

Abstract
Polycystic liver diseases (PCLDs) are a heterogeneous group of genetic disorders characterized by the development of multiple fluid-filled cysts in the liver, which derive from cholangiocytes, the epithelial cells lining the bile ducts. When these cysts grow, symptoms such as abdominal distension, nausea, and abdominal pain may occur. PCLDs may exist isolated (i.e., autosomal dominant polycystic liver disease, ADPLD) or in combination with renal cystogenesis (i.e., autosomal dominant polycystic kidney disease and autosomal recessive polycystic liver disease). The exact prevalence of PCLDs is unknown, but is estimated to occur in approximately 1:1000 persons. Although the pathogenesis of each form of PCLD appears to be different, increasing evidences indicate that hepatic cystogenesis is a phenomenon that may involve somatic loss of heterozygosity (LOH) in those pathological conditions inherited in a dominant form. A recent report, using highly sophisticated methodology, demonstrated that ADPLD patients with a germline mutation in the protein kinase C substrate 80K-H (PRKCSH) gene mostly develop hepatic cystogenesis through a second somatic mutation. While hepatocystin, the PRKCSH-encoding protein, was absent in the hepatic cysts with LOH, it was still expressed in the heterozygous cysts. On the other hand, no additional trans-heterozygous mutations on the SEC63 homolog (S. cerevisiae/SEC63) gene (also involved in the development of PCLDs) were observed. These data indicate that PCLD is recessive at the cellular level, and point out the important role of hepatocystin loss in cystogenesis. In this commentary, we discuss the knowledge regarding the role of somatic second-hit mutations in the development of PCLDs, and the most relevant findings have been highlighted.
Keywords: Polycystic liver diseases, Cholangiocyte, Cystogenesis, Loss of heterozygosity, Protein kinase C substrate 80K-H, SEC63
TO THE EDITOR
We have read with great interest the recently publication by Janssen et al[1] in Gastroenterology. The manuscript nicely demonstrate that cystogenesis in polycystic liver diseases is mostly associated with a loss of heterozygosity (LOH) in the biliary epithelial cells, termed cholangiocytes[2]. In this study, the group of professor Drenth employed blood and liver cystic tissue from 8 female ADPLD patients, who had a heterozygous germline mutation in the protein kinase C substrate 80K-H (PRKCSH) gene (5 patients with a c.1341-2A>G mutation and 3 with a c.292+1G>C mutation) resulting in premature stop codons. Moreover, no additional germline mutations in SEC63 were detected in any of these patients. By using laser microdissection of hepatic cysts, they found that 76% of the liver cysts presented a secondary somatic mutation in the normal PRKCSH allele (i.e., LOH). In contrast, the surrounding liver tissue and blood of the patients showed heterozygosity for this gene (Figure 1). Additionally, no detectable somatic changes in the SEC63 homolog-(S. cerevisiae/SEC63) gene sequence were found in cystic epithelium compared to non-epithelial cells or blood DNA, excluding trans-heterozygous SEC63 somatic mutations as a cause of cyst formation in cysts without somatic PRKCSH mutations.
Figure 1.
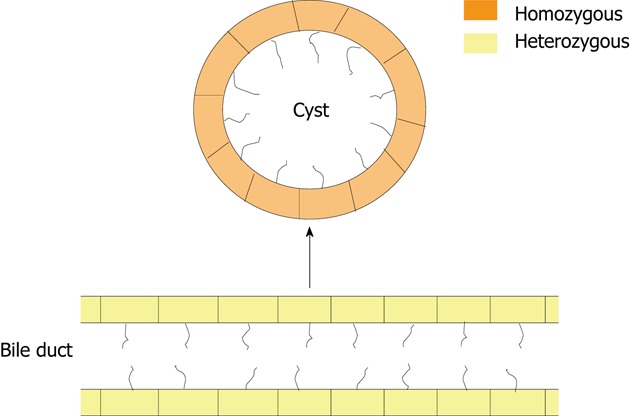
Chart representing the second somatic mutations occurring in the majority of cysts from autosomal dominant polycystic liver disease patients. Most of autosomal dominant polycystic liver disease cysts with a protein kinase C substrate 80K-H germline mutation develop over time a second somatic mutations in the same gene that result in the loss of heterozygosity and cyst formation.
The authors also indicated that hepatic cysts presented different somatic mutations extended through out the 70-kilo-base region in the majority (97%) of the cysts. These data support the concept that cyst formation results from a specific genetic event in a subset of cells. Although all known mutations in ADPLD are heterozygous, the fact that ADPLD bile ducts express hepatocystin and ADPLD cystic cholangiocytes do not express this protein via a 2-hit somatic genomic mutation support the hypothesis that this disease in recessive on a cellular level. Interestingly, sequencing of full PRKCSH gene in those 12 cysts without LOH showed that 2 of them, despite expressing hepatocystin, developed a somatic missense mutation. Therefore, the other cysts without LOH in the PRKCSH gene may represent all the other mutations in other genes that may lead to cyst formation.
We agree with the authors that it is important to remark that although somatic 2-hit mutations in PRKCSH may develop anywhere in the body, cyst formation is only seen in the liver. This observation indicates that the liver-specific phenotype of ADPLD patients is a consequence of a more sensitive response of cholangiocytes to the lack of hepatocystin. Because liver cysts display cholangiocyte features and no malignant transformation, the possibility that a somatic 2-hit mutation originating from a liver progenitor cell would explain the cyst development and the hyperproliferative properties of the cysts. There have been reports on the presence of 2-hit mechanisms in other non-malignant diseases including autosomal dominant polycystic kidney disease (ADPKD), where a somatic LOH was found in a proportion of both kidney and liver cysts[3,4].
Polycystic liver diseases (PCLDs) are genetic disorders characterized by the development of fluid-filled cysts in the liver derived from cholangiocytes[5-7]. The exact prevalence of PCLDs is unknown, but is estimated to occur in approximately 1:1000 persons. The most common symptoms are caused by enlargement of the liver, and include abdominal distension, dyspepsia, abdominal pain, and early satiety. Although not found frequently, serious complications such as portal hypertension and haemorrhage, or infections of cysts, may also occur[5-7]. The liver phenotype may exist isolated (i.e., ADPLD) or in combination with renal cystogenesis (i.e., autosomal dominant polycystic liver disease ADPLD, and autosomal recessive polycystic liver disease, ARPKD)[5-7]. ADPLD is a rare disease that can be caused by mutations in PRKCSH or SEC63 genes, which encode for the endoplasmic reticulum (ER)-associated proteins hepatocystin and Sec63, involved in glycosylation and transport of glycoproteins into and out of the ER, respectively[5]. On the other hand, the genes responsible for the development of ADPKD (1:1000 prevalence) are PKD1 and PKD2, which encode for polycystin-1 and polycystin-2, respectively[6,7]. Polycystin-1 is a mechanoreceptor involved in calcium signalling, and polycystin-2 a non-selective calcium channel. Finally, ARPKD (1:20.000 prevalence) is caused by mutations in the PKHD1 gene, which encodes for fibrocystin, a protein with unknown function[6,7]. The pathogenesis of each form of PCLD appears to be different. However, alterations in different mechanisms such as proliferation, fluid secretion, and cell-matrix interactions seem to play an important role in all of them. Moreover, these altered events seem to be also associated with changes in the microRNA expression pattern, with increased levels of cyclic adenosine monophosphate levels, and with downregulation of the intracellular calcium levels[8,9]. Currently, there is no standard treatment for PCLDs. Pharmacological approaches include somatostatin analogues and mammalian target of rapamycin-inhibitors, but most clinical trials have only shown a small reduction of liver volume. On the other hand, the most frequently used surgical procedures are aspiration and sclerotherapy, fenestration, segmental hepatic resection, and liver transplantation, which have a better short term effect, but high recurrence and complication rate preclude wide spread use[5].
In summary, this article represents a milestone in the understanding of the molecular mechanisms involved in liver cystogenesis and leads to several questions such as the unknown mechanisms causing LOH in PCLDs, as well as the role of 2-hit somatic mutations in all forms of PCLDs, including ADPLD patients with a germline mutation in SEC63.
Footnotes
P- Reviewer Levesque H S- Editor Jia F L- Editor A E- Editor Xiong L
References
- 1.Janssen MJ, Waanders E, Te Morsche RH, Xing R, Dijkman HB, Woudenberg J, Drenth JP. Secondary, somatic mutations might promote cyst formation in patients with autosomal dominant polycystic liver disease. Gastroenterology. 2011;141:2056–2063.e2. doi: 10.1053/j.gastro.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 2.Banales JM, Prieto J, Medina JF. Cholangiocyte anion exchange and biliary bicarbonate excretion. World J Gastroenterol. 2006;12:3496–3511. doi: 10.3748/wjg.v12.i22.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qian F, Germino GG. “Mistakes happen”: somatic mutation and disease. Am J Hum Genet. 1997;61:1000–1005. doi: 10.1086/301618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qian F, Watnick TJ, Onuchic LF, Germino GG. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell. 1996;87:979–987. doi: 10.1016/s0092-8674(00)81793-6. [DOI] [PubMed] [Google Scholar]
- 5.Drenth JP, Chrispijn M, Nagorney DM, Kamath PS, Torres VE. Medical and surgical treatment options for polycystic liver disease. Hepatology. 2010;52:2223–2230. doi: 10.1002/hep.24036. [DOI] [PubMed] [Google Scholar]
- 6.Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369:1287–1301. doi: 10.1016/S0140-6736(07)60601-1. [DOI] [PubMed] [Google Scholar]
- 7.Strazzabosco M, Somlo S. Polycystic liver diseases: congenital disorders of cholangiocyte signaling. Gastroenterology. 2011;140:1855–189, 1859.e1. doi: 10.1053/j.gastro.2011.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banales JM, Masyuk TV, Gradilone SA, Masyuk AI, Medina JF, LaRusso NF. The cAMP effectors Epac and protein kinase a (PKA) are involved in the hepatic cystogenesis of an animal model of autosomal recessive polycystic kidney disease (ARPKD) Hepatology. 2009;49:160–174. doi: 10.1002/hep.22636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gradilone SA, Masyuk TV, Huang BQ, Banales JM, Lehmann GL, Radtke BN, Stroope A, Masyuk AI, Splinter PL, LaRusso NF. Activation of Trpv4 reduces the hyperproliferative phenotype of cystic cholangiocytes from an animal model of ARPKD. Gastroenterology. 2010;139:304–14.e2. doi: 10.1053/j.gastro.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]