)90778-6 [DOI] [PubMed] [Google Scholar]
- Steckbeck J. D., Sun C., Sturgeon T. J., Montelaro R. C. (2010). Topology of the C-terminal tail of HIV-1 gp41: differential exposure of the Kennedy epitope on cell and viral membranes. PLoS ONE 5, e15261 10.1371/journal.pone.0015261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steckbeck J. D., Craigo J. K., Barnes C. O., Montelaro R. C. (2011). Highly conserved structural properties of the C-terminal tail of HIV-1 gp41 protein despite substantial sequence variation among diverse clades: implications for functions in viral replication. J Biol Chem 286, 27156–27166 10.1074/jbc.M111.258855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tencza S. B., Miller M. A., Islam K., Mietzner T. A., Montelaro R. C. (1995). Effect of amino acid substitutions on calmodulin binding and cytolytic properties of the LLP-1 peptide segment of human immunodeficiency virus type 1 transmembrane protein. J Virol 69, 5199–5202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillman T. S., Cascio M. (2003). Effects of membrane lipids on ion channel structure and function. Cell Biochem Biophys 38, 161–190 10.1385/CBB:38:2:161 [DOI] [PubMed] [Google Scholar]
- Tristram-Nagle S., Nagle J. F. (2007). HIV-1 fusion peptide decreases bending energy and promotes curved fusion intermediates. Biophys J 93, 2048–2055 10.1529/biophysj.107.109181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tristram-Nagle S., Chan R., Kooijman E., Uppamoochikkal P., Qiang W., Weliky D. P., Nagle J. F. (2010). HIV fusion peptide penetrates, disorders, and softens T-cell membrane mimics. J Mol Biol 402, 139–153 10.1016/j.jmb.2010.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung C.-H., Weissleder R. (2003). Arginine containing peptides as delivery vectors. Adv Drug Deliv Rev 55, 281–294 10.1016/S0169-409X(02)00183-7 [DOI] [PubMed] [Google Scholar]
- van den Brink-van der Laan E., Killian J. A., de Kruijff B. (2004). Nonbilayer lipids affect peripheral and integral membrane proteins via changes in the lateral pressure profile. Biochim Biophys Acta 1666, 275–288 10.1016/j.bbamem.2004.06.010 [DOI] [PubMed] [Google Scholar]
- Vella C., Ferguson M., Dunn G., Meloen R., Langedijk H., Evans D., Minor P. D. (1993). Characterization and primary structure of a human immunodeficiency virus type 1 (HIV-1) neutralization domain as presented by a poliovirus type 1/HIV-1 chimera. J Gen Virol 74, 2603–2607 10.1099/0022-1317-74-12-2603 [DOI] [PubMed] [Google Scholar]
- Viard M., Ablan S. D., Zhou M., Veenstra T. D., Freed E. O., Raviv Y., Blumenthal R. (2008). Photoinduced reactivity of the HIV-1 envelope glycoprotein with a membrane-embedded probe reveals insertion of portions of the HIV-1 gp41 cytoplasmic tail into the viral membrane. Biochemistry 47, 1977–1983 10.1021/bi701920f [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waheed A. A., Ablan S. D., Roser J. D., Sowder R. C., Schaffner C. P., Chertova E., Freed E. O. (2007). HIV-1 escape from the entry-inhibiting effects of a cholesterol-binding compound via cleavage of gp41 by the viral protease. Proc Natl Acad Sci U S A 104, 8467–8471 10.1073/pnas.0701443104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waheed A. A., Ablan S. D., Sowder R. C., Roser J. D., Schaffner C. P., Chertova E., Freed E. O. (2010). Effect of mutations in the human immunodeficiency virus type 1 protease on cleavage of the gp41 cytoplasmic tail. J Virol 84, 3121–3126 10.1128/JVI.02002-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissenhorn W., Dessen A., Harrison S. C., Skehel J. J., Wiley D. C. (1997). Atomic structure of the ectodomain from HIV-1 gp41. Nature 387, 426–430 10.1038/387426a0 [DOI] [PubMed] [Google Scholar]
- White S. H., Wimley W. C. (1999). Membrane protein folding and stability: physical principles. Annu Rev Biophys Biomol Struct 28, 319–365 10.1146/annurev.biophys.28.1.319 [DOI] [PubMed] [Google Scholar]
- White T. A., Bartesaghi A., Borgnia M. J., Meyerson J. R., de la Cruz M. J. V., Bess J. W., Nandwani R., Hoxie J. A., Lifson J. D. & other authors (2010). Molecular architectures of trimeric SIV and HIV-1 envelope glycoproteins on intact viruses: strain-dependent variation in quaternary structure. PLoS Pathog 6, e1001249 10.1371/journal.ppat.1001249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willey R. L., Rutledge R. A., Dias S., Folks T., Theodore T., Buckler C. E., Martin M. A. (1986). Identification of conserved and divergent domains within the envelope gene of the acquired immunodeficiency syndrome retrovirus. Proc Natl Acad Sci U S A 83, 5038–5042 10.1073/pnas.83.14.5038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Yang Z.-Y., Li Y., Hogerkorp C.-M., Schief W. R., Seaman M. S., Zhou T., Schmidt S. D., Wu L. & other authors (2010). Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 329, 856–861 10.1126/science.1187659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyma D. J., Kotov A., Aiken C. (2000). Evidence for a stable interaction of gp41 with Pr55Gag in immature human immunodeficiency virus type 1 particles. J Virol 74, 9381–9387 10.1128/JVI.74.20.9381-9387.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyma D. J., Jiang J., Shi J., Zhou J., Lineberger J. E., Miller M. D., Aiken C. (2004). Coupling of human immunodeficiency virus type 1 fusion to virion maturation: a novel role of the gp41 cytoplasmic tail. J Virol 78, 3429–3435 10.1128/JVI.78.7.3429-3435.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss S., Dimitrov A. S., Baribaud F., Edwards T. G., Blumenthal R., Hoxie J. A. (2005). Regulation of human immunodeficiency virus type 1 envelope glycoprotein fusion by a membrane-interactive domain in the gp41 cytoplasmic tail. J Virol 79, 12231–12241 10.1128/JVI.79.19.12231-12241.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue L., Shang L., Hunter E. (2009). Truncation of the membrane-spanning domain of human immunodeficiency virus type 1 envelope glycoprotein defines elements required for fusion, incorporation, and infectivity. J Virol 83, 11588–11598 10.1128/JVI.00914-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Wang L., Kao S., Whitehead I. P., Hart M. J., Liu B., Duus K., Burridge K., Der C. J., Su L. (1999). Functional interaction between the cytoplasmic leucine-zipper domain of HIV-1 gp41 and p115-RhoGEF. Curr Biol 9, 1271–1274 10.1016/S0960-9822(99)80511-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T., Xu L., Dey B., Hessell A. J., Van Ryk D., Xiang S.-H., Yang X., Zhang M.-Y., Zwick M. B. & other authors (2007). Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature 445, 732–737 10.1038/nature05580 [DOI] [PMC free article] [PubMed] [Google Scholar]