

Abstract
A single exercise bout stimulates skeletal muscle glucose transport (GT) in the absence or presence of insulin. It has been suggested that the Kallikrein-Kinin System (KKS) may contribute to exercise effects on both insulin-independent and insulin-dependent glucose transport. Plasma kininogen, a key KKS component, is a protein substrate for the enzyme kallikrein and the source of the peptide bradykinin.
Purpose
To determine if the post-exercise (PEX) increase in insulin-dependent or insulin-independent GT is reduced in rats deficient in plasma kininogen vs. normal rats.
Methods
Male Brown Norway (BN) and Brown Norway Katholiek (BNK; plasma kininogen deficient) rats were studied. BN and BNK rats were assigned to exercise (4 × 30 minute swim) or sedentary (SED) groups. Rats were anesthetized immediately (0hPEX) or 3 hours (3hPEX) after exercise. For 0hPEX and 0hSED rats, one epitrochlearis muscle per rat was used for AMPK phosphorylation and muscle glycogen analyses. The contralateral muscle was incubated with [3H]-3-O-methylglucose (3MG) for GT assay. For 3hPEX and 3hSED rats, one muscle from each rat was incubated without insulin, and the contralateral muscle was incubated with 60μU/ml insulin and both muscles were incubated with 3MG for GT measurement.
Results
For 0hPEX vs. 0hSED, both BN and BNK rats had greater insulin-independent GT and AMPK phosphorylation with reduced glycogen post-exercise. No genotype effects were found 0hPEX. There was a significant main effect of exercise (3hPEX > 3hSED) and no interaction between exercise and genotype for basal or insulin-stimulated GT.
Conclusion
Plasma kininogen deficiency did not alter insulin-independent GT, AMPK phosphorylation or glycogen depletion 0hPEX or insulin-dependent GT 3hPEX suggesting that normal plasma kininogen is not essential for these important exercise effects.
Keywords: insulin sensitivity, insulin resistance, bradykinin, physical activity
INTRODUCTION
Skeletal muscle glucose transport is stimulated both by insulin and exercise. A single exercise session can stimulate glucose transport in the absence of insulin (insulin-independent), and it can also increase insulin’s ability to increase glucose transport (insulin-dependent) (7,8,10). Most of the insulin independent effect is lost ~3 hours post-exercise in rat muscle, at which time glucose transport in the presence of physiological insulin concentration is elevated, i.e., insulin sensitivity is increased (7,8).
The mechanisms leading to the increased insulin-independent immediately after exercise and increased insulin-dependent glucose transport several hours after exercise are not fully understood. Several lines of evidence have led to the proposal that the Kallikrein-Kinin System (KKS) might play a role in the effects of exercise on skeletal muscle glucose transport (13,24,38). Exercise or skeletal muscle contraction can activate the KKS as evidenced by increased circulating levels of bradykinin, a nonapeptide that is produced as the result of the interaction between two plasma proteins (kallikrein and kininogen) which are essential elements of the KKS (3,26,37). Exercise has been shown to increase the activity of kallikrein (39), the enzyme which catalyzes the proteolytic cleavage of bradykinin from plasma kininogen (2). Bradykinin binding to the B2 receptor of bradykinin has been implicated as a mediator of increased glucose transport and improved insulin sensitivity (9,14,19–21,25,29).
Gao et al. (17) demonstrated that electrically stimulated contraction by isolated rat epitrochlearis muscle in the presence of serum, but not in the absence of serum, can lead to a subsequent improvement in insulin sensitivity. They also provided convincing evidence that the enhanced insulin-stimulated glucose transport in this model requires the presence of a serum protein with a molecular mass greater than 10 kDa. The molecular masses of kallikrein and kininogen are greater than 10 kDa, raising the possibility that KKS proteins might be required for the post-contraction increase in glucose transport. Dumke et al. (15) found that muscles stimulated to contract in kallikrein-deficient plasma compared to muscles that performed contractions in normal plasma had reduced insulin-stimulated glucose transport. Although electrically stimulated contraction is a valuable model, it is not a perfect replica of in vivo exercise. Accordingly, we used in vivo exercise for the current study.
Brown Norway Katholiek (BNK) rats have a spontaneous point mutation in the kininogen gene which causes circulating kininogen deficiency by interfering with secretion of the liver synthesized kininogen (12). Plasma kininogen values of BNK rats have been reported to be less than 1% of the normal plasma values for Brown Norway (BN) rats (32). Urinary kinin release is readily measured in normal rats and barely detectable in BNK rats (27,28,30). BNK rats have also been reported to have moderate in vivo insulin resistance and fasting normoglycemia compared to BN rats (12). The primary hypothesis tested by this study was that BNK compared to BN rats are characterized by a reduction in post-exercise insulin-stimulated glucose transport. It has also been suggested that activation of the KKS may play a role in the exercise-induced increase in insulin-independent glucose transport (24,38). Therefore, a secondary aim was to compare the post-exercise increase in insulin-independent glucose transport in skeletal muscles from BN and BNK rats.
METHODS
Materials
Human recombinant insulin was obtained from Eli Lilly (Indianapolis, IN). Reagents and apparatus for SDS-PAGE and immunoblotting were purchased from Bio-Rad (Hercules, CA). Bicinchoninic acid protein assay reagent (no. 23227), T-PER tissue protein extraction reagent (no. 78510) and West Dura Extended Duration Substrate (no. 34075) were purchased from Pierce Biotechnology (Rockford, IL). Goat anti-rabbit IgG horseradish peroxidase conjugate (no. 7074) and Anti-phospho-Thr172 AMPKα (pAMPK-Thr172; no. 2531) were purchased from Cell Signaling Technology (Danvers, MA). 3-O-methyl[3H]glucose ([3H]3-MG) was purchased from Sigma-Aldrich (St. Louis, MO). [14C]Mannitol was from PerkinElmer (Waltham, MA). Other reagents were purchased from Sigma-Aldrich and Fisher Scientific (Pittsburgh, PA).
Animal treatment
Procedures for animal care were approved by the University of Michigan Committee on Use and Care of Animals, and the experiments were performed under adherence to American College of Sports Medicine animal care standards. Male Brown Norway (BN) rats (7–10 wk-old; 160 ± 3g; Charles River Laboratories, Wilmington, MA) and male Brown Norway Katholiek (BNK) rats (7–10 wk-old; 168 ± 8g; generously provided by Dr. Oscar Carretero) had ad libitum access to rodent chow (PMI Nutritional International, Brentwood, MO) and water until 1700 on the night before the experiments, when food was removed from their cages, and all of the rats remained fasted for the remainder of the study. On the following day, rats were randomly assigned to a post-exercise (PEX) or sedentary (SED) treatment. Beginning at ~0900, PEX rats swam in a barrel filled with water (35°C) to a depth of ~60 cm (7 or 8 rats/barrel) for 4 × 30 minute bouts, with a 5 minute rest period between each bout.
Experimental Design
PEX rats of each genotype (BN and BNK) were studied either immediately after exercise (0hPEX; n=7 for BN and n=7 for BNK) or 3 hours after exercise (3hPEX; n=16 for BN and n=15 for BNK). Sedentary rats remained in their cages while the PEX rats were swimming and were studied at times that matched either the 0hPEX group (0hSED; n=8 for BN and n=8 for BNK) or the 3hPEX group (3hSED; n=15 for BN and n=15 for BNK).
Muscle incubations
Rats were anesthetized with an intraperitoneal injection of pentobarbital sodium (5mg/100 g body wt) either immediately post-exercise (0hPEX) or 3–4 hours post-exercise (3hPEX) along with time-matched sedentary rats. For the 0hPEX experiment, following anesthetization, one epitrochlearis muscle from each rat was rapidly dissected out, trimmed, freeze-clamped using aluminum clamps cooled to the temperature of liquid N2, and stored at −80°C until analyzed. After dissection, the contralateral muscle in the 0hPEX study was incubated for 10 minutes in flasks containing Krebs-Henseleit Buffer (KHB) with 0.1% bovine serum albumin (BSA), 2 mM pyruvate, and 6 mM mannitol for 10 minutes. The muscles were then transferred to flasks containing KHB, 0.1% BSA, 8 mM 3-MG (including 0.25 mCi/mmol [3H]3-MG), and 2 mM mannitol (including 0.1 mCi/mmol [14C]mannitol). For both incubation steps, flasks were continuously gassed from above with 95% O2-5% CO2 and shaken in a heated water bath (30°C).
For the 3hPEX experiment, rats were dried following the final exercise bout and returned to their cage for 3 hours before being anesthetized. After anesthetization, both epitrochlearis muscles from each animal were dissected out. One muscle from each rat was incubated in KHB, 0.1% BSA, 8 mM glucose, 2 mM mannitol supplemented with 60μU/ml insulin (a physiological concentration), and the contralateral muscle was incubated in the same solution without insulin. Both muscles were incubated for 30 minutes in a shaking water bath at 35°C. Insulin remained present at the same concentration throughout all subsequent incubations. After the initial incubation, muscles were transferred to vials containing KHB, 0.1% BSA, 2 mM pyruvate, and 6mM mannitol with or without 60μU/ml insulin at 30°C for 10 minutes. Finally, muscles were transferred to flasks containing KHB, 0.1% BSA with 8 mM 3-MG (including 0.25 mCi/mmol [3H]3-MG), and 2 mM mannitol (including 0.1 mCi/mmol [14C]mannitol) with or without 60μU/ml insulin for determination of glucose transport rate. After incubation with 3-MG for 15 minutes, the muscles were rapidly blotted on filter paper dampened with incubation medium, trimmed, freeze-clamped, and stored at −80°C.
Homogenization, glucose transport measurement, and protein concentration determination
Frozen muscles used for glucose transport and immunoblotting (pAMPK-Thr172) were homogenized in 1 ml ice-cold homogenization buffer (2 mM activated Na3VO4, 2 mM EDTA, 2 mM EGTA, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM phenylmethanesulfonyl fluoride, and 1 μg/ml leupeptin in T-PER) using tissue grinder tubes (Kontes, Vineland, NJ). Homogenates were subsequently rotated at 4°C at 50 rpm for 1 hour before being centrifuged (15,000 g for 15 minutes at 4°C). Aliquots of the supernatant from muscles used for the 3-MG transport measurement were pipetted into vials with scintillation cocktail for scintillation counting, and 3-MG transport was determined as previously described (6). A portion of the supernatant was used to determine protein concentration by the bicinchoninic acid assay according to the manufacturer’s instructions (Pierce Biotechnology catalog no. 23227). The remaining supernatant was stored at −80°C until further analyzed.
Immunoblotting
Aliquots of supernatants from homogenized muscle lysates were combined with sodium dodecyl sulfate (SDS) loading buffer and separated by SDS-PAGE before being electrophoretically transferred to nitrocellulose. Samples were then rinsed with Tris-buffered saline plus Tween-20 (TBST) (20mM Tris base, 150mM NaCl, pH 7.6, and 0.1% Tween-20), blocked with 5% nonfat dry milk in TBST for 1 hour at room temperature, washed 3 × 5 minutes at room temperature, and treated with the primary antibody (anti-pAMPK-Thr172 at 1:1,000 in TBST with 5% BSA) overnight at 4°C. Blots were then washed 3 × 5 minutes with TBST, incubated with the secondary antibody, goat anti-rabbit IgG horseradish peroxidase conjugate (1:20,000 in TBST with 5% milk), for 1 hour at room temperature, washed again 3 × 5 minutes with TBST, and developed with West Dura SuperSignal enhanced chemiluminescence reagent (Thermo Fisher Scientific, Waltham, MA). Protein bands were quantified by densitometry (Cell Biosciences, Santa Clara, CA). The mean values for sedentary samples without insulin on each blot were normalized to equal 1.0, and then all samples on the blot were expressed relative to the normalized sedentary without insulin value.
Muscle glycogen concentration
Muscles used for measurement of glycogen were frozen immediately after dissection, weighed, and homogenized in ice-cold 0.3 M perchloric acid. An aliquot of the homogenate was stored at −80°C for later determination of glycogen concentration by the amyloglucosidase method (31).
Statistical analyses
Statistical analyses used Sigma Stat version 2.0 (San Rafael, CA). Data are expressed as means ± SE. Two-way ANOVA was used to determine significant differences between the main effects of exercise and genotype, and a Tukey post hoc test was used to identify the source of significant variance. A P value ≤0.05 was considered statistically significant.
RESULTS
Muscle Glycogen
Epitrochlearis muscle glycogen was reduced in 0hPEX vs. 0hSED rats, and it was not significantly different for BNK compared to BN rats (Fig. 1).
Figure 1.
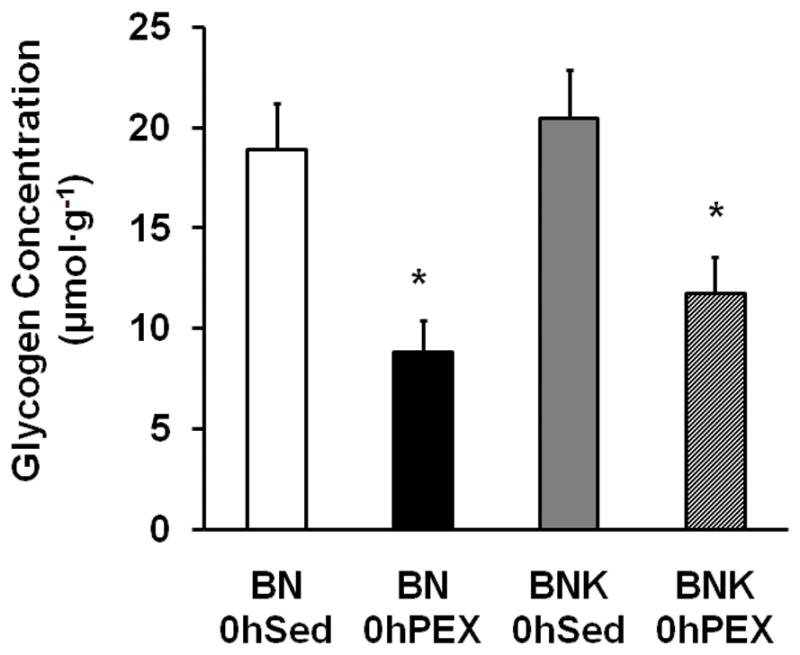
Muscle glycogen concentration in isolated epitrochlearis from BN and BNK rats that were sedentary (0hSED) or exercised (0hPEX). Values are mean ±SE for 7–8 muscles per group. *P < 0.05.
AMPK Thr172 Phosphorylation
Epitrochlearis AMPK Thr172 phosphorylation was increased in 0hPEX vs. 0hSED rats, and it was not significantly different in BNK vs. BN rats (Fig. 2).
Figure 2.
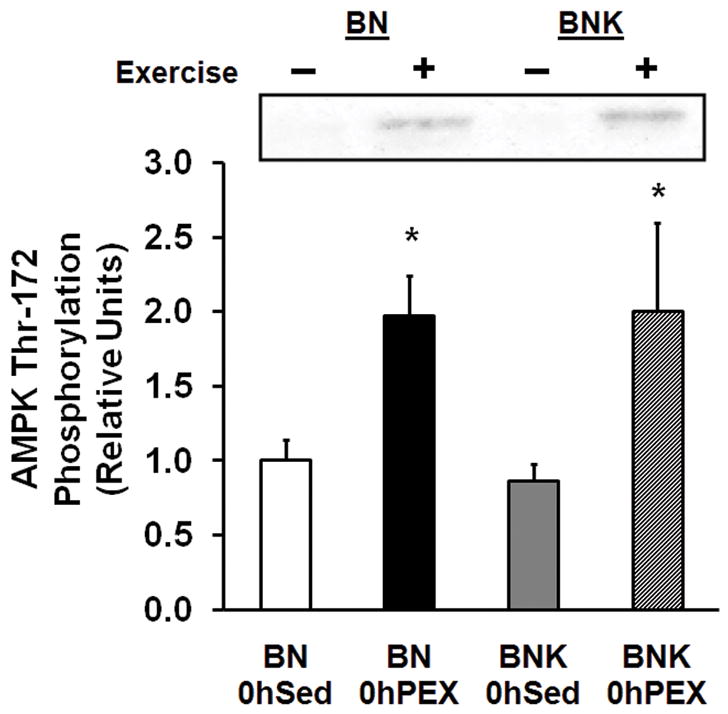
AMPK threonine172 phosphorylation in isolated epitrochlearis from BN and BNK rats that were sedentary (0hSED) or exercised (0hPEX). Values are mean ±SE for 7–8 muscles per group. *P < 0.05.
Muscle Glucose Transport
There was an increase in glucose transport in 0hPEX vs. 0hSED in both BN and BNK rats (Fig. 3). There was not a difference between BNK and BN rats for the exercise-induced increase in insulin-independent glucose transport.
Figure 3.
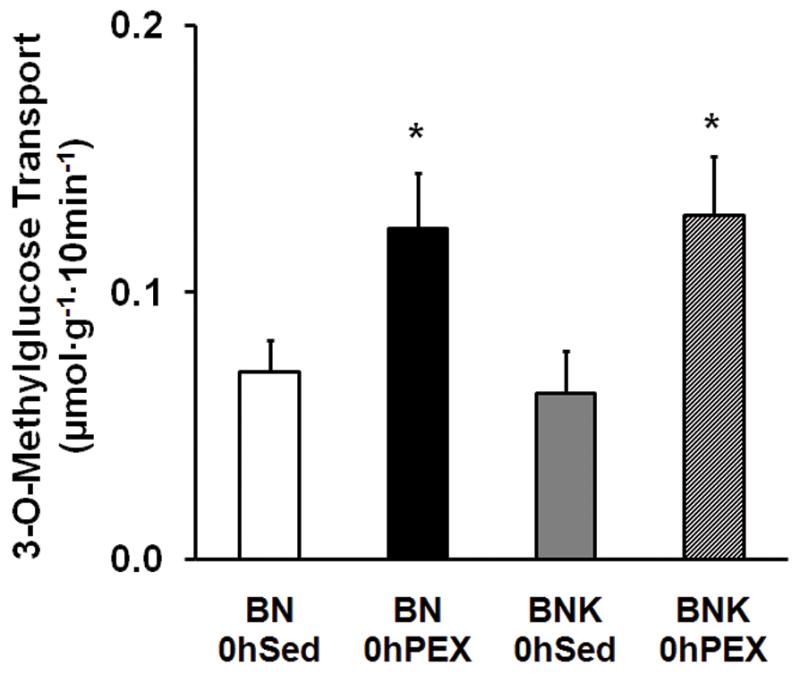
Glucose transport by isolated epitrochlearis muscles without insulin from BN and BNK rats that were sedentary (0hSED) or exercised (0hPEX). Values are mean ± SE for 7–8 muscles per group. *P < 0.05.
Glucose transport without insulin was significantly greater for 3hPEX vs. 3hSED rats in (P < 0.05; Fig. 4). There was not a significant effect of genotype (BNK vs. BN) on glucose transport without insulin in 3hSED and 3hPEX rats (Fig. 4).
Figure 4.
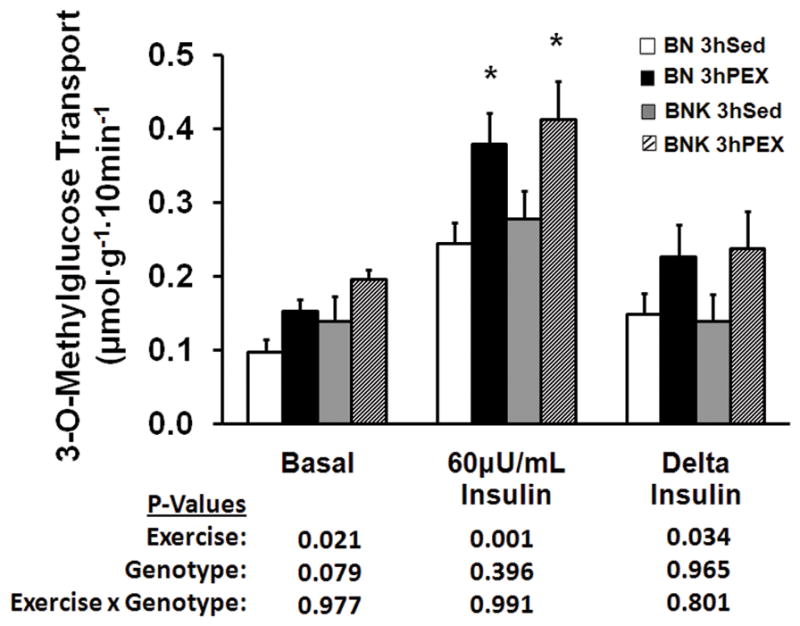
Glucose transport by isolated epitrochlearis muscles without or with 60μU/mL insulin from BN and BNK rats that had undergone two hours of swimming exercise and rested for three hours (3hPEX) or time-matched sedentary controls (3hSED). Delta insulin represents the insulin-stimulated increase above basal in glucose transport calculated by subtracting the basal value from the insulin-stimulated value of paired muscles. Values are mean ± SE for 15–16 muscles per group. *P < 0.05. Exercise, main effect of exercise treatment; Genotype, main effect of genotype; Exercise x Genotype, interaction between main effects.
In the insulin-stimulated epitrochlearis, glucose transport was significantly greater for 3hPEX vs. 3hSED rats (P < 0.001; Fig. 4). Glucose transport with insulin was not different for BNK vs. BN rats in 3hSED and 3hPEX groups. Paired muscles were used for glucose transport measurements, with one muscle from each rat incubated without insulin and the contralateral muscle incubated with insulin. We calculated the insulin-stimulated increase in glucose transport (delta insulin) by subtracting the basal value from the insulin-stimulated value for each rat. Delta insulin was also significantly greater for 3hPEX vs. 3hSED rats (P < 0.05). Delta insulin was not different for BNK compared to BN rats in the 3hSED and 3hPEX groups.
There were no significant interactions (exercise x genotype) for any of the measurements made.
DISCUSSION
A single exercise session can cause an increase in both insulin-dependent and insulin-independent glucose uptake by skeletal muscle in mice (4,5,18), rats (7,8,33,40), and humans (34). Several previous studies have demonstrated that the Kallikrein-Kinin System (KKS) can be activated by exercise or muscle contractions (24,37,39), and it has been suggested that this activation might play a role in elevated glucose uptake by skeletal muscle (13,15,38). The most important new findings of this study were that unexercised BN and BNK rats (which are known to be deficient in plasma kininogen) did not differ for insulin-stimulated glucose transport, and that the effects of exercise on both insulin-independent and insulin-dependent glucose transport were undiminished in the BNK rats.
Results of several published studies suggest that the KKS is linked to insulin-dependent glucose transport in the absence of exercise, but the results of other published studies are not consistent with this idea. In support, incubation with bradykinin has been reported to increase insulin-stimulation of GLUT4 translocation and glucose transport of L6 myocytes, 3T3-L1 adipocytes or isolated dog adipocytes (1,23,29). In addition, previous studies found that insulin-stimulated glucose transport was greater in isolated epitrochlearis muscles that were dissected from obese Zucker rats that had been injected with bradykinin either acutely (2 hours prior to muscle dissection) or chronically (twice daily for 14 days) (21,22). However, in vivo bradykinin treatment did not enhance insulin-stimulated glucose uptake by isolated epitrochlearis muscles from lean Zucker rats (21). Most relevant to the results of the current study, BNK vs. BN rats had a significantly reduced glucose disposal rate during a hyperinsulinemic-euglycemic clamp (12). In contrast, the current study did not provide evidence for insulin resistance in the isolated epitrochlearis of BNK vs. BN rats. The results from the in vivo clamp procedure would be directly influenced by systemic factors that were absent from the isolated muscle preparation, including blood flow, circulating hormones and neural regulation. In addition, glucose disposal in vivo depends on multiple tissues, including the liver, adipose tissue and skeletal muscle. It is notable that Shimojo et al. (36) also did not find an effect of bradykinin on in vitro, insulin-stimulated glucose uptake by the predominantly type I soleus muscle. The current results clearly demonstrate that there is not intrinsic insulin resistance of the isolated epitrochlearis (composed of predominantly type II fibers) of BNK rats.
A number of studies have assessed the possible role of the KKS system in insulin-independent increase in skeletal muscle glucose transport with exercise or contraction. Dietze and Wicklmayr (13) found that most of the isometric exercise-induced increase in glucose uptake by the human forearm could be blocked by infusion of aprotinin (a kallikrein inhibitor). They also found that bradykinin infusion could eliminate aprotinin’s inhibition of exercise-induced glucose uptake. Bradykinin can elevate blood flow, so the results for forearm glucose uptake during in vivo exercise were likely partly attributable to changes in muscle blood flow which was observed to be decreased by aprotinin and increased by bradykinin infusion. Consistent with this interpretation, studying isolated rat epitrochlearis muscles, Constable et al. (11) found that a range of bradykinin concentrations did not result in increased glucose transport, nor was contraction-stimulated glucose transport attenuated in the presence of aprotinin. Insulin-independent glucose uptake by L6 myotubes, 3T3-L1 adipocytes, and Chinese hamster ovary cells was greater for cells that stably overexpressed both GLUT4 and B2 receptor of bradykinin compared to cells that overexpressed only GLUT4 (24). However, in L6 cells or isolated muscle strips from dogs without overexpression of B2 receptors of bradykinin, insulin-independent glucose transport was not elevated by bradykinin (29). Taguchi et al. (38) measured the sarcolemmal GLUT4 content and glucose uptake by sarcolemmal vesicles prepared from skeletal muscle that was sampled immediately post-exercise (1 hour swim by rats). Exercised rats were studied with or without 5 days of infusion of a B2 receptor of bradykinin inhibitor. The inhibitor caused an ~50% decline in the immediate post-exercise increases in membrane GLUT4 and glucose uptake. It seems possible that chronic treatment with a bradykinin B2 receptor inhibitor might induce adaptations that altered the effects of exercise on GLUT4 and glucose uptake. In the current study, using isolated epitrochlearis muscles, BNK vs. BN rats were not different for insulin-independent glucose transport measured immediately post-exercise. Although the KKS can influence blood flow and thereby indirectly alter in vivo glucose uptake during exercise, data from studies that have evaluated glucose transport by isolated skeletal muscle (in which blood flow is not a factor) after in vivo exercise or in vitro contractions provide no support for the idea that KKS is responsible for increased insulin-independent glucose transport after exercise or contractions
The current results indicated that exercise effects on subsequent insulin-stimulated glucose transport are essentially the same for BN and BNK rats. We recently performed a study using the bradykinin B2 receptor null mouse as a different model to assess the possible role of the KKS in post-exercise glucose transport (35). Isolated skeletal muscles from the mice that were null for the B2 receptor of bradykinin and normal control mice did not differ for their post-exercise increase in insulin-dependent glucose transport after exercise. The results of experiments using two different animal models for important components of the KKS provide evidence that a normal KKS is not essential to attain a normal elevation in insulin-stimulated glucose transport by isolated skeletal muscles after in vivo exercise.
Electrical stimulation of isolated skeletal muscle has often been used as model for in vivo exercise. Dumke et al. (15) reported that rat epitrochlearis muscles electrically stimulated to contract in kallikrein-deficient plasma compared to muscles stimulated to contract in normal plasma had lower insulin-stimulated glucose transport. However, in vivo exercise and in vitro contractions in serum may not lead to increased insulin-stimulated glucose transport by an identical mechanism. Funai et al. (16) found that when in vivo exercise is followed by rapid removal of the epitrochlearis and in vitro contraction of the isolated muscle, the two stimuli induced an additive effect on the subsequent increase in insulin-stimulated glucose transport. These results suggested that the increase in insulin-stimulated glucose transport induced after in vitro contraction and after in vivo exercise may be caused by distinct mechanisms. The current study focused on the effects of in vivo exercise because this intervention is more physiologically relevant than electrical stimulation.
In conclusion, insulin-stimulated glucose transport by isolated epitrochlearis muscles was similar for sedentary BN and BNK rats. Rats deficient in plasma kininogen compared to normal rats had similar post-exercise increases in both insulin-independent and insulin-dependent glucose transport by isolated epitrochlearis muscle. It is well-established that exercise has robust effects on insulin-independent and insulin-dependent glucose transport that persist in isolated muscles, and the current data indicate that these important exercise effects are undiminished in plasma kininogen-deficient rats.
Acknowledgments
The study was supported by R01-DK077171 from the National Institutes of Health. The authors thank Dr. Oscar Carretero for generously providing the Brown Norway Katholiek rats and Dr. Edward Shesely for assistance in transferring the rats to the University of Michigan.
Footnotes
The authors have no conflicts of interest to disclose.
The results of the present study do not constitute endorsement by the American College of Sports Medicine.
References
- 1.Beard KM, Lu H, Ho K, Fantus IG. Bradykinin augments insulin-stimulated glucose transport in rat adipocytes via endothelial nitric oxide synthase-mediated inhibition of Jun NH2-terminal kinase. Diabetes. 2006;55(10):2678–87. doi: 10.2337/db05-1538. [DOI] [PubMed] [Google Scholar]
- 2.Bhoola KD, Figueroa CD, Worthy K. Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacol Rev. 1992;44(1):1–80. [PubMed] [Google Scholar]
- 3.Blais C, Jr, Adam A, Massicotte D, Peronnet F. Increase in blood bradykinin concentration after eccentric weight-training exercise in men. J Appl Physiol. 1999;87(3):1197–201. doi: 10.1152/jappl.1999.87.3.1197. [DOI] [PubMed] [Google Scholar]
- 4.Bonen A, Tan MH. Dissociation between insulin binding and glucose utilization after intense exercise in mouse skeletal muscles. Horm Metab Res. 1989;21(4):172–8. doi: 10.1055/s-2007-1009184. [DOI] [PubMed] [Google Scholar]
- 5.Bonen A, Tan MH, Watson-Wright WM. Effects of exercise on insulin binding and glucose metabolism in muscle. Can J Physiol Pharmacol. 1984;62(12):1500–4. doi: 10.1139/y84-248. [DOI] [PubMed] [Google Scholar]
- 6.Cartee GD, Bohn EE. Growth hormone reduces glucose transport but not GLUT-1 or GLUT-4 in adult and old rats. Am J Physiol. 1995;268(5 Pt 1):E902–9. doi: 10.1152/ajpendo.1995.268.5.E902. [DOI] [PubMed] [Google Scholar]
- 7.Cartee GD, Holloszy JO. Exercise increases susceptibility of muscle glucose transport to activation by various stimuli. Am J Physiol. 1990;258(2 Pt 1):E390–3. doi: 10.1152/ajpendo.1990.258.2.E390. [DOI] [PubMed] [Google Scholar]
- 8.Cartee GD, Young DA, Sleeper MD, Zierath J, Wallberg-Henriksson H, Holloszy JO. Prolonged increase in insulin-stimulated glucose transport in muscle after exercise. Am J Physiol. 1989;256(4 Pt 1):E494–9. doi: 10.1152/ajpendo.1989.256.4.E494. [DOI] [PubMed] [Google Scholar]
- 9.Carvalho CR, Thirone AC, Gontijo JA, Velloso LA, Saad MJ. Effect of captopril, losartan, and bradykinin on early steps of insulin action. Diabetes. 1997;46(12):1950–7. doi: 10.2337/diab.46.12.1950. [DOI] [PubMed] [Google Scholar]
- 10.Constable SH, Favier RJ, Cartee GD, Young DA, Holloszy JO. Muscle glucose transport: interactions of in vitro contractions, insulin, and exercise. J Appl Physiol. 1988;64(6):2329–32. doi: 10.1152/jappl.1988.64.6.2329. [DOI] [PubMed] [Google Scholar]
- 11.Constable SH, Favier RJ, Uhl J, Holloszy JO. Bradykinin does not mediate activation of glucose transport by muscle contraction. J Appl Physiol. 1986;61(3):881–4. doi: 10.1152/jappl.1986.61.3.881. [DOI] [PubMed] [Google Scholar]
- 12.Damas J, Bourdon V, Lefebvre PJ. Insulin sensitivity, clearance and release in kininogen-deficient rats. Exp Physiol. 1999;84(3):549–57. doi: 10.1111/j.1469-445x.1999.01812.x. [DOI] [PubMed] [Google Scholar]
- 13.Dietze G, Wicklmayr M. Evidence for a participation of the kallikrein-kinin system in the regulation of muscle metabolism during muscular work. FEBS Lett. 1977;74(2):205–8. doi: 10.1016/0014-5793(77)80847-8. [DOI] [PubMed] [Google Scholar]
- 14.Duka I, Shenouda S, Johns C, Kintsurashvili E, Gavras I, Gavras H. Role of the B(2) receptor of bradykinin in insulin sensitivity. Hypertension. 2001;38(6):1355–60. doi: 10.1161/hy1201.096574. [DOI] [PubMed] [Google Scholar]
- 15.Dumke CL, Kim J, Arias EB, Cartee GD. Role of kallikrein-kininogen system in insulin-stimulated glucose transport after muscle contractions. J Appl Physiol. 2002;92(2):657–64. doi: 10.1152/japplphysiol.00854.2001. [DOI] [PubMed] [Google Scholar]
- 16.Funai K, Schweitzer GG, Castorena CM, Kanzaki M, Cartee GD. In vivo exercise followed by in vitro contraction additively elevates subsequent insulin-stimulated glucose transport by rat skeletal muscle. Am J Physiol Endocrinol Metab. 2010;298(5):E999–1010. doi: 10.1152/ajpendo.00758.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao J, Gulve EA, Holloszy JO. Contraction-induced increase in muscle insulin sensitivity: requirement for a serum factor. Am J Physiol. 1994;266(2 Pt 1):E186–92. doi: 10.1152/ajpendo.1994.266.2.E186. [DOI] [PubMed] [Google Scholar]
- 18.Hamada T, Arias EB, Cartee GD. Increased submaximal insulin-stimulated glucose uptake in mouse skeletal muscle after treadmill exercise. J Appl Physiol. 2006;101(5):1368–76. doi: 10.1152/japplphysiol.00416.2006. [DOI] [PubMed] [Google Scholar]
- 19.Haring HU, Tippmer S, Kellerer M, Mosthaf L, Kroder G, Bossenmaier B, Berti L. Modulation of insulin receptor signaling. Potential mechanisms of a cross talk between bradykinin and the insulin receptor. Diabetes. 1996;45(Suppl 1):S115–9. doi: 10.2337/diab.45.1.s115. [DOI] [PubMed] [Google Scholar]
- 20.Henriksen EJ, Jacob S, Augustin HJ, Dietze GJ. Glucose transport activity in insulin-resistant rat muscle. Effects of angiotensin-converting enzyme inhibitors and bradykinin antagonism. Diabetes. 1996;45(Suppl 1):S125–8. doi: 10.2337/diab.45.1.s125. [DOI] [PubMed] [Google Scholar]
- 21.Henriksen EJ, Jacob S, Fogt DL, Dietze GJ. Effect of chronic bradykinin administration on insulin action in an animal model of insulin resistance. Am J Physiol. 1998;275(1 Pt 2):R40–5. doi: 10.1152/ajpregu.1998.275.1.R40. [DOI] [PubMed] [Google Scholar]
- 22.Henriksen EJ, Jacob S, Kinnick TR, Youngblood EB, Schmit MB, Dietze GJ. ACE inhibition and glucose transport in insulinresistant muscle: roles of bradykinin and nitric oxide. Am J Physiol. 1999;277(1 Pt 2):R332–6. doi: 10.1152/ajpregu.1999.277.1.R332. [DOI] [PubMed] [Google Scholar]
- 23.Isami S, Kishikawa H, Araki E, Uehara M, Kaneko K, Shirotani T, Todaka M, Ura S, Motoyoshi S, Matsumoto K, Miyamura N, Shichiri M. Bradykinin enhances GLUT4 translocation through the increase of insulin receptor tyrosine kinase in primary adipocytes: evidence that bradykinin stimulates the insulin signalling pathway. Diabetologia. 1996;39(4):412–20. doi: 10.1007/BF00400672. [DOI] [PubMed] [Google Scholar]
- 24.Kishi K, Muromoto N, Nakaya Y, Miyata I, Hagi A, Hayashi H, Ebina Y. Bradykinin directly triggers GLUT4 translocation via an insulin-independent pathway. Diabetes. 1998;47(4):550–8. doi: 10.2337/diabetes.47.4.550. [DOI] [PubMed] [Google Scholar]
- 25.Kohlman O, Jr, de Neves FA, Ginoza M, Tavares A, Cezaretti ML, Zanella MT, Ribeiro AB, Gavras I, Gavras H. Role of bradykinin in insulin sensitivity and blood pressure regulation during hyperinsulinemia. Hypertension. 1995;25(5):1003–7. doi: 10.1161/01.hyp.25.5.1003. [DOI] [PubMed] [Google Scholar]
- 26.Langberg H, Bjorn C, Boushel R, Hellsten Y, Kjaer M. Exercise-induced increase in interstitial bradykinin and adenosine concentrations in skeletal muscle and peritendinous tissue in humans. J Physiol. 2002;542(Pt 3):977–83. doi: 10.1113/jphysiol.2002.018077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Majima M, Mizogami S, Kuribayashi Y, Katori M, Oh-ishi S. Hypertension induced by a nonpressor dose of angiotensin II in kininogen-deficient rats. Hypertension. 1994;24(1):111–9. doi: 10.1161/01.hyp.24.1.111. [DOI] [PubMed] [Google Scholar]
- 28.Majima M, Yoshida O, Mihara H, Muto T, Mizogami S, Kuribayashi Y, Katori M, Oh-ishi S. High sensitivity to salt in kininogen-deficient brown Norway Katholiek rats. Hypertension. 1993;22(5):705–14. doi: 10.1161/01.hyp.22.5.705. [DOI] [PubMed] [Google Scholar]
- 29.Miyata T, Taguchi T, Uehara M, Isami S, Kishikawa H, Kaneko K, Araki E, Shichiri M. Bradykinin potentiates insulin-stimulated glucose uptake and enhances insulin signal through the bradykinin B2 receptor in dog skeletal muscle and rat L6 myoblasts. Eur J Endocrinol. 1998;138(3):344–52. doi: 10.1530/eje.0.1380344. [DOI] [PubMed] [Google Scholar]
- 30.Nakajima S, Ito H, Hayashi I, Kuribayashi Y, Okumura T, Yajima Y, Katori M, Majima M. Inhibition of kinin degradation on the luminal side of renal tubules reduces high blood pressure in deoxycorticosterone acetate salt-treated rats. Clin Exp Pharmacol Physiol. 2000;27(1–2):80–7. doi: 10.1046/j.1440-1681.2000.03209.x. [DOI] [PubMed] [Google Scholar]
- 31.Passoneau JV, Lauderdale VR. A comparison of three methods of glycogen measurement. Anal Biochem. 1974;60:405–12. doi: 10.1016/0003-2697(74)90248-6. [DOI] [PubMed] [Google Scholar]
- 32.Rhaleb NE, Yang XP, Nanba M, Shesely EG, Carretero OA. Effect of Chronic Blockade of the Kallikrein-Kinin System on the Development of Hypertension in Rats. Hypertension. 2001;37(1):121–28. doi: 10.1161/01.hyp.37.1.121. [DOI] [PubMed] [Google Scholar]
- 33.Richter EA, Garetto LP, Goodman MN, Ruderman NB. Muscle glucose metabolism following exercise in the rat: increased sensitivity to insulin. J Clin Invest. 1982;69(4):785–93. doi: 10.1172/JCI110517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richter EA, Mikines KJ, Galbo H, Kiens B. Effect of exercise on insulin action in human skeletal muscle. J Appl Physiol. 1989;66(2):876–85. doi: 10.1152/jappl.1989.66.2.876. [DOI] [PubMed] [Google Scholar]
- 35.Schweitzer GG, Castorena CM, Hamada T, Arias EB, Cartee GD. The B2 receptor of bradykinin is not essential for the increase in insulin-stimulated glucose uptake following acute exercise. Diabetes. 2008;(Supplement 1A):LB-11. [Google Scholar]
- 36.Shimojo N, Pickens TG, Margolius HS, Mayfield RK. Tissue kallikrein and bradykinin do not have direct insulin-like actions on skeletal muscle glucose utilization. Biol Chem Hoppe Seyler. 1987;368(10):1355–61. doi: 10.1515/bchm3.1987.368.2.1355. [DOI] [PubMed] [Google Scholar]
- 37.Stebbins CL, Carretero OA, Mindroiu T, Longhurst JC. Bradykinin release from contracting skeletal muscle of the cat. J Appl Physiol. 1990;69(4):1225–30. doi: 10.1152/jappl.1990.69.4.1225. [DOI] [PubMed] [Google Scholar]
- 38.Taguchi T, Kishikawa H, Motoshima H, Sakai K, Nishiyama T, Yoshizato K, Shirakami A, Toyonaga T, Shirontani T, Araki E, Shichiri M. Involvement of bradykinin in acute exercise-induced increase of glucose uptake and GLUT-4 translocation in skeletal muscle: studies in normal and diabetic humans and rats. Metabolism. 2000;49(7):920–30. doi: 10.1053/meta.2000.6755. [DOI] [PubMed] [Google Scholar]
- 39.Vettor R, De Palo C, Calo L, De Carlo E, Sicolo N, Martini C, Federspil G. Effect of exercise on plasma kallikrein and muscular phospholipase A2 activity in rats. Mol Cell Endocrinol. 1986;45(1):65–70. doi: 10.1016/0303-7207(86)90083-3. [DOI] [PubMed] [Google Scholar]
- 40.Wallberg-Henriksson H, Constable SH, Young DA, Holloszy JO. Glucose transport into rat skeletal muscle: interaction between exercise and insulin. J Appl Physiol. 1988;65(2):909–13. doi: 10.1152/jappl.1988.65.2.909. [DOI] [PubMed] [Google Scholar]