

Abstract
The poultry disease coccidiosis, caused by infection with Eimeria spp. apicomplexan parasites, is responsible for enormous economic losses to the global poultry industry. The rapid increase of resistance to therapeutic agents, as well as the expense of vaccination with live attenuated vaccines, requires the development of new effective treatments for coccidiosis. Because of their key regulatory function in the eukaryotic cell cycle, cyclin-dependent kinases (CDKs) are prominent drug targets. The Eimeria tenella CDC2-related kinase 2 (EtCRK2) is a validated drug target that can be activated in vitro by the CDK activator XlRINGO (Xenopus laevis rapid inducer of G2/M progression in oocytes). Bioinformatics analyses revealed four putative E. tenella cyclins (EtCYCs) that are closely related to cyclins found in the human apicomplexan parasite Plasmodium falciparum. EtCYC3a was cloned, expressed in Escherichia coli and purified in a complex with EtCRK2. Using the non-radioactive time-resolved fluorescence energy transfer (TR-FRET) assay, we demonstrated the ability of EtCYC3a to activate EtCRK2 as shown previously for XlRINGO. The EtCRK2/EtCYC3a complex was used for a combined in vitro and in silico high-throughput screening approach, which resulted in three lead structures, a naphthoquinone, an 8-hydroxyquinoline and a 2-pyrimidinyl-aminopiperidine-propane-2-ol. This constitutes a promising starting point for the subsequent lead optimization phase and the development of novel anticoccidial drugs.
Introduction
The obligate intracellular apicomplexan parasites cause devastating diseases in humans and domestic animals. Well-known members of this phylum are Plasmodium falciparum, Toxoplasma gondii, Cryptosporidium parvum, Theileria annulata and Eimeria tenella (Mehlhorn, 2008; Morrison, 2009). The poultry intestinal disease known as coccidiosis is caused by Eimeria spp. such as Eimeria acervulina, Eimeria necatrix, E. tenella and Eimeria mivati. The most pathogenic species, E. tenella, provokes a haemorrhagic diarrhoea in young chickens, leading to a loss of weight and appetite, resorption problems, bacterial secondary infections and often also to the bird’s death (Chauhan & Roy, 2007; Mehlhorn, 2008; Shirley et al., 2007). Moreover, coccidiosis causes tremendous economic losses, in excess of three billion US Dollars annually, to the world poultry industry (Dalloul & Lillehoj, 2006; Shirley et al., 2007). Although prophylaxis using live-attenuated vaccines is efficient, it may have transitory adverse effects on chicken growth rate and is economically restrictive due to the high costs of these vaccines (Shirley & Bedrník, 1997; Shirley, 2000; Shirley et al., 2005; Wallach et al., 2008). Several drugs are available for the treatment of coccidiosis; however, resistance develops frequently (Chapman, 1997; Williams et al., 1999). Therefore, there is a clear need for new, affordable and effective anticoccidial drugs (Williams et al., 1999; Dalloul & Lillehoj, 2006; Shirley et al., 2007).
The apicomplexan life cycle involves both asexual (sporogony and schizogony) and sexual (gamogony) reproduction (Mehlhorn, 2008). During schizogony the Eimeria parasite proliferates asexually with a very high cell division rate within the host cells (Kinnaird et al., 2004; Mehlhorn, 2008). Evidence for the presence of cyclin-dependent kinases (CDKs) in apicomplexa (Doerig et al., 2000) including Eimeria (Kinnaird et al., 2004) suggests a similar CDK-dependent mode of cell cycle regulation to that found in higher eukaryotes (Malumbres & Barbacid, 2009). There are a high number of anti-cancer drugs that interfere with cell cycle regulation, leading to the death of the rapidly dividing cells (Tamamori et al., 1998; Doerig et al., 2002; Monaco & Vallano, 2003; Malumbres & Barbacid, 2009). Therefore, we, like others, predict that the disruption of the parasite’s cell cycle will lead to its death (Engels et al., 2010; Waters & Geyer, 2003).
CDKs play a key role in cell cycle progression, transcription and neuronal function (Morgan, 1997), and are therefore extremely interesting for the development of novel pharmaceuticals (Sielecki et al., 2000; Waters & Geyer, 2003; Bruno et al., 1992; Schang et al., 2006). E. tenella CDC2-related kinase 2 (EtCRK2) is by analogy assumed to play a similar key role in Eimeria (Kinnaird et al., 2004; Engels et al., 2010). It has been chemically validated using the synthetic flavone flavopiridole, a specific CDK inhibitor (Dai & Grant, 2003). Flavopiridole inhibits EtCRK2 in enzyme assays with an IC50 of 33±10 nM and a Ki of 11±3 nM, and fully inhibits E. tenella schizont development at concentrations of 150 and 300 nM. Concentrations below 80 nm show no inhibitory effects, and host cell toxicity is observed at concentrations above 600 nM. Therefore, E. tenella CDKs are considered to be chemically validated drug targets (Engels et al., 2010).
As the name implies, CDKs are dependent on activation through binding of the respective cyclin (Olashaw & Pledger, 2002). To the best of our knowledge, in contrast to other apicomplexan parasites such as Plasmodium, no eimerian cyclins have been reported so far.
Here, we describe the bioinformatic discovery of two partial and two complete predicted cyclin-like proteins out of the publicly available genome sequence of E. tenella. We were able to clone one of the identified E. tenella cyclins (EtCYCs) (EtCYC3a) and demonstrated that its protein product was able to activate EtCRK2, in a similar manner to that shown with the non-cyclin activator Xenopus laevis rapid inducer of G2/M progression in oocytes (XlRINGO) (Engels et al., 2010; Ferby et al., 1999). In a combined in vitro and in silico high-throughput screening approach, using real (3514 compounds) and virtual (approx. 6 000 000 compounds) compound libraries, we identified numerous hit compound structures. The most promising hits were further analysed by IC50 and Ki determinations, resulting in three lead structures. The three lead structures have similar activities towards EtCRK2/EtCYC3a and EtCRK2/XlRINGO. From our data we conclude that most likely EtCYC3a is a natural activator of EtCRK2.
Methods
Bioinformatic analysis.
Bioinformatic analyses were carried out in analogy to Engels et al. (2010) and run on Silicon Graphics (SGI) computers (models Origin 3200, O2, Octane2, Fuel) running the SGI operating system IRIX6.5 as well as on Dell Precision workstations (models 390 and T3400) running Red Hat Enterprise Linux 5 (RHEL 5). Publicly available E. tenella genome data were downloaded from the Wellcome Trust Sanger Institute (http://www.sanger.ac.uk/resources/downloads/protozoa/eimeria-tenella.html).
Chemoinformatic analysis.
Chemoinformatic analyses were run according to Engels et al. (2010). At the time of the analysis the virtual compound library comprised approximately 6×106 compounds. Molecular docking was done using the docking software gold as described in Engels et al. (2010). Molecular clustering was done using the hierarchical clustering method of the software suite Spotfire Decision Site 9.1.1 (Tibco Software) based on the MDL Keys (MDL Information Systems; nowadays Accelrys).
Chemicals
Standard CDK inhibitors.
The purity of all screening compounds used was ≥90 %, if not stated otherwise. Flavopiridole was ordered as flavopiridole hydrochloride hydrate from Sigma-Aldrich; IUPAC name 2-(2-chlorophenyl)-5,7-dihydroxy-8-[(3S,4R)-3-hydroxy-1-methyl-4-piperidinyl]-4H-1-benzopyran-4-one; purity ≥98 %. Indirubin-5-sulfonate was ordered from Biomol International; IUPAC name (3Z)-2-oxo-3-(3-oxo-1H-indol-2-ylidene)-1H-indole-5-sulfonic acid; purity ≥98 %. 10-Z-Hymenialdisine was ordered from Axxora; IUPAC name (4Z)-4-(2-amino-4-oxo-1H-imidazol-5-ylidene)-2-bromo-1,5,6,7-tetrahydropyrrolo[2,3-c]azepin-8-one; purity ≥95 %. Purvalanol A was ordered from Axxora; IUPAC name 2-[[6-[(3-chlorophenyl)amino]-9-isopropyl-purin-2-yl]amino]-3-methyl-butan-1-ol; purity ≥95 %. Staurosporine was ordered from Sigma-Aldrich; IUPAC name (9S,10R,11R,13R)-2,3,10,11,12,13-hexahydro-10-methoxy-9-methyl-11-(methylamino)-9,13-epoxy-1H,9H-diindolo[1,2,3-gh:3′,2′,1′-lm]pyrrolo[3,4-j][1,7]benzodiazonin-1-one; purity ≥98 %. Alsterpaullone was ordered from Axxora; IUPAC name 9-nitro-7,12-dihydro-5H-indolo[3,2-d][1]benzazepin-6-one; purity ≥95 %.
Novel identified inhibitors.
BES124764 was ordered from Maybridge Ltd; IUPAC name 1-phenylsulfanyl-3-[4-[[4-(trifluoromethyl)pyrimidin-2-yl]amino]-1-piperidyl]propane-2-ol; purity ≥85 %. BES312351 was ordered from Bridge Chem Pvt. Ltd., Mumbai, India; IUPAC name 7-amino-8-hydroxy-quinoline-5-sulfonic acid hydrate; purity ≥95 %. BES130131 was ordered from InterBioScreen Ltd, Chernogolovka, Russia; IUPAC name 2,3-dichloro-5,8-dihydroxy-6-methyl-naphthalene-1,4-dione; purity ≥95 %. BES153950 was ordered from SPECS GmbH; IUPAC name (4Z,5Z,6E)-4,5-bis(hydroxyimino)-6-phenacylidene-hexahydropyrimidin-2-one; purity ≥90 %. BES154393 was ordered from SPECS GmbH; IUPAC name 4-chloro-1H-pyrazolo[4,3-c]quinolin-3-amine; purity ≥90 %.
Biochemical analysis
Isolation of E. tenella sporozoites.
Sporulated oocysts of E. tenella Houghton strain (9.6×105 oocysts ml−1 in 4 % potassium dichromate solution) were used as the source of parasite material. Sporozoites were obtained as described earlier (Hofmann & Raether, 1990). A 200 ml volume of oocysts in potassium dichromate solution was centrifuged at 6 °C (2500 g, 3 min), resuspended in 100 ml sodium hypochlorite (Honeywell Riedel-de Haën) and stirred in this solution for no more than 10 min until a deformation in the parasite cell wall was visible (monitored by microscopy) (Hofmann & Raether, 1990). Following centrifugation (2500 g, 3 min), floating oocysts were aspirated with a vacuum pump, diluted in distilled water and again centrifuged (2500 g, 3 min) (Hofmann & Raether, 1990). This step was repeated several times to remove the residual chloride. Oocysts were diluted in Hank’s Balanced Salt Solution (HBSS; Adcock-Scientific) and fractured by mixing with glass beads (1 mm diameter, Sigma-Aldrich) on a vortex mixer until a disruption of 80 % of oocysts was detected microscopically (Hofmann & Raether, 1990). The glass beads were washed several times with buffer, and after centrifugation the sporocyst pellet was resuspended in HBSS and stored at 4 °C (Hofmann & Raether, 1990).
RNA isolation and cDNA production.
The isolated sporocysts were centrifuged in 2 ml Eppendorf tubes at 13 000 g for 10 min at 4 °C in a tabletop centrifuge, and the pellets were collected. A 1 ml volume of TRI Reagent (Invitrogen) was added to 100 mg of sporocysts and placed on ice. The sample was immediately homogenized with a Precellys 24 homogenizer (MO-BIO Laboratories), and the disruption of sporocysts and sporozoites was verified microscopically. The total RNA from sporozoites was prepared following the manufacturer’s instructions.
In order to produce cDNA, RT-PCRs were performed with 10 µg of total RNA using the SuperScript First-Strand Synthesis system for RT-PCR (Invitrogen) according to the manufacturer’s instructions.
PCR amplification of EtCYC-like 3a (EtCYC3a) from cDNA.
A full-length EtCYC3a protein sequence was identified in silico by blast-searching the E. tenella proteome with several known human and protozoan cyclins. The Etcyc3a gene was amplified in vitro from 2 µg E. tenella cDNA using Platinum Taq DNA Polymerase High Fidelity (Invitrogen) in the presence of 2 mM MgSO4 and 150 pmol of each primer (SRD, Oberursel, Germany). The primers contained BamHI (forward primer, 5′-TAGGATCCATGTTGGAGGCATCCCGAGAC-3′) or HindIII (reverse primer, 5′-GGCAAGCTTAGCGCAGTGATGGTTGTG-3′) restriction sites (recognition sites underlined). The reaction was incubated at 94 °C for 2 min, 40 cycles at 94 °C for 30s, 62 °C for 1 min and 68 °C for 1 min, followed by 10 min at 68 °C. Amplified fragments were separated by agarose gel electrophoresis and isolated using the QIAquick Gel Extraction kit (Qiagen) following the manufacturer’s instructions.
After fragment isolation, the PCR product was digested with BamHI and HindIII (both enzymes from Roche Diagnostics) prior to insertion into the pMALc2X vector (New England Biolabs). The resulting expression plasmid pMALc2X-Etcyc3a was amplified in Escherichia coli TOP10 cells (Invitrogen) and the insert was verified by sequencing. For protein expression, Escherichia coli BL21(DE3) competent cells (Invitrogen) were transformed with the plasmid pMALc2X-Etcyc3a.
Expression and purification of recombinant EtCRK2 and XlRINGO.
Recombinant EtCRK2 and XlRINGO were expressed and purified according to Engels et al. (2010).
Expression and co-purification of recombinant EtCRK2-His/maltose binding protein (MBP)-EtCYC3a complex.
Both plasmids (pQE60-Etcrk2 and pMALc2X-Etcyc3a) were transformed in Escherichia coli BL21(DE3) (Invitrogen). The cells were used to express the recombinant EtCRK2–His and MBP–EtCYC3a fusion proteins separately overnight at 24 °C using 0.1 mM and 1 mM IPTG, respectively. Cell pellets were harvested by centrifugation at 9000 g for 40 min. Pellets from both transformations [Escherichia coli BL21(DE3) pLysS/pQE60-Etcrk2 and Escherichia coli BL21(DE3)/pMALc2X-Etcyc3a] were pooled and disrupted together using a French press. The cell lysate was incubated in a shaker for 1 h at 4 °C to allow for the formation of an EtCRK2–His/MBP-EtCYC3a complex. The resulting complex was co-purified by amylose affinity chromatography using the MBP-tag of EtCYC3a. Free MBP was subsequently removed using size exclusion chromatography as described for MBP–XlRINGO by Engels et al. (2010).
IC50, Ki determination, and compound solubility.
IC50 and Ki determination based on the time-resolved fluorescence energy transfer (TR-FRET) assay as well as compound solubility testing was done according to Engels et al. (2010).
Liquid chromatography/mass spectrometry (LC/MS) and NMR analyses for compound quality determination.
LC/MS and NMR analyses for compound quality control were conducted as described in Engels et al. (2010).
Results and Discussion
In silico identification of EtCYCs
In order to identify potential EtCYCs we ran a number of blast database searches on the currently unfinished genomic dataset of E. tenella using a wide set of cyclins, including Plasmodium falciparum cyclin (PfCYC)1 (Le Roch et al., 2000), PfCYC2, PfCYC3 and PfCYC4 (Merckx et al., 2003) from P. falciparum, as query sequences in analogy to Engels et al. (2010). The database queries revealed four potential E. tenella gene sequences which might encode cyclins (Fig. 1a).
Fig. 1.

Bioinformatic identification of cyclin-like proteins of E. tenella. (a) Multiple sequence alignment of the potential cyclin-like E. tenella proteins (EtCYC1, EtCYC3a, EtCYC3b and EtCYC4). For EtCYC1 and EtCYC3b, only incomplete sequences could be identified. Blue-shaded residues are conserved among the four sequences, yellow-shaded residues among any three of the four sequences, grey-shaded residues among any two only. (b) Comparison of known cyclin-like sequences of P. falciparum and E. tenella. The potential cyclin-like proteins are homologous to the known cyclin-like proteins of P. falciparum. The characteristic cyclin box sequence motif is conserved among the apicomplexan cyclin-like protein sequences and is coloured blue.
(1) Query sequence PfCYC1 identified a potentially homologous gene product fragment of 141 aa, which we named EtCYC1. It was not possible to identify the full-length sequence of this hypothetical protein fragment.
(2) Query sequence PfCYC2 led to no results, i.e. no E. tenella gene homologous to the corresponding Pfcyc2 gene could be found.
(3) Query sequence PfCYC3 led to two significant hits, which we named EtCYC3a and EtCYC3b. While the full-length (358 aa) EtCYC3a sequence was available, only a partial sequence for EtCYC3b (138 aa) could be retrieved. The two sequences EtCYC3a and EtCYC3b share a distinct sequence identity of 66 % at the protein level over the whole available length of the EtCYC3b sequence. However, because the available EtCYC3b sequence was only about one-third the length of the corresponding EtCYC3a sequence and because the N-terminal end as well as the C-terminal end of the sequence were missing, we pursued our investigation using the full-length sequence only. The molecular size of the EtCYC3a protein was predicted to be 38 kDa.
(4) Query sequence PfCYC4 identified a full-length sequence (420 aa) which we named EtCYC4. The molecular size of this potential protein was predicted to be 44 kDa.
The four hypothetical proteins and protein fragments EtCYC1, EtCYC3a, EtCYC3b and EtCYC4 were subjected to an InterproScan analysis. A cyclin-box motif which forms the first of five α-helical repeats of the canonical cyclin fold (Jeffrey et al., 1995) was identified for each of them (Fig. 1b). Other typical cyclin motifs such as the MRAIL motif, the RXL motif and the destruction box (Russo et al., 1996; Schulman et al., 1998; Morgan, 1997) were not found in any of the hypothetical proteins.
PfCYC4 is unable to activate PfPK5 in vitro and has been proposed to have roles distinct from cell cycle regulation (Nairn & Greengard, 2001; Merckx et al., 2003). Although PfCYC4 and EtCYC4 are homologous to each other, an orthologous relation of the two proteins has not been proven so far. Nevertheless, in the absence of other guides to a decision, we discounted EtCYC4 from our analysis and concentrated on EtCYC1 and EtCYC3a as being potential activating partners of EtCRK2. As we were unable to access full-length EtCYC1, we focused attention on EtCYC3a as the potential activating partner of EtCRK2.
In vitro activation of EtCRK2
Activation of EtCRK2 by XlRINGO.
XlRINGO is known to be able to fully activate mammalian CDKs (Karaiskou et al., 2001) as well as PfPK5 (Merckx et al., 2003; Nebreda 2006). Therefore, we tested XlRINGO as an activator of EtCRK2. Using a TR-FRET assay with a fluorescein-labelled histone H1 substrate we determined the Km value for ATP to be 9.39±1.21 µM (data not shown) and its specific activity to be 1.29±0.24 µmol min−1 mg−1 (Fig. 2a). From these data we conclude that XlRINGO is able to activate EtCRK2.
Fig. 2.

Comparison of the activity and inhibition of EtCRK2–His activated either by MBP–EtCYC3a or by MBP–XlRINGO. (a) Both activator proteins were able to activate EtCRK2 in a similar manner. Values are depicted as mean±sd EtCRK2 activity values from at least four independent experiments (n≥4). (b) Percentage activity of the two protein–activator complexes in the presence of increasing concentrations of 10-Z-hymenialdisine. Eleven inhibitor concentrations from 0.02 to 200 µM were tested in the TR-FRET assay. Points represent mean values±sd from at least three independent experiments (n≥3). The IC50 values of the inhibitor for EtCRK2/XlRINGO and EtCRK2/EtCYC3a were calculated to be 0.44±0.08 and 0.93±0.40 µM, respectively.
Activation of EtCRK2 by EtCYC3a.
To further study the biochemical properties of EtCYC3a and its ability to activate EtCRK2, we cloned the entire coding region into the pMALc2X vector, allowing for the expression of an EtCYC3a–MBP fusion protein (MBP–EtCYC3a) in Escherichia coli. Cells expressing MBP–EtCYC3a and cells expressing EtCRK2–His were pooled and disrupted together, and the cell lysate was incubated to allow for the formation of an EtCRK2–His/MBP–EtCYC3a complex. The resulting complex was co-purified by amylase affinity chromatography using the MBP-tag of EtCYC3a. If EtCRK2–His and MBP–EtCYC3a did indeed form a stable complex, this affinity chromatography step should have retained the protein complex as well as monomeric MBP–EtCYC3a, while all other components not having an MBP-tag should have been removed in the wash. A gel electrophoresis under denaturing conditions (SDS-PAGE) of the eluate fraction showed multiple proteins, including two proteins at 32 and 80 kDa, corresponding to the sizes expected for EtCRK2–His and MBP–EtCYC3a, respectively (Fig. 3, lane 1, arrows). Size exclusion chromatography was then performed and the 112 kDa complex purified (Fig. 3, lane 2). Proteins between 60 and 80 kDa correspond most likely to degradation products of MBP–EtCYC3a.
Fig. 3.
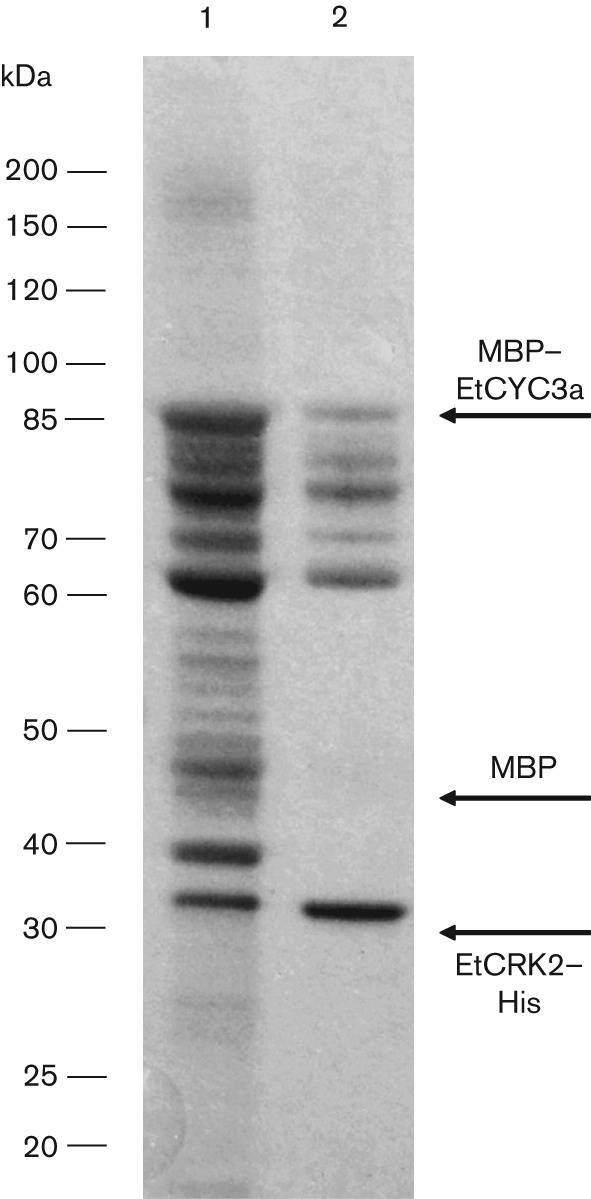
SDS-PAGE of the co-purification of the EtCRK2–His/MPB–EtCYC3a complex. Lane 1, amylase affinity chromatography, protein complex eluate fraction; lane 2, size exclusion chromatography, eluate fraction above 100 kDa.
The purified EtCRK2–His/MBP–EtCYC3a (EtCRK2/EtCYC3a) complex was active in the TR-FRET assay. EtCYC3a, like XlRINGO, was able to fully activate EtCRK2 (Fig. 2a). Moreover, both complexes showed comparable IC50 and Ki values for standard inhibitors (Fig. 2b, Table 1). The Km value for ATP of EtCRK2 in the EtCRK2/EtCYC3a complex was 9.05±0.76 µM (n = 3) and is very similar to the Km value for ATP of EtCRK2 in the EtCRK2/XlRINGO complex (Table 2). However, its specific activity in the EtCRK2/EtCYC3a complex of 0.15±0.02 µmol min−1 mg−1 is about nine times lower than its specific activity of 1.29±0.24 µmol min−1 mg−1 in the EtCRK2/XlRINGO complex (Table 2). This is in good agreement with the findings of Merckx et al. (2003) that XlRINGO is a stronger activator for PfPK5 than P. falciparum cyclins. We therefore hypothesize that EtCYC3a is a natural activator of EtCRK2. Whether EtCYC1, EtCYC3b and EtCYC4 are also able to activate EtCRK2 has to be proven in future experiments.
Table 1. Comparison of IC50 and Ki values of standard CDK inhibitors.
Results are expressed as mean values±sd of at least three independent experiments (n = 3–6). See also Fig. 2(b).
| CDK inhibitor | IC50 (nM) | Ki (nM) | ||||
| EtCRK2/XlRINGO | EtCRK2/EtCYC3a | HsCDK2/HsCYCA | EtCRK2/XlRINGO | EtCRK2/EtCYC3a | HsCDK2/HsCYCA | |
| Staurosporine | 90±10 | 190±90 | 3±1 | 20±10 | 530±200 | 1±0 |
| Indirubin-5′-sulfonic acid | 670±180 | 940±310 | 230±110 | 170±30 | 240±100 | 80±10 |
| Purvalanol A | 800±110 | 1030±180 | 140±40 | 80±10 | 360±130 | 40±1 |
| Alsterpaullone | 220±60 | 640±340 | 160±80 | 70±2 | 210±40 | 60±0 |
| Flavopiridole | 40±10 | 260±70 | 50±10 | 10±3 | 260±20 | 20±4 |
| 10-Z-Hymendialdisine | 440±80 | 930±40 | 120±20 | 50±20 | 190±10 | 30±1 |
Table 2. Comparison of Km values and specific activity of EtCRK2 in the two protein–activator complexes.
Results are expressed as mean values±sd of at least three independent experiments (n = 3).
| Parameter | EtCRK2/XlRINGO | EtCRK2/EtCYC3a |
| Specific activity (µmol min−1 mg−1) | 1.29±0.24 | 0.15±0.02 |
| Km for ATP (µM) | 9.39±1.21 | 9.05±0.76 |
| Km for fluorescein-labelled substrate (µM) | 1.72±1.10 | 2.05±1.18 |
Inhibition assays using EtCRK2 /XlRINGO and EtCRK2/EtCYC3a complexes
In order to discover specific inhibitors of EtCRK2 we performed an in vitro screening campaign supported by an in silico hit enrichment (Fig. 4). In a primary screening we assayed approximately 3500 compounds from a highly diverse excerpt of a vendor compound library on the active EtCRK2/XlRINGO complex. The hit threshold for this primary screen was set to 30 % inhibition at a concentration of 30 µM, achieving a hit rate of 1.5 % (53 primary hits). The primary hits were then confirmed in a secondary screen using the EtCRK2/XlRINGO complex as well as the EtCRK2/EtCYC3a complex, resulting in 23 compounds showing an IC50 value equal to or below 100 µM (IC50 ≤100 µM) on both EtCRK2 complexes. For the final confirmation step, the 23 compounds were freshly dissolved from solid stock and tested on both EtCRK2 complexes (IC50 ≤100 µM), and the structure of the compounds was verified using LC/MS and NMR. The activity and the structural integrity of 14 compounds were verified.
Fig. 4.

Filtering steps during in vitro screening and in silico hit enrichment. Three lead compounds, each being the most active representative of their respective cluster of structurally related compounds, were selected.
In order to allow for the discovery of additional compounds belonging to the same chemical classes we performed an in silico hit enrichment. Each of the 14 actives was used as a starting structure for a fingerprint-based similarity search using a Tanimoto similarity metric and a scaffold-based substructure search. The resulting virtual compound library of 2208 compounds was then screened using molecular docking and post processing as described by Engels et al. (2010), leading to 64 potential inhibitors of EtCRK2. These 64 compounds were purchased and tested in our in vitro set-up. Five compounds were confirmed to have an IC50 value below 100 µM (IC50 <100 µM) on both EtCRK2 complexes. Together with the 14 hits coming from the initial in vitro screen we found an overall hit set of 19 compounds. In order to identify compound families whose members belonged either to the same chemical class or to a common scaffold with similar pharmacophore features we performed a clustering based on the MDL Keys fingerprint. The clustering identified three compound clusters: the naphthoquinone cluster, the 8-hydroxyquinoline cluster, and the 2-pyrimidinyl-aminopiperidine-propane-2-ol cluster, which we named after the most active representative of the individual cluster. Naphthoquinones, 8-hydroxyquinolines as well as 2-pyrimidinyl-aminopiperidine-propane-2-ols (Table 3) have already been described in the literature as inhibitors of different kinases (Aziz et al., 2008, Al-Sha’er & Taha, 2010; Kim et al., 2011).
Table 3. Comparison of IC50 and Ki values of the lead structures (most active representatives of the three clusters).
Results are expressed as mean values±sd of at least three independent experiments (n = 3–6).
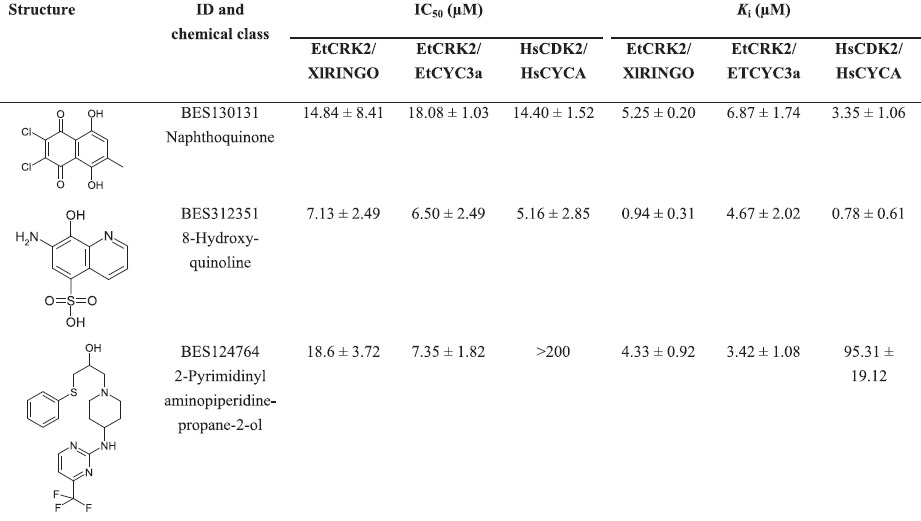
The most active representative of each of the three clusters was subjected to Ki value and IC50 determinations on both EtCRK2/XlRINGO and EtCRK2/EtCYC3a, as well as on the complex of human CDK2 activated by human cyclin A: HsCDK2/HsCYCA (Table 3). While the naphthoquinone representative and the 8-hydroxyquinoline representative displayed similar activities towards the EtCRK2 complexes and HsCDK2/HsCYCA, BES124764, the 2-pyrimidinyl-aminopiperidine-propane-2-ol representative, was approximately 25–30 times more active towards the two E. tenella complexes than towards the HsCDK2/HsCYCA complex. The Ki determination suggests ATP competitiveness, which in addition is backed by the molecular binding mode found in docking experiments (Fig. 5). From its molecular binding mode, BES124764 is an atypical kinase inhibitor forming no H-bonds to the hinge region residues (Fig. 5). Moreover, the selectivity site E88K reported by Engels et al. (2010) is not addressed by this inhibitor, and it is therefore not clear how the selectivity for the eimerian enzyme is conferred. Therefore, we will focus future work towards the molecular mode of action of BES124764 and its derivatives.
Fig. 5.
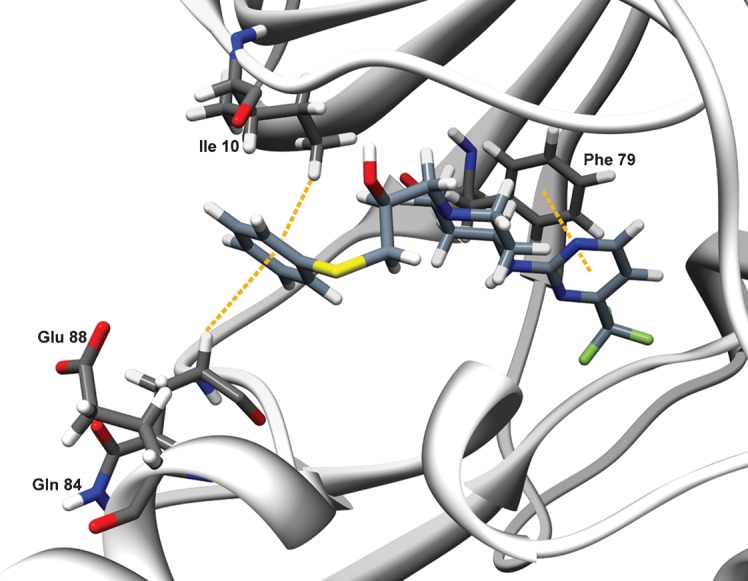
Top ranked docking solution for BES124764. BES124764 is docked into the predicted ATP binding pocket of EtCRK2. Arene–H interactions to Gln84 and Ile10 and the arene–arene interaction to Phe79 are shown as dashed orange lines.
Conclusion
EtCRK2 is a valid drug target for the apicomplexan parasite E. tenella (Engels et al., 2010). Although those authors were able to assay EtCRK2 using the non-native activator XlRINGO, the native activator of EtCRK2 was not known. Therefore, one part of our work was devoted to the identification of the putative native activator of EtCRK2. Using bioinformatics and molecular biology methods we successfully identified a number of potential cyclin-like proteins which could be the native activators. The most promising of these proteins, EtCYC3a, was studied further and it was proven that it is able to activate EtCRK2.
In a subsequent in vitro screening on EtCRK2/XlRINGO and EtCRK2/EtCYC3a supported by an in silico hit enrichment, we were able to identify 19 active compounds. We subsequently identified three clusters of structurally related compounds, which we named after the most active representative of the cluster as the naphthoquinone cluster, 8-hydroxyquinoline cluster and 2-pyrimidinyl-aminopiperidine-propane-2-ol cluster. The most active representative of each of the three clusters was picked as a lead structure and was assayed for its selectivity for EtCRK2 versus HsCDK2. It turned out that BES124764, the representative of the 2-pyrimidinyl-aminopiperidine-propane-2-ol cluster, was selective for EtCRK2. Therefore, BES124764 and derivatives thereof will be introduced into lead optimization, the next step of the stage-gated drug discovery process as described by Rohwer et al. (2011). In the next steps of this process, the compounds will be chemically modified and carefully evaluated for their specificity against several CDK and CDK-like proteins of mammalians, and for their activity towards parasites in cell culture and in vivo.
Acknowledgements
This work was supported by the ‘German Academic Exchange Service’ (DAAD) and the Spanish foundation ‘La Caixa’ research fellowships. J. C. M. was supported by an SRDG grant (HR04013) from the Scottish Funding Council. The Wellcome Trust Centre for Molecular Parasitology is supported by core funding from the Wellcome Trust (085349). Our special thanks to Dr Jane H. Kinnaird (University of Glasgow, UK) and Dr Ario de Marco (European Molecular Biology Laboratory, Heidelberg, Germany) for kindly providing the plasmids pQE60-EtCRK2 and pMALc2X-XlRINGO, respectively. We are grateful to Dr Christian Schorn for the quality assessment of the compounds.
Abbreviations:
- CDK
cyclin-dependent kinase
- EtCRK2
E. tenella CDC2-related kinase 2
- EtCYC
E. tenella cyclin
- LC/MS
liquid chromatography/mass spectrometry
- MBP
maltose binding protein
- PfCYC
Plasmodium falciparum cyclin
- TR-FRET
time-resolved fluorescence energy transfer
- XlRINGO
Xenopus laevis rapid inducer of G2/M progression in oocytes
References
- Al-Sha’er M. A., Taha M. O. (2010). Discovery of novel CDK1 inhibitors by combining pharmacophore modeling, QSAR analysis and in silico screening followed by in vitro bioassay. Eur J Med Chem 45, 4316–4330. 10.1016/j.ejmech.2010.06.034 [DOI] [PubMed] [Google Scholar]
- Aziz M. H., Dreckschmidt N. E., Verma A. K. (2008). Plumbagin, a medicinal plant-derived naphthoquinone, is a novel inhibitor of the growth and invasion of hormone-refractory prostate cancer. Cancer Res 68, 9024–9032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno S., Ardelt B., Skierski J. S., Traganos F., Darzynkiewicz Z. (1992). Different effects of staurosporine, an inhibitor of protein kinases, on the cell cycle and chromatin structure of normal and leukemic lymphocytes. Cancer Res 52, 470–473. [PubMed] [Google Scholar]
- Chapman H. D. (1997). Biochemical, genetic and applied aspects of drug resistance in Eimeria parasites of the fowl. Avian Pathol 26, 221–244. 10.1080/03079459708419208 [DOI] [PubMed] [Google Scholar]
- Chauhan H. V. S., Roy S. (2007). Poultry Diseases, Diagnosis and Treatment, 3rd edn New Delhi: New Age Publications. [Google Scholar]
- Dai Y., Grant S. (2003). Cyclin-dependent kinase inhibitors. Curr Opin Pharmacol 3, 362–370. 10.1016/S1471-4892(03)00079-1 [DOI] [PubMed] [Google Scholar]
- Dalloul R. A., Lillehoj H. S. (2006). Poultry coccidiosis: recent advancements in control measures and vaccine development. Expert Rev Vaccines 5, 143–163. 10.1586/14760584.5.1.143 [DOI] [PubMed] [Google Scholar]
- Doerig C., Chakrabarti D., Kappes B., Matthews K. (2000). The cell cycle in protozoan parasites. Prog Cell Cycle Res 4, 163–183. 10.1007/978-1-4615-4253-7_15 [DOI] [PubMed] [Google Scholar]
- Doerig C., Meijer L., Mottram J. C. (2002). Protein kinases as drug targets in parasitic protozoa. Trends Parasitol 18, 366–371. 10.1016/S1471-4922(02)02321-8 [DOI] [PubMed] [Google Scholar]
- Engels K., Beyer C., Suárez Fernández M. L., Bender F., Gaßel M., Unden G., Marhöfer R. J., Mottram J. C., Selzer P. M. (2010). Inhibition of Eimeria tenella CDK-related kinase 2: from target identification to lead compounds. ChemMedChem 5, 1259–1271. 10.1002/cmdc.201000157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferby I., Blazquez M., Palmer A., Eritja R., Nebreda A. R. (1999). A novel p34(cdc2)-binding and activating protein that is necessary and sufficient to trigger G(2)/M progression in Xenopus oocytes. Genes Dev 13, 2177–2189. 10.1101/gad.13.16.2177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann J., Raether W. (1990). Improved techniques for the in vitro cultivation of Eimeria tenella in primary chick kidney cells. Parasitol Res 76, 479–486. 10.1007/BF00931053 [DOI] [PubMed] [Google Scholar]
- Jeffrey P. D., Russo A. A., Polyak K., Gibbs E., Hurwitz J., Massagué J., Pavletich N. P. (1995). Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature 376, 313–320. 10.1038/376313a0 [DOI] [PubMed] [Google Scholar]
- Karaiskou A., Perez L. H., Ferby I., Ozon R., Jessus C., Nebreda A. R. (2001). Differential regulation of Cdc2 and Cdk2 by RINGO and cyclins. J Biol Chem 276, 36028–36034. 10.1074/jbc.M104722200 [DOI] [PubMed] [Google Scholar]
- Kim S., Jung J. K., Lee H. S., Kim Y., Kim J., Choi K., Baek D.-J., Moon B., Oh K.-S. & other authors (2011). Discovery of piperidinyl aminopyrimidine derivatives as IKK-2 inhibitors. Bioorg Med Chem Lett 21, 3002–3006. 10.1016/j.bmcl.2011.03.044 [DOI] [PubMed] [Google Scholar]
- Kinnaird J. H., Bumstead J. M., Mann D. J., Ryan R., Shirley M. W., Shiels B. R., Tomley F. M. (2004). EtCRK2, a cyclin-dependent kinase gene expressed during the sexual and asexual phases of the Eimeria tenella life cycle. Int J Parasitol 34, 683–692. 10.1016/j.ijpara.2004.01.003 [DOI] [PubMed] [Google Scholar]
- Le Roch K., Sestier C., Dorin D., Waters N., Kappes B., Chakrabarti D., Meijer L., Doerig C. (2000). Activation of a Plasmodium falciparum cdc2-related kinase by heterologous p25 and cyclin H. Functional characterization of a P. falciparum cyclin homologue. J Biol Chem 275, 8952–8958. 10.1074/jbc.275.12.8952 [DOI] [PubMed] [Google Scholar]
- Malumbres M., Barbacid M. (2009). Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 9, 153–166. 10.1038/nrc2602 [DOI] [PubMed] [Google Scholar]
- Mehlhorn H. (2008). Encyclopedia of Parasitology, 3rd edn Berlin: Springer; 10.1007/978-3-540-48996-2 [DOI] [Google Scholar]
- Merckx A., Le Roch K., Nivez M. P., Dorin D., Alano P., Gutierrez G. J., Nebreda A. R., Goldring D., Whittle C. & other authors (2003). Identification and initial characterization of three novel cyclin-related proteins of the human malaria parasite Plasmodium falciparum. J Biol Chem 278, 39839–39850. 10.1074/jbc.M301625200 [DOI] [PubMed] [Google Scholar]
- Monaco E. A., III, Vallano M. L. (2003). Cyclin-dependent kinase inhibitors: cancer killers to neuronal guardians. Curr Med Chem 10, 367–379. [DOI] [PubMed] [Google Scholar]
- Morgan D. O. (1997). Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol 13, 261–291. 10.1146/annurev.cellbio.13.1.261 [DOI] [PubMed] [Google Scholar]
- Morrison D. A. (2009). Evolution of the Apicomplexa: where are we now? Trends Parasitol 25, 375–382. 10.1016/j.pt.2009.05.010 [DOI] [PubMed] [Google Scholar]
- Nairn A. C., Greengard P. (2001). A novel cyclin provides a link between dopamine and RNA processing. Neuron 32, 174–176. 10.1016/S0896-6273(01)00469-X [DOI] [PubMed] [Google Scholar]
- Nebreda A. R. (2006). CDK activation by non-cyclin proteins. Curr Opin Cell Biol 18, 192–198. 10.1016/j.ceb.2006.01.001 [DOI] [PubMed] [Google Scholar]
- Olashaw N., Pledger W. J. (2002). Paradigms of growth control: relation to Cdk activation. Sci STKE 2002, re7. 10.1126/stke.2002.134.re7 [DOI] [PubMed] [Google Scholar]
- Rohwer A., Marhöfer R. J., Caffrey C. R., Selzer P. M. (2011). Drug discovery approaches toward anti-parasitic agents. In Drug Discovery in Infectious Diseases, Apicomplexan Parasites – Molecular Approaches toward Targeted Drug Development, pp. 3–20. Edited by Becker K., Selzer P. M. Weinheim: Wiley-VCH; 10.1002/9783527633883.ch1 [DOI] [Google Scholar]
- Russo A. A., Jeffrey P. D., Pavletich N. P. (1996). Structural basis of cyclin-dependent kinase activation by phosphorylation. Nat Struct Biol 3, 696–700. 10.1038/nsb0896-696 [DOI] [PubMed] [Google Scholar]
- Schang L. M., St Vincent M. R., Lacasse J. J. (2006). Five years of progress on cyclin-dependent kinases and other cellular proteins as potential targets for antiviral drugs. Antivir Chem Chemother 17, 293–320. [DOI] [PubMed] [Google Scholar]
- Schulman B. A., Lindstrom D. L., Harlow E. (1998). Substrate recruitment to cyclin-dependent kinase 2 by a multipurpose docking site on cyclin A. Proc Natl Acad Sci U S A 95, 10453–10458. 10.1073/pnas.95.18.10453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirley M. W. (2000). The genome of Eimeria spp., with special reference to Eimeria tenella—a coccidium from the chicken. Int J Parasitol 30, 485–493. 10.1016/S0020-7519(99)00183-6 [DOI] [PubMed] [Google Scholar]
- Shirley M. W., Bedrník P. (1997). Live attenuated vaccines against avian coccidiosis: success with precocious and egg-adapted lines of Eimeria. Parasitol Today 13, 481–484. 10.1016/S0169-4758(97)01153-8 [DOI] [PubMed] [Google Scholar]
- Shirley M. W., Smith A. L., Tomley F. M. (2005). The biology of avian Eimeria with an emphasis on their control by vaccination. Adv Parasitol 60, 285–330. 10.1016/S0065-308X(05)60005-X [DOI] [PubMed] [Google Scholar]
- Shirley M. W., Smith A. L., Blake D. P. (2007). Challenges in the successful control of the avian coccidia. Vaccine 25, 5540–5547. 10.1016/j.vaccine.2006.12.030 [DOI] [PubMed] [Google Scholar]
- Sielecki T. M., Boylan J. F., Benfield P. A., Trainor G. L. (2000). Cyclin-dependent kinase inhibitors: useful targets in cell cycle regulation. J Med Chem 43, 1–18. 10.1021/jm990256j [DOI] [PubMed] [Google Scholar]
- Tamamori M., Ito H., Hiroe M., Terada Y., Marumo F., Ikeda M. A. (1998). Essential roles for G1 cyclin-dependent kinase activity in development of cardiomyocyte hypertrophy. Am J Physiol 275, H2036–H2040. [DOI] [PubMed] [Google Scholar]
- Wallach M. G., Ashash U., Michael A., Smith N. C. (2008). Field application of a subunit vaccine against an enteric protozoan disease. PLoS ONE 3, e3948. 10.1371/journal.pone.0003948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters N. C., Geyer J. A. (2003). Cyclin-dependent protein kinases as therapeutic drug targets for antimalarial drug development. Expert Opin Ther Targets 7, 7–17. 10.1517/14728222.7.1.7 [DOI] [PubMed] [Google Scholar]
- Williams R. B., Carlyle W. W., Bond D. R., Brown I. A. (1999). The efficacy and economic benefits of Paracox, a live attenuated anticoccidial vaccine, in commercial trials with standard broiler chickens in the United Kingdom. Int J Parasitol 29, 341–355. 10.1016/S0020-7519(98)00212-4 [DOI] [PubMed] [Google Scholar]