

Abstract
The role of the CcpC regulatory protein as a repressor of the genes encoding the tricarboxylic acid branch enzymes of the Krebs cycle (citrate synthase, citZ; aconitase, citB; and isocitrate dehydrogenase, citC) has been established for both Bacillus subtilis and Listeria monocytogenes. In addition, hyperexpression of citB–lacZ reporter constructs in an aconitase null mutant strain has been reported for B. subtilis. We show here that such hyperexpression of citB occurs in L. monocytogenes as well as in B. subtilis and that in both species the hyperexpression is unexpectedly dependent on CcpC. We propose a revision of the existing CcpC–citB regulatory scheme and suggest a mechanism of regulation in which CcpC represses citB expression at low citrate levels and activates citB expression when citrate levels are high.
Introduction
In the Gram-positive bacterium Bacillus subtilis, the coordinated expression of the first three enzymes of the Krebs cycle (citrate synthase, citZ; aconitase, citB; and isocitrate dehydrogenase, citC) is controlled by three regulatory proteins: CodY, CcpA and CcpC (reviewed by Sonenshein, 2007). CcpA and CodY are global regulatory proteins that respond to the intracellular pools of fructose-1,6-bisphosphate and ATP for CcpA (Jault et al., 2000) and the combination of GTP and the branched-chain amino acids for CodY (Handke et al., 2008; Villapakkam et al., 2009). CcpA exerts both direct (through citZ and ccpC) and indirect effects on tricarboxylic acid (TCA) branch enzyme expression (Kim et al., 2002b), while CodY binds to the citB promoter region and represses citB transcription (Kim et al., 2003a). Unlike these global regulators, CcpC exclusively regulates the TCA branch enzymes by responding to a pathway-specific metabolite, citrate. Citrate-antagonized repression of the B. subtilis citZ, citB and citC genes by CcpC has been described in detail (Jourlin-Castelli et al., 2000; Kim et al., 2003a, b). In the closely related intracellular pathogen Listeria monocytogenes, the citZ and citB genes are regulated by CcpC and CodY, but not by CcpA (Kim et al., 2006; Mittal, 2008; Mittal et al., 2009).
CcpC is a member of the LysR-type transcriptional regulator (LTTR) family of proteins (Jourlin-Castelli et al., 2000; Kim et al., 2003b), a group that includes B. subtilis GltC, the regulator of the glutamate synthase genes (Belitsky et al., 1995) and Escherichia coli OxyR (Maddocks & Oyston, 2008). B. subtilis CcpC dimers bind to two sites within the citB promoter region (Fig. 1), a dyad symmetry element centred at position −66 with respect to the transcriptional start site and a half-dyad element located at positions −27 to −33 (Fouet et al., 1990; Fouet & Sonenshein, 1990; Jourlin-Castelli et al., 2000). Both sites are required for repression; interaction between the two CcpC dimers bound at these sites results in bending of the DNA, blocking access of RNA polymerase to the promoter and resulting in repression of citB expression (Jourlin-Castelli et al., 2000; Kim et al., 2003b). As with other LTTR proteins, CcpC repression is relieved by the interaction of the protein with a metabolite acting as an inducer; in this case, citrate induces the expression of citB (Blencke et al., 2006). Citrate disrupts CcpC binding to the −27 site in the citB promoter; while CcpC remains bound to the −66 dyad symmetry element, the loss of binding at the −27 half-dyad element relaxes the bending of the DNA and allows RNA polymerase to interact with the promoter (Kim et al., 2003b). Similar citrate-dependent derepression has also been described for the citB gene of L. monocytogenes (Kim et al., 2006). In L. monocytogenes, CcpC represses transcription of citB as well as of citZ and the lmo0847 gene, which encodes a putative glutamine transporter (Kim et al., 2006; Mittal et al., 2009). The organization of the CcpC binding sites in the L. monocytogenes citB regulatory region is almost identical to that in B. subtilis, with a dyad symmetry element centred at position −68 and a half-dyad at positions −28 to −32 (Kim et al., 2006). In vitro, in the presence of citrate, binding to the full dyad is maintained, but binding to the half-dyad is reduced (Kim et al., 2006). Therefore, in both species, citrate synthesis is necessary for full expression of the TCA branch enzymes. Because citrate is produced uniquely by the activity of citrate synthase, this regulatory loop provides a mechanism for the sequential expression of these central metabolic enzymes. However, in this report we present data indicating that this model is incomplete.
Fig. 1.

CcpC binding sites in the citB promoter region. Two CcpC binding sites are present in the citB regulatory regions of L. monocytogenes and B. subtilis. The B. subtilis −66 dyad symmetry element and −27 half-dyad element are shown with their respective sequences. The −35 and −10 promoter elements are also indicated (distances are not to scale).
Previous results hinted that the nature of the CcpC–citB interaction is more complex than a simple repression model. B. subtilis cells that lack a functional aconitase (citB null mutants) accumulate a vast excess of citrate during growth (Craig et al., 1997). Concomitant with unusually high citrate accumulation is hyperexpression from the citB promoter; the expression of a citB–lacZ promoter fusion is enhanced in a citB null mutant to levels five- to tenfold higher than those seen in wild-type cells, and this overexpression is dependent on a functional citrate synthase (Kim et al., 2003a), reinforcing the notion that citrate is the factor that hyperinduces citB expression. We show here that hyperexpression of citB–lacZ in a citB null mutant is also seen in L. monocytogenes and that this phenotype is again correlated with a vast overaccumulation of citrate and is dependent on the synthesis of citrate. In searching for the regulatory mechanism responsible for this citrate-dependent hyperexpression in these related species, we found to our surprise that CcpC, previously known only as a repressor, becomes an activator of citB transcription under conditions of high citrate accumulation. Thus, CcpC in both species switches from a negative regulator to a positive regulator when citrate accumulates.
Methods
Bacterial strains and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. B. subtilis strains were grown at 37 °C with aeration in DS medium [0.8 % nutrient broth, 0.1 % KCl, 0.025 % MgSO4 . 7H2O, 1 mM Ca(NO3)2, 10 µM MnCl2, 1 µM FeSO4 (Fouet & Sonenshein, 1990)] and supplemented with chloramphenicol (2.5 µg ml−1), erythromycin (1.0 µg ml−1), neomycin (2.5 µg ml−1), phleomycin (0.25 µg ml−1) or spectinomycin (50 µg ml−1) when necessary. L. monocytogenes strains were grown in brain heart infusion medium (BHI; Difco) at 37 °C. In general, cultures were grown in Erlenmeyer flasks with a medium-to-flask volume ratio of 10 and a circular agitation speed of 200 r.p.m.
Table 1. Bacterial strains used in this study.
| Strain | Genotype | Source or reference |
| B. subtilis | ||
| SMY | Prototroph | P. Schaeffer |
| JH642 | trpC2 pheA1 | Brehm et al. (1973), Dean et al. (1977) |
| AF21 | ΔamyE : : Φ(citBp21–lacZ cat) | SMY × pAF1. Fouet & Sonenshein (1990) |
| MAB160 | trpC2 pheA1 ΩcitB : : spc | Craig et al. (1997) |
| AWS96 | trpC2 pheA1 | MAB160 × JH642 DNA. Serio et al. (2006) |
| PS258 | trpC2 ΔcodY : : erm | P. Serror |
| HKB125 | ΔamyE : : Φ(citBp23–lacZ cat) ΔcodY : : erm | Kim et al. (2003a) |
| HKB126 | ΔamyE : : Φ(citBp24–lacZ cat) ΔcodY : : erm | Kim et al. (2003a) |
| HKB165 | ΔamyE : : Φ(citBp21–lacZ cat) ΩcitB : : spc | Kim et al. (2003a) |
| HKB186 | ΔamyE : : Φ(citBp21–lacZ cat) ΔccpCBS : : ble | Kim et al. (2006) |
| KBP26 | trpC2 pheA1 ΔamyE : : Φ(citBp21–lacZ cat) | AWS96 × AF21 DNA |
| KBP51 | trpC2 pheA1 ΔamyE : : Φ(citBp21–lacZ cat) ΩcitB : : spc | KBP26 × MAB160 DNA |
| KBP52 | trpC2 pheA1 ΔamyE : : Φ(citBp21–lacZ cat) ΔccpC : : ble | KBP26 × HKB186 DNA |
| KBP54 | trpC2 pheA1 ΔamyE : : Φ(citBp21–lacZ cat) ΩcitB : : spc ΔccpC : : ble | KBP51 × HKB186 DNA |
| KBP56 | trpC2 pheA1 ΔamyE : : Φ(citBp23–lacZ cat) | AWS96 × HKB125 DNA |
| KBP57 | trpC2 pheA1 ΔamyE : : Φ(citBp24–lacZ cat) | AWS96 × HKB126 DNA |
| KBP62 | trpC2 pheA1 ΔamyE : : Φ(citBp23–lacZ cat) ΩcitB : : spc | MAB160 × KBP56 DNA |
| KBP63 | trpC2 pheA1 ΔamyE : : Φ(citBp24–lacZ cat) ΩcitB : : spc | MAB160 × KBP57 DNA |
| KBP141 | trpC2 pheA1 ΔamyE : : Φ(citBp21–lacZ cat) ΔcodY : : erm | KBP26 × PS258 DNA |
| KBP142 | trpC2 pheA1 ΔamyE : : Φ(citBp21–lacZ cat) ΔcodY : : erm ΩcitB : : spc | KBP51 × PS258 DNA |
| KBP143 | trpC2 pheA1 ΔamyE : : Φ(citBp21–lacZ cat) ΔcodY : : erm ΔccpC : : ble | KBP52 × PS258 DNA |
| KBP144 | trpC2 pheA1 ΔamyE : : Φ(citBp21–lacZ cat) ΔcodY : : erm ΩcitB : : spc ΔccpC : : ble | KBP54 × PS258 DNA |
| L. monocytogenes | ||
| HKB214 | Δint : : Φ(citB–lacZ neo) | Kim et al. (2006) |
| HKB217 | Δint : : Φ(citB–lacZ neo) ΔccpC : : spc | Kim et al. (2006) |
| LMM12 | Δint : : Φ(citB–lacZ neo) ΔcitB : : tet | HKB214 × pEMM20 |
| LMM25 | Δint : : Φ(citB–lacZ neo) ΔcitB : : tet ΔccpC : : spc | HKB217 × pEMM20 |
| LMM33 | Δint : : Φ(citB–lacZ neo) ΔcitZ | Mittal et al. (2009) |
| LMM34 | Δint : : Φ(citB–lacZ neo) ΔcitB : : tet ΔcitZ | LMM12 × pEMM47 DNA |
Construction of a citB null mutant of L. monocytogenes.
Using oligonucleotide primers specific to each DNA segment, the first 960 bp of the citB gene, a tetracycline resistance gene and the last 1019 bp of citB were amplified by PCR and ligated sequentially to pPS34, a derivative of pSK- (Stratagene) that was modified to carry an erythromycin-resistance gene active in Gram-positive bacteria (P. Serror, personal communication). The resulting plasmid was digested with SacI and KpnI and the insert (citB′-tet-′citB) was ligated to pCON-1 (Behari & Youngman, 1998). The final plasmid, pEMM20, was introduced by transformation into HKB214, a L. monocytogenes strain carrying a citB–lacZ fusion at the nonessential int locus (Kim et al., 2006). Transformants were isolated and passaged to obtain a double-crossover at the citB locus, resulting in strain LMM12 [Δint : : (Φ citB–lacZ neo) ΔcitB : : tet]. Plasmid pEMM20 was also introduced into strain HKB217 to create strain LMM25 [Δint : : (Φ citB–lacZ neo) ΔcitB : : tet ΔccpC : : spc].
β-Galactosidase assay.
For β-galactosidase activity assays, samples (1 ml) were removed from B. subtilis or L. monocytogenes broth cultures during growth after determining the OD600 of the culture at that time point, and cell pellets were frozen on dry ice. Cells were permeabilized and assayed as described previously (Belitsky et al., 1995). β-Galactosidase activity (Miller units) was calculated as described previously (Miller, 1972); however, a volume correction factor of 1.25 was used to account for the increase in the reaction volume due to addition of sodium carbonate to stop the reaction.
Assay of intracellular citrate concentrations.
Cultures (25 ml) of L. monocytogenes strains were grown in BHI at 37 °C until they reached OD600 0.8–1.0. After collection of the cells by centrifugation, the pellet was washed with 20 mM Tris/HCl, pH 8, containing 1 mM EDTA, and resuspended in 4 ml 0.3 M perchloric acid. After incubation on ice for 10 min, the cell debris was removed by centrifugation and the supernatant fluid was mixed with 2 ml 0.75 M K2CO3 and kept on ice for 15 min. The concentration of citrate in the supernatant fluid after subsequent centrifugation was determined using a kit (R-Biopharm) in which citrate lyase activity is coupled to malate dehydrogenase, lactate dehydrogenase and oxidation of NADH.
Assay of aconitase enzyme activity.
L. monocytogenes cells were grown in BHI broth and harvested by centrifugation at the beginning of stationary phase, washed with a buffer containing 20 mM Tris-citrate (pH 7.35), 150 mM KCl and 0.5 mM PMSF and stored frozen at −80 °C. Cell pellets were thawed in the same buffer and treated with mutanolysin as described by Fliss et al. (1991). Cell extracts were clarified by centrifugation and stored at 4 °C. Protein concentrations were determined using the Bradford protein assay reagent (Bio-Rad). The aconitase enzyme activity was determined as described previously (Dingman & Sonenshein, 1987). When assaying crude extracts, it is difficult to estimate very low specific activities accurately. One unit of aconitase activity is equal to a change in A240 of 0.0033 min−1; the change in absorbance directly measures the production of 1 nmol cis-aconitate ml−1. Therefore, when 3 µg of extract is assayed, a change in A240 of 0.001 beyond background over 10 min corresponds to a specific activity of 10 units mg−1.
Results
Hyperexpression of the citB promoter in a L. monocytogenes citB null mutant
To determine whether the hyperexpression of the citB gene previously observed in a citB null mutant of B. subtilis (Kim et al., 2003a) also occurs in L. monocytogenes, a related, medically relevant pathogen, we introduced a citB null mutation into a L. monocytogenes strain carrying a citB–lacZ fusion. The L. monocytogenes citB null strain was a glutamate auxotroph; it did not grow in minimal medium unless a source of glutamate (e.g. glutamine) was provided, indicating that it does not possess a complete TCA branch of the Krebs cycle. To confirm the loss of aconitase enzyme activity in the citB null strain, cell extracts of wild-type (HKB214) and citB null mutant (LMM12) strains were prepared by mutanolysin treatment and assayed for aconitase activity. The wild-type extract had an aconitase specific activity [195 units (mg protein)−1] that was 10-fold higher than that of the citB null mutant extract [18 units (mg protein)−1]. The apparent presence of residual aconitase activity in the null mutant strain is due to the high background of the assay (see Methods).
To determine the effect of the citB null mutation on citB–lacZ expression, the wild-type and citB null strains grown in BHI were assayed for β-galactosidase activity. Wild-type L. monocytogenes yielded a low level of β-galactosidase activity (Fig. 2a). In the citB null strain, however, citB–lacZ expression was increased 10-fold compared with the wild-type strain (Fig. 2a).
Fig. 2.
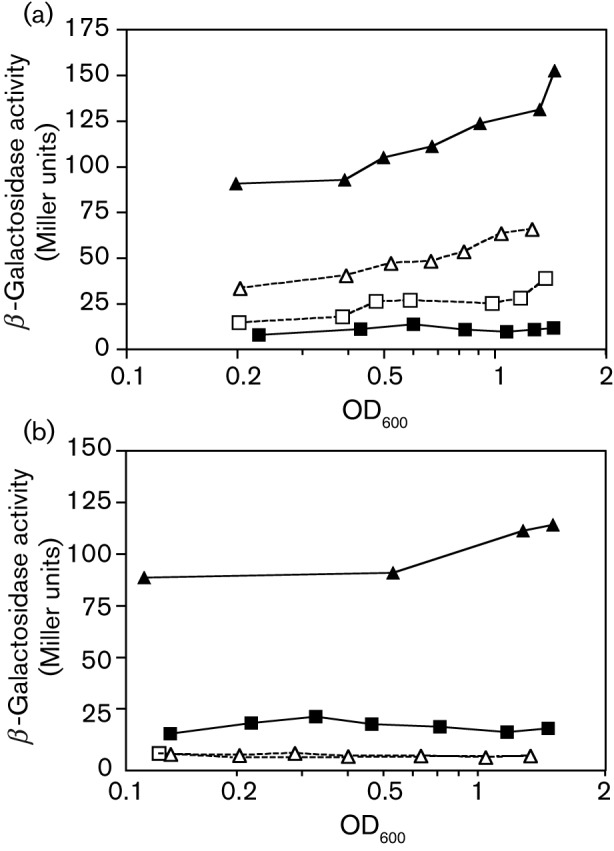
Expression of a citB–lacZ fusion in citB and ccpC null mutant L. monocytogenes strains. β-Galactosidase activity was measured at various points during growth of L. monocytogenes cells carrying a citB′–lacZ fusion linked to a neo resistance marker and integrated at the non-essential int′–′comK locus (Kim et al., 2006). For each of the strains, the end of exponential growth phase (OD600 ~ 0.8) occurred 2.25–2.75 h after the strains reached OD600 = 0.2. (a) Strains HKB214 (citB+ ccpC+; ▪), LMM12 (ΔcitB : : tet ccpC+; ▴), HKB217 (citB+ ΔccpC : : spc; □) and LMM25 (ΔcitB : : tet ΔccpC : : spc; ▵) were compared. (b) Strains HKB214 (citB+ citZ+; ▪), LMM12 (ΔcitB : : tet citZ+; ▴), LMM33 (ΔcitZ; □) and LMM34 (ΔcitB : : tet ΔcitZ; ▵) were compared. The end of exponential growth phase occurred 3–4 h after the strains reached OD600 0.1.
Hyperexpression is dependent on the accumulation of citrate
To verify that citrate synthesis is necessary for hyperexpression of citB–lacZ in an L. monocytogenes citB null strain, as it is in B. subtilis, we created a citB citZ double mutant strain. The citZ gene encodes the sole citrate synthase in L. monocytogenes. A previously described insertion–deletion mutation in the citZ gene (Mittal et al., 2009) was utilized; genomic DNA from this strain, LMM33, was introduced into strain LMM12 (citB null) by double crossover recombination, producing strain LMM34. Importantly, the citB citZ double mutant strain exhibited very low levels of citB–lacZ activity compared with the citB single mutant (Fig. 2b), indicating that citrate synthesis is required for the hyperexpression of citB in L. monocytogenes as well as in B. subtilis.
To verify that the high level of citB–lacZ expression seen in the citB mutant strain was correlated with a significant change in the pool of citrate within the cell, we assayed intracellular citrate (see Methods). The concentration of citrate in citB mutant cells was about 600-fold higher than that in wild-type cells (Table 2).
Table 2. Intracellular pool of citrate in L. monocytogenes mutant and wild-type cells.
Cultures of the indicated strains were assayed for intracellular citrate as described in Methods.
| Strain | Genotype | Intracellular citrate (mM) |
| HKB214 | Wild-type | 0.016±0.007 |
| HKB217 | ccpC null | Not detectable |
| LMM12 | citB null | 9.5±0.75 |
| LMM25 | citB ccpC null | 8.9±0.61 |
CcpC is necessary for hyperexpression of citB–lacZ in a L. monocytogenes citB null mutant
Given that high levels of citrate correlate with citB hyperexpression in L. monocytogenes (Fig. 2b) and that CcpC is known to be inactivated as a repressor by citrate, we attempted to rule out CcpC as the agent of hyperexpression. As previously reported (Kim et al., 2006), a ccpC null mutation leads to a slight increase in citB–lacZ expression in cells grown in BHI medium. To our surprise, however, the introduction of a ccpC null mutation into the citB null mutant background resulted in loss of the hyperexpression phenotype (Fig. 2a). The citB ccpC null mutant had a level of citB–lacZ expression similar to that of the ccpC single mutant strain. Thus, L. monocytogenes CcpC is required for the hyperexpression of citB–lacZ caused by a citB null mutation, implying that CcpC acts as a positive regulator of citB expression when citrate accumulates to high levels.
In addition, inactivating CcpC overcame the hyperexpression phenotype without reducing the citrate pool. Whereas the concentration of citrate in ccpC single mutant cells was below the level of detection, presumably reflecting a high rate of citrate metabolism in the ccpC mutant strain, the intracellular pool of citrate in the ccpC citB double mutant was high, similar to those levels found in the citB single mutant (Table 2). Thus, eliminating CcpC suppresses the high-level citB–lacZ expression caused by inactivating citB without reducing the accumulation of citrate. This indicates that the high citrate pool alone does not cause citB hyperexpression in the absence of CcpC.
CcpC is necessary for hyperexpression of citB–lacZ in a B. subtilis citB null mutant
To study the role of CcpC in citB hyperexpression in a bacterium that is more highly tractable than L. monocytogenes and in which our understanding of citB regulation is more detailed, we sought to determine whether CcpC is necessary for hyperexpression of a citB–lacZ fusion in B. subtilis. To do so, we created a citB ccpC double null mutant carrying a citB–lacZ fusion (KBP54). Wild-type (KBP26), citB null (KBP51), ccpC null (KBP52) and citB ccpC double mutant strains were grown in DS medium and β-galactosidase activity was measured during growth. [DS medium was used because the effect of a citB null mutation is more pronounced in that medium than in glucose-minimal medium (data not shown).] As reported previously (Kim et al., 2003a), the citB null mutation resulted in hyperexpression of citB–lacZ, while the ccpC null mutation caused a slight increase in citB–lacZ expression (Fig. 3a). As was the case in L. monocytogenes, the ccpC citB double mutant strain of B. subtilis behaved like a ccpC null strain; that is, the ccpC null mutation suppressed the citB hyperexpression phenotype (Fig. 3a). Therefore, CcpC appears to be a positive regulator of citB expression in B. subtilis as well as in L. monocytogenes.
Fig. 3.
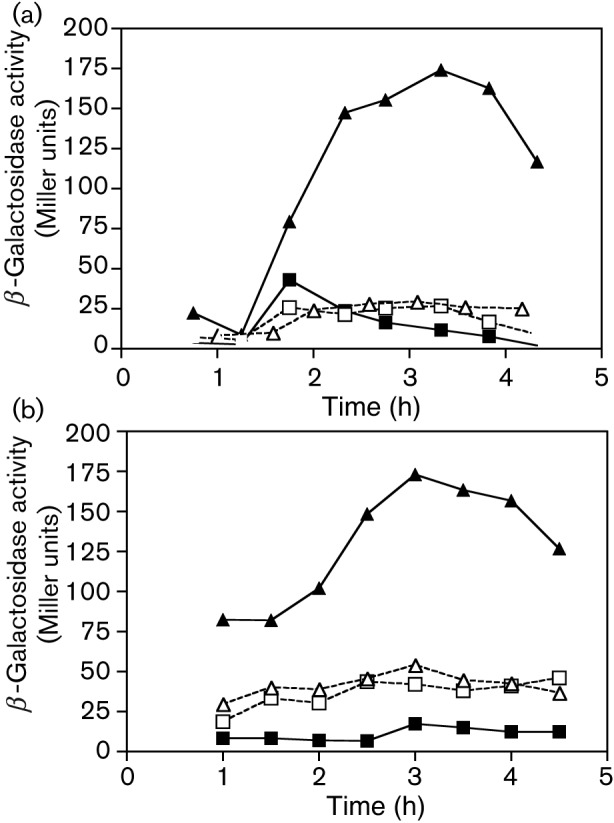
Expression of a citB–lacZ fusion in citB, ccpC and codY mutant strains of B. subtilis. β-Galactosidase activity was measured at various points during growth of B. subtilis strains carrying a citB′–lacZ fusion integrated at the non-essential amyE locus. (a) Strains KBP26 (citB+; ▪), KBP51 (citB null; ▴), KBP52 (ccpC null; □) and KBP54 (citB ccpC null; ▵) strains were compared. The end of exponential growth phase was reached at 3.3 h for strains KBP26, KBP51 and KBP52 and at 2.67 h for strain KBP54. (b) Strains KBP141 (codY null; ▪), KBP142 (codY citB null; ▴), KBP143 (codY ccpC null; □) and KBP144 (codY citB ccpC null; ▵) were compared. The end of exponential growth phase was reached at 3.5 h for strains KBP141, KBP142 and KBP144 and at 4.0 h for strain KBP143.
The −66 dyad symmetry element is necessary for the activation of citB expression by B. subtilis CcpC
As described above, CcpC represses citB in B. subtilis by binding to a dyad element centred at position −66 and a half-dyad element at position −27 (Jourlin-Castelli et al., 2000). When CcpC interacts with citrate in vitro, the protein releases from the −27 half-dyad element, but remains bound to the −66 site (Jourlin-Castelli et al., 2000). To elucidate the mechanism by which CcpC activates citB in B. subtilis and to provide clues for the potential mechanism in L. monocytogenes, we utilized a citB promoter fusion lacking the upstream arm of the −66 dyad symmetry element (Fouet & Sonenshein, 1990). This fusion, citBp24–lacZ, and a fusion that contains the intact −66 site, citBp23–lacZ, were independently introduced into B. subtilis wild-type and citB null mutant strains, and expression was monitored during growth. In the presence of the intact −66 site, the hyperexpression of citBp was seen in a citB null strain as expected (Fig. 4). However, the loss of the upstream arm of the dyad resulted in loss of the hyperexpression phenotype; activity levels of the citBp24–lacZ fusion in the citB null strain were similar to those observed in the wild-type strain. This result indicates that the intact −66 dyad symmetry element is required for CcpC-dependent activation of B. subtilis citB, implying a direct role for CcpC in such hyperexpression.
Fig. 4.
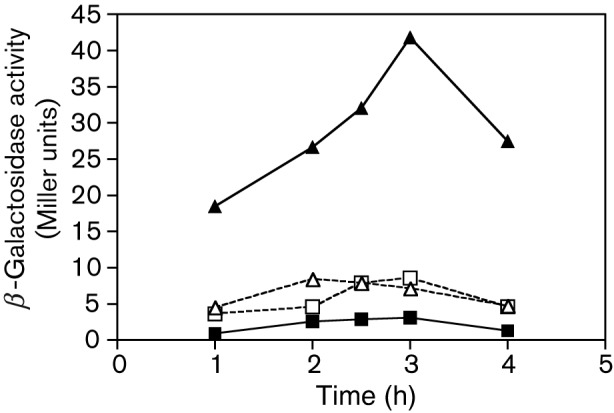
The −66 dyad symmetry element is necessary for CcpC-dependent hyperexpression of citB–lacZ in B. subtilis. β-Galactosidase activity was measured at various times during growth of B. subtilis cells carrying two different citB′–lacZ fusions: one containing the intact −66 dyad symmetry element on the citB promoter (citBp23–lacZ) and the other missing the upstream arm of the dyad (citBp24–lacZ) (Fouet & Sonenshein, 1990). Expression of the two fusions was compared in citB+ and citB null cells: for citBp23–lacZ, strains KBP56 (citB+; ▪) and KBP62 (citB null; ▴); for citBp24–lacZ, strains KBP57 (citB+; □) and KBP63 (citB null; ▵). The end of exponential growth phase was reached between 3 and 4 h of growth for all strains.
Competition with CodY is not the basis for positive regulation of citB by CcpC
In previous work, we have shown that CodY acts as a negative regulator of citB and does so by binding to a site that overlaps with the CcpC binding site (Kim et al., 2003a). Therefore, a possible mechanism by which CcpC might act as a positive regulator would be by blocking the binding of CodY. However, the introduction of a codY mutation into the various B. subtilis strains carrying the citB–lacZ fusion had only a small effect, if any, on expression (Fig. 3b). Most tellingly, inactivation of codY did not alter the phenotype of the citB ccpC double mutant, indicating that the ccpC mutation suppresses the citB hyperexpression phenotype without causing greater repression by CodY.
Discussion
We report here evidence that CcpC acts as both a negative and a positive regulator of the citB promoter in the related bacteria B. subtilis and L. monocytogenes. In both organisms, the high citrate levels found in citB null mutants result in CcpC-dependent activation of a citB–lacZ reporter gene. Further exploration in B. subtilis revealed that this activation is independent of CodY. In addition, in B. subtilis (and presumably in L. monocytogenes) this activation is dependent on the CcpC binding site centred at position −66 with respect to the citB transcriptional start point. The evidence of CcpC-dependent activation presented here was generated using a citB null mutant strain, and thus we were unable to assess the contribution of CcpC activation of the citB promoter to aconitase protein levels. However, recent results shed light on this issue. Two B. subtilis citB point mutants (citB2, citB7) that result in increased citrate levels lead to the CcpC-dependent activation of the citB promoter; this effect is accompanied by overaccumulation of aconitase protein (K. B. Pechter and others, unpublished data).
Combining these new data with previously published work, we can propose a model for CcpC as a complex, citrate-responsive regulator of citB (Fig. 5). When the citrate pool is low, CcpC dimers are unliganded and they are able to interact with both the full −66 dyad symmetry element and the −27 half-dyad element to repress citB transcription (Fig. 2b) (Kim et al., 2006, 2003b). As citrate levels rise, citrate binds to the CcpC dimer and alters its conformation, leading to release from the −27 site. This is supported by in vitro data; when citrate is present at 7–17 mM (0.2–0.5 % Na2-citrate . 2H2O), equivalent to the high concentration of citrate in citB mutant cells, CcpC binding to the −27 site is reduced and a new hypersensitive band is created at the upstream end of the element, suggesting an alteration in the bending of the DNA (Kim et al., 2003b). However, binding to the −66 dyad symmetry element is not affected by citrate, and due to the interaction between the two dimers, they remain tethered to the −66 site as a tetramer. This orientation is likely to permit interaction of CcpC with RNA polymerase to increase the efficiency of transcription from the citB promoter.
Fig. 5.

An updated model of CcpC binding to the citB promoter. CcpC binds to the citB promoter and acts as a repressor or an activator in response to citrate. In the presence of no or very low levels of citrate, CcpC (grey circles) binds as a dimer to both the −66 and −27 binding sites, blocking access of RNA polymerase to the promoter and resulting in repression of citB expression. In the case of a ccpC null mutant, derepression of citB occurs and a low level of transcript is made. In the presence of high levels of citrate (as in a citB null mutant), binding to the −27 site is lost; however, the CcpC complex remains bound at the −66 site. We hypothesize that a direct interaction between CcpC and RNA polymerase allows CcpC to activate citB gene expression, resulting in the production of high levels of transcript.
Mechanistically, there are two possibilities for how citrate converts CcpC to an activator. First, binding of the citrate ligand to a single site on CcpC could convert CcpC from a repressing to an activating conformation, as depicted in Fig. 5. This would result in an on/off mechanism of regulation, and in this case the moderate level of induction of citB expression seen in wild-type cells in the late-exponential growth phase would be the consequence of modest accumulation of citrate and conversion of only a fraction of the CcpC molecules to the activating conformation. In addition, the level of citB expression seen in a ccpC null mutant would reflect the intrinsic activity of the citB promoter in the absence of both repression and activation. (The citB promoters of B. subtilis and L. monocytogenes have only 8/12 and 9/12 matches, respectively, with the −10 and −35 consensus sequences.) The second possibility is that CcpC has two binding sites for citrate with different affinities and that CcpC interacts with the citB promoter in three ways: repression, derepression and activation. CcpC bound in the unliganded state would repress citB, as described above. When citrate accumulates to a moderate level, it would bind to a relatively high affinity site on CcpC, changing the CcpC conformation to cause release from the −27 half-dyad element along with derepression of citB. When citrate accumulates to a very high level, as in a citB null strain or in conditions when aconitase is inactive (see below), citrate would bind to both the high affinity site and a second, lower affinity site on CcpC, converting the protein to a transcriptional activator and causing citB to be hyperexpressed. The effect of citrate on CcpC binding to the citB promoters of B. subtilis and L. monocytogenes in vitro is consistent with both models. It is important to note, however, that it is also possible that CcpC influences citB transcription indirectly by regulating the synthesis of another factor.
We also note that hyperactivation of B. subtilis citB–lacZ expression in a citB null mutant seems to overcome repression by CodY. In fact, introduction of a codY mutation does not increase citB–lacZ expression substantially in a citB null mutant. One interpretation of these results is that binding of CcpC in its activating conformation interferes with CodY binding. This would not be surprising inasmuch as the two proteins bind to overlapping sequences in the citB promoter region (Kim et al., 2003a). However, if this interpretation were correct, the level of β-galactosidase activity in the citB ccpC codY triple mutant would be considerably higher than in the citB ccpC double mutant. It is not, indicating that the contribution of CodY to citB regulation under the conditions tested is relatively small.
CcpC is not the only LTTR family member capable of switching between multiple regulatory modes. The B. subtilis GltC protein has two metabolite ligands and each induces opposing effects; GltC activates the gltAB locus when bound to 2-oxoglutarate and represses gltAB when complexed with glutamate (Picossi et al., 2007). The unliganded and ligand-bound forms of GltC bind differentially to three sites within the gltAB promoter region to effect this regulatory switch. In addition, the glutamate dehydrogenase enzyme, RocG, also acts as a negative regulator of gltAB transcription, apparently by interacting with GltC (Gunka et al., 2010). In other cases, LTTR family proteins (such as NahR from Pseudomonas putida and TrpI from P. aeruginosa) bind to a primary site in the absence of effector, but, in the presence of effector, binding is extended to a second site and transcription is activated (Chang & Crawford, 1990; Huang & Schell, 1991). In yet another group of LTTR proteins (Toledano et al., 1994; van Keulen et al., 2003; Wang & Winans, 1995), the presence of the inducer shifts the protein from one binding site on the promoter to another, causing activation. In the case of the CbbR regulatory protein from Xanthobacter flavus, which controls the expression of Calvin cycle genes, three regulatory sites are found at the cbb promoter, two of which (IR2 and IR3) are overlapping. CbbR binds to IR1 and IR3 in the absence of inducer, but, in the presence of the inducer, NADPH, CbbR shifts from IR3 to IR2, activating expression of the cbb operon (van Keulen et al., 2003).
From a physiological point of view, the dual regulatory role of CcpC at the citB promoter creates an efficient system to control the intracellular abundance of citrate. Importantly, the action of CcpC as an activator is specific to aconitase; in other work we have shown that CcpC acts solely as a repressor of the citZ operon (K. B. Pechter and others, unpublished data). As is the case for the E. coli lac operon, which is induced by allolactose (Meiss et al., 1969), and the Salmonella typhimurium hut operon, which is induced by urocanate (Jobe & Bourgeois, 1972), expression of the TCA branch of the Krebs cycle is induced by the product of the first enzyme of the pathway. When citrate synthase substrates acetyl-CoA and oxaloacetate are present, any leaky citZ expression, despite repression by CcpC, will allow the accumulation of a small amount of citrate that will alleviate some CcpC repression and increase the levels of citrate synthase. If citrate levels rise faster than they can be reduced by aconitase, as would occur in cells stressed by iron limitation or exposure to reactive oxygen species, CcpC will convert to an activator, increasing aconitase expression dramatically. Moreover, the relief of autorepression of the ccpC gene as citrate levels increase ensures that the concentration of CcpC will also increase (Kim et al., 2002a). This mechanism not only allows the cell to dissipate internally produced citrate and to prevent the hyperaccumulation of citrate acquired from the environment but also endows the cell with a high concentration of CcpC that is available to reimpose repression when the citrate pool decreases. Citrate is not only a key metabolic intermediate but also a carrier for iron uptake and a chelator of iron and other cations. To balance these roles, the cell needs a robust mechanism to avoid excessive intracellular citrate accumulation. While other bacterial species can metabolize citrate via citrate lyase, aconitase is the only citrate-metabolizing enzyme in B. subtilis and L. monocytogenes; thus, its activity is critical to maintaining this fine balance. In addition, it is noteworthy that the metabolism of citrate to 2-oxoglutarate by the combined activities of aconitase and isocitrate dehydrogenase leads to conversion of NADP+ to NADPH; high-level citrate metabolism would potentially exhaust the NADP+ pool. Such an outcome would be deleterious to the cell, but could be attenuated by rapid conversion of 2-oxoglutarate and NADPH (the products of the isocitrate dehydrogenase reaction) to glutamate and NADP+ by glutamate synthase.
Acknowledgements
We thank Boris Belitsky (Tufts University, Boston, MA, USA) for many helpful discussions and criticism of the manuscript, Jane Craig King (University of Manchester, UK), who was the first to observe hyperexpression of B. subtilis citB in a citB null mutant, for sharing her unpublished results, and Pascale Serror (INRA, Jouy en Josas, France) for making strain PS258 and plasmid pPS34 available. Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award numbers R01 GM036718 and R01 GM042219. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations:
- TCA
tricarboxylic acid
References
- Behari J., Youngman P. (1998). A homolog of CcpA mediates catabolite control in Listeria monocytogenes but not carbon source regulation of virulence genes. J Bacteriol 180, 6316–6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belitsky B. R., Janssen P. J., Sonenshein A. L. (1995). Sites required for GltC-dependent regulation of Bacillus subtilis glutamate synthase expression. J Bacteriol 177, 5686–5695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blencke H.-M., Reif I., Commichau F. M., Detsch C., Wacker I., Ludwig H., Stülke J. (2006). Regulation of citB expression in Bacillus subtilis: integration of multiple metabolic signals in the citrate pool and by the general nitrogen regulatory system. Arch Microbiol 185, 136–146. 10.1007/s00203-005-0078-0 [DOI] [PubMed] [Google Scholar]
- Brehm S. P., Staal S. P., Hoch J. A. (1973). Phenotypes of pleiotropic-negative sporulation mutants of Bacillus subtilis. J Bacteriol 115, 1063–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M., Crawford I. P. (1990). The roles of indoleglycerol phosphate and the TrpI protein in the expression of trpBA from Pseudomonas aeruginosa. Nucleic Acids Res 18, 979–988. 10.1093/nar/18.4.979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig J. E., Ford M. J., Blaydon D. C., Sonenshein A. L. (1997). A null mutation in the Bacillus subtilis aconitase gene causes a block in Spo0A-phosphate-dependent gene expression. J Bacteriol 179, 7351–7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean D. R., Hoch J. A., Aronson A. I. (1977). Alteration of the Bacillus subtilis glutamine synthetase results in overproduction of the enzyme. J Bacteriol 131, 981–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingman D. W., Sonenshein A. L. (1987). Purification of aconitase from Bacillus subtilis and correlation of its N-terminal amino acid sequence with the sequence of the citB gene. J Bacteriol 169, 3062–3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliss I., Emond E., Simard R. E., Pandian S. (1991). A rapid and efficient method of lysis of Listeria and other Gram-positive bacteria using mutanolysin. Biotechniques 11, 453–, 456–457.. [PubMed] [Google Scholar]
- Fouet A., Sonenshein A. L. (1990). A target for carbon source-dependent negative regulation of the citB promoter of Bacillus subtilis. J Bacteriol 172, 835–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouet A., Jin S. F., Raffel G., Sonenshein A. L. (1990). Multiple regulatory sites in the Bacillus subtilis citB promoter region. J Bacteriol 172, 5408–5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunka K., Newman J. A., Commichau F. M., Herzberg C., Rodrigues C., Hewitt L., Lewis R. J., Stülke J. (2010). Functional dissection of a trigger enzyme: mutations of the Bacillus subtilis glutamate dehydrogenase RocG that affect differentially its catalytic activity and regulatory properties. J Mol Biol 400, 815–827. 10.1016/j.jmb.2010.05.055 [DOI] [PubMed] [Google Scholar]
- Handke L. D., Shivers R. P., Sonenshein A. L. (2008). Interaction of Bacillus subtilis CodY with GTP. J Bacteriol 190, 798–806. 10.1128/JB.01115-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J. Z., Schell M. A. (1991). In vivo interactions of the NahR transcriptional activator with its target sequences. Inducer-mediated changes resulting in transcription activation. J Biol Chem 266, 10830–10838. [PubMed] [Google Scholar]
- Jault J. M., Fieulaine S., Nessler S., Gonzalo P., Di Pietro A., Deutscher J., Galinier A. (2000). The HPr kinase from Bacillus subtilis is a homo-oligomeric enzyme which exhibits strong positive cooperativity for nucleotide and fructose 1,6-bisphosphate binding. J Biol Chem 275, 1773–1780. 10.1074/jbc.275.3.1773 [DOI] [PubMed] [Google Scholar]
- Jobe A., Bourgeois S. (1972). lac Repressor–operator interaction. VI. The natural inducer of the lac operon. J Mol Biol 69, 397–408. 10.1016/0022-2836(72)90253-7 [DOI] [PubMed] [Google Scholar]
- Jourlin-Castelli C., Mani N., Nakano M. M., Sonenshein A. L. (2000). CcpC, a novel regulator of the LysR family required for glucose repression of the citB gene in Bacillus subtilis. J Mol Biol 295, 865–878. 10.1006/jmbi.1999.3420 [DOI] [PubMed] [Google Scholar]
- Kim H. J., Jourlin-Castelli C., Kim S. I., Sonenshein A. L. (2002a). Regulation of the bacillus subtilis ccpC gene by ccpA and ccpC. Mol Microbiol 43, 399–410. 10.1046/j.1365-2958.2002.02751.x [DOI] [PubMed] [Google Scholar]
- Kim H. J., Roux A., Sonenshein A. L. (2002b). Direct and indirect roles of CcpA in regulation of Bacillus subtilis Krebs cycle genes. Mol Microbiol 45, 179–190. 10.1046/j.1365-2958.2002.03003.x [DOI] [PubMed] [Google Scholar]
- Kim H. J., Kim S. I., Ratnayake-Lecamwasam M., Tachikawa K., Sonenshein A. L., Strauch M. (2003a). Complex regulation of the Bacillus subtilis aconitase gene. J Bacteriol 185, 1672–1680. 10.1128/JB.185.5.1672-1680.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. I., Jourlin-Castelli C., Wellington S. R., Sonenshein A. L. (2003b). Mechanism of repression by Bacillus subtilis CcpC, a LysR family regulator. J Mol Biol 334, 609–624. 10.1016/j.jmb.2003.09.078 [DOI] [PubMed] [Google Scholar]
- Kim H. J., Mittal M., Sonenshein A. L. (2006). CcpC-dependent regulation of citB and lmo0847 in Listeria monocytogenes. J Bacteriol 188, 179–190. 10.1128/JB.188.1.179-190.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddocks S. E., Oyston P. C. (2008). Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology 154, 3609–3623. 10.1099/mic.0.2008/022772-0 [DOI] [PubMed] [Google Scholar]
- Meiss H. K., Brill W. J., Magasanik B. (1969). Genetic control of histidine degradation in Salmonella typhimurium, strain LT-2. J Biol Chem 244, 5382–5391. [PubMed] [Google Scholar]
- Miller J. (1972). Experiments in Molecular Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory. [Google Scholar]
- Mittal M. (2008). The roles of CcpC and CodY in regulation of Krebs cycle genes and virulence of Listeria monocytogenes. PhD thesis, Tufts University, Boston, MA. [Google Scholar]
- Mittal M., Picossi S., Sonenshein A. L. (2009). CcpC-dependent regulation of citrate synthase gene expression in Listeria monocytogenes. J Bacteriol 191, 862–872. 10.1128/JB.01384-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picossi S., Belitsky B. R., Sonenshein A. L. (2007). Molecular mechanism of the regulation of Bacillus subtilis gltAB expression by GltC. J Mol Biol 365, 1298–1313. 10.1016/j.jmb.2006.10.100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serio A. W., Pechter K. B., Sonenshein A. L. (2006). Bacillus subtilis aconitase is required for efficient late-sporulation gene expression. J Bacteriol 188, 6396–6405. 10.1128/JB.00249-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenshein A. L. (2007). Control of key metabolic intersections in Bacillus subtilis. Nat Rev Microbiol 5, 917–927. 10.1038/nrmicro1772 [DOI] [PubMed] [Google Scholar]
- Toledano M. B., Kullik I., Trinh F., Baird P. T., Schneider T. D., Storz G. (1994). Redox-dependent shift of OxyR–DNA contacts along an extended DNA-binding site: a mechanism for differential promoter selection. Cell 78, 897–909. 10.1016/S0092-8674(94)90702-1 [DOI] [PubMed] [Google Scholar]
- van Keulen G., Ridder A. N., Dijkhuizen L., Meijer W. G. (2003). Analysis of DNA binding and transcriptional activation by the LysR-type transcriptional regulator CbbR of Xanthobacter flavus. J Bacteriol 185, 1245–1252. 10.1128/JB.185.4.1245-1252.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villapakkam A. C., Handke L. D., Belitsky B. R., Levdikov V. M., Wilkinson A. J., Sonenshein A. L. (2009). Genetic and biochemical analysis of the interaction of Bacillus subtilis CodY with branched-chain amino acids. J Bacteriol 191, 6865–6876. 10.1128/JB.00818-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Winans S. C. (1995). The sixty nucleotide OccR operator contains a subsite essential and sufficient for OccR binding and a second subsite required for ligand-responsive DNA bending. J Mol Biol 253, 691–702. 10.1006/jmbi.1995.0583 [DOI] [PubMed] [Google Scholar]