

Abstract
Phenolic glycolipids (PGLs) are non-covalently bound components of the outer membrane of many clinically relevant mycobacterial pathogens, and play important roles in pathogen biology. We report a mutational analysis that conclusively demonstrates that the conserved acyltransferase-encoding gene papA5 is essential for PGL production. In addition, we provide an in vitro acyltransferase activity analysis that establishes proof of principle for the competency of PapA5 to utilize diol-containing polyketide compounds of mycobacterial origin as acyl-acceptor substrates. Overall, the results reported herein are in line with a model in which PapA5 catalyses the acylation of diol-containing polyketides to form PGLs. These studies advance our understanding of the biosynthesis of an important group of mycobacterial glycolipids and suggest that PapA5 might be an attractive target for exploring the development of antivirulence drugs.
Introduction
The mycobacterial cell envelope contains a distinctive array of complex free lipids and glycolipids associated with the mycolic acid layer of the cell wall (Brennan & Nikaido, 1995; Crick et al., 2008; Onwueme et al., 2005a). These lipids and glycolipids are thought to be located in the membrane outer leaflet that partners with the mycolic acid-based membrane inner leaflet to form the asymmetric lipid bilayer that constitutes the characteristic mycobacterial outer membrane. Among the free lipids found in the outer membrane of Mycobacterium leprae, various strains of Mycobacterium tuberculosis and several opportunistic mycobacterial human pathogens are two structurally related groups of diesters, both composed of glycol-containing long-chain aliphatic polyketides and polyketide synthase-derived methyl-branched fatty acids (Onwueme et al., 2005a). One of these groups of unusual lipid diesters is represented by phenolphthiocerol dimycocerosates, which are glycosylated compounds generally known as phenolic glycolipids (PGLs). The other group is represented by phthiocerol dimycocerosates (PDIMs), which lack the aromatic ring and glycosidic moieties characteristic of PGLs (Fig. 1).
Fig. 1.

Representative structures of mycobacterial PGLs and PDIMs.
An overwhelming body of evidence has accumulated that demonstrates that PGLs and PDIMs play important roles in host–pathogen interaction and/or virulence in various mycobacterial pathogens (Alibaud et al., 2011; Astarie-Dequeker et al., 2009; Brodin et al., 2010; Camacho et al., 1999; Collins et al., 2005; Cox et al., 1999; Dhungel et al., 2008; Murry et al., 2009; Ng et al., 2000; Rambukkana et al., 2002; Reed et al., 2004; Robinson et al., 2008; Ruley et al., 2004; Sinsimer et al., 2008; Tsenova et al., 2005). Moreover, production of these compounds has been shown to correlate with decreased antimicrobial drug susceptibility (Alibaud et al., 2011; Chavadi et al., 2011b). PGL production has also been suggested as a trait that predisposes M. tuberculosis strains of the W-Beijing family to their characteristic epidemic spread and increased likelihood of developing drug resistance (Reed et al., 2007). Thus, dissecting the molecular logic of PGL and PDIM biosynthesis not only will expand our general understanding of cell wall biosynthesis in mycobacteria of clinical significance, but also may reveal potential avenues for exploring the development of alternative therapeutics against selected mycobacterial infections (Ferreras et al., 2008; Quadri, 2007). These views have directed our previous studies of the biosynthesis of PGLs and PDIMs (Buglino et al., 2004; Chavadi et al., 2011b; Ferreras et al., 2008; He et al., 2009; Onwueme et al., 2004, 2005a, b), which include the development of the first PGL biosynthesis inhibitor (Ferreras et al., 2008). This inhibitor drastically reduces PGL production in several Mycobacterium species (Ferreras et al., 2008) using a mechanistic principle analogous to that of the first reported inhibitor of mycobacterial siderophore (iron chelator) production (Ferreras et al., 2005; Quadri, 2007).
Despite significant progress made towards understanding the partially overlapping biosynthetic routes of PGLs and PDIMs (Onwueme et al., 2005a), many aspects of these pathways remain unclear. One such aspect is whether the conserved gene papA5 is required for PGL production. An orthologue of papA5 is clustered with genes that have confirmed or suspected involvement in PGL and/or PDIM production in each PDIM/PGL producer for which the genome has been analysed (Onwueme et al., 2005a). We have previously established that papA5 is required for PDIM production, demonstrated that PapA5 has acyltransferase activity and reported the crystal structure of PapA5 from M. tuberculosis (Buglino et al., 2004; Onwueme et al., 2004). The earlier mutational work involved the characterization of a papA5 knockout engineered in the Erdman strain of M. tuberculosis, a strain that is PGL-deficient due to a natural mutation in a polyketide synthase gene required for PGL synthesis (Constant et al., 2002; He et al., 2009). The study of PapA5 was conducted using non-physiological aliphatic alcohols as surrogate substrates. Thus, it remains unknown whether papA5 is required for PGL production, and demonstration of the ability of PapA5 to utilize mycobacterial alcohols as acyl acceptor substrates is lacking.
In this study, we report a mutational analysis that conclusively establishes that papA5 is required for production of PGLs in the opportunistic human pathogen Mycobacterium marinum. We also present in vitro biochemical evidence to demonstrate that PapA5 has the capacity to utilize mycobacterial glycosyl-phenolphthiocerols as acyl acceptor substrates. Overall, the studies reported herein advance our understanding of the biosynthesis of an important group of mycobacterial cell wall glycolipids.
Methods
Chemicals and reagents.
Solvents and non-radiolabelled chemical reagents were acquired from Sigma-Aldrich. [1-14C]Palmitoyl-CoA thioester (specific activity 55–57 mCi mmol−1; 2.04–2.11 GBq mmol−1) was purchased from Perkin Elmer, Amersham Biosciences or American Radiolabelled Chemicals (ARC). [1-14C]-propionate (specific activity 54 mCi mmol−1; 2.00 GBq mmol−1) was acquired from ARC. Molecular biology reagents were obtained from Sigma, Invitrogen, New England Biolabs, Novagen, Qiagen or Stratagene.
Bacterial culturing and recombinant DNA manipulations.
M. marinum (strain M; ATCC BAA-535) was cultured at 30 °C in Middlebrook 7H9 medium (Difco) supplemented with 10 % ADN (5 % BSA, 2 % glucose, 0.85 % NaCl) (Difco) and 0.05 % Tween-80 (supplemented 7H9) or Middlebrook 7H11 (Difco) supplemented with ADN (supplemented 7H11) (Chavadi et al., 2011b). Escherichia coli DH5α (Invitrogen) was cultured in Luria–Bertani media under standard conditions (Sambrook et al., 1989). When required, kanamycin (30 µg ml−1), hygromycin (50 µg ml−1), sucrose (2 %) and/or X-Gal (70 µg ml−1) were added to the media. General recombinant DNA manipulations were carried out by standard methods and using E. coli DH5α as the primary cloning host (Sambrook et al., 1989). PCR-generated DNA fragments used in plasmid constructions were sequenced to verify fidelity. Genomic DNA isolation from and plasmid electroporation into mycobacteria were carried out as reported elsewhere (Parish & Stoker, 1998). The plasmids and oligonucleotides used in this study are shown in Table 1.
Table 1. Plasmids and oligonucleotide primers used in this study.
Restriction sites are underlined.
| Plasmid | Characteristics | Source or reference |
| pCR2.1Topo | Cloning vector, kanamycin and ampicillin resistance | Invitrogen |
| pCP0 | E. coli–mycobacteria shuttle vector for gene expression in mycobacteria, kanamycin resistance | Ferreras et al. (2008) |
| pCP0-papA5mm | pCP0 expressing M. marinum papA5 (papA5mm) | This study |
| p2NIL | Mutagenesis vector, kanamycin resistance | Parish & Stoker (2000) |
| pGOAL19 | Mutagenesis vector, hygromycin resistance, sacB–lacZ PacI cassette | Parish & Stoker (2000) |
| p2NIL-GOALc-ΔpapA5c | Suicide delivery vector carrying papA5 deletion cassette (ΔpapA5c) | This study |
| Oligonucleotide | Sequence (5′–3′) | Characteristics |
| papA5OF | AAGCTTATCCTGGCGGACGCGGTTCGGATTCTGTTCA | HindIII site |
| papA5IR | CGCAGGTCATTCCATGAACACGTTAAGACTCTCCGTTCT | SOE primer |
| papA5IF | AGTCTTAACGTGTTCATGGAATGACCTGCGCCTGCG | SOE primer |
| papA5OR | TTAATTAAGTGGGCTGAGATGTTCCAGTCCAGTCCAGCGGTCAA | PacI site |
| papA5F | CTGCAGGAAGAACGGAGAGTCTTAACGTGT | PstI site |
| papA5R | AAGCTTGCAGGCGCAGGTCATTCCATGAAC | HindIII site |
Construction of M. marinum ΔpapA5.
The mutant was engineered using the p2NIL/pGOAL19-based flexible cassette method (Parish & Stoker, 2000) as reported previously (Chavadi et al., 2011a, b; Ferreras et al., 2008; Onwueme et al., 2004). A suicide delivery vector (p2NIL-GOALc-ΔpapA5c, see below) carrying a papA5 (MMAR_1768) deletion cassette (ΔpapA5c) was used to generate M. marinum ΔpapA5. Electroporation of p2NIL-GOALc-ΔpapA5c into M. marinum and selection of potential single- and double-crossover mutants were conducted as previously reported (Chavadi et al., 2011b). The papA5 deletion in potential double-crossover mutants was screened for and confirmed by PCR using two independent primer pairs (papA5OF and papA5OR, papA5F and papA5R).
Construction of p2NIL-GOALc-ΔpapA5c.
The construction of the plasmid is outlined in Supplementary Fig. S1 (available with the online version of this paper). The deletion cassette ΔpapA5c was generated using splicing by overlap extension (SOE) PCR (Horton et al., 1989). ΔpapA5c contained a 5′ arm (485 bp = 479 bp segment upstream of papA5+papA5 first two codons) and a 3′ arm (554 bp = papA5 last two codons+stop codon+545 bp segment downstream of papA5). Each arm was PCR-generated from genomic DNA. Primer pair papA5OF and papA5IR and primer pair papA5IF and papA5OR were used to generate the 5′ and 3′ arms, respectively. The arms were then used as a template for PCR with primers papA5OF and papA5OR to fuse the arms and generate ΔpapA5c (1039 bp). The PCR-generated ΔpapA5c was cloned into pCR2.1Topo (Invitrogen). ΔpapA5c was subsequently excised from the pCR2.1Topo construct using HindIII and PacI, and the excerpt was ligated to plasmid p2NIL (Parish & Stoker, 2000) linearized by HindIII/PacI digestion. The resulting p2NIL-ΔpapA5c plasmid and plasmid pGOAL19 (Parish & Stoker, 2000) were digested with PacI, and the PacI cassette (GOALc, 7939 bp) of pGOAL19 was ligated to the linearized p2NIL-ΔpapA5c backbone to create p2NIL-GOALc-ΔpapA5c.
Construction of pCP0-papA5mm.
A DNA fragment (1275 bp) encompassing papA5 of M. marinum (papA5mm) and its predicted ribosome-binding site (RBS) was PCR-generated from genomic DNA using primer pair papA5F (starts and ends 20 nt upstream and 1 nt downstream, respectively, of the start codon) and papA5R (starts and ends 11 nt upstream and 10 nt downstream, respectively, of the stop codon). The PCR product was cloned into pCR2.1Topo. The RBS-papA5mm insert was recovered from the pCR2.1Topo construct as a PstI–HindIII fragment and the excerpt was subcloned into the expression vector pCP0 linearized by PstI/HindIII digestion. The cloned papA5mm was placed under the control of the hsp60 promoter of pCP0 for expression in mycobacteria.
Analysis of PGLs and PDIMs.
Five-day-old mycobacterial cultures were diluted to OD595 0.6 in supplemented 7H9 and loaded into 12-well plates (1 ml per well). [14C]Propionate was added to each well at 0.2 µCi ml−1 (7.4 MBq ml−1) and the plates were incubated at 30 °C with rotary agitation (170 r.p.m.) for 24 h. After incubation, the OD595 of the cultures was measured in a DTX Plate Reader (Beckman Coulter), and the cells were harvested for apolar lipid fraction extraction with a biphasic mixture of methanolic saline and petroleum ether, as reported previously (Chavadi et al., 2011b; Ferreras et al., 2008). The lipid extracts were subjected to radio-TLC for analysis of 14C-labelled PGLs (using CHCl3/CH3OH, 95 : 5) and PDIMs (using petroleum ether/diethyl ether, 9 : 1) as described previously (Ferreras et al., 2008). Developed TLC plates were exposed to phosphor screens, which were scanned using a Cyclone Plus Storage Phosphor System (PerkinElmer Life and Analytical Sciences).
Purification of GPPOLs.
PGLs were purified from M. leprae-infected Armadillo tissues. Briefly, homogenized M. leprae infected liver (10 g) supernatants were lyophilized and extracted with CHCl3/CH3OH (2 : 1) at 50 °C for 18 h. The homogenate was centrifuged and the organic layer (lower phase) was collected. This organic layer served as the main source of PGLs. The organic layer was washed with water, concentrated and extracted with diethyl ether. The ether-soluble dried lipids were resuspended in CHCl3, applied to a silica gel G60 column (2.5×60 cm), and successively eluted from the column with CHCl3 containing increasing concentrations of CH3OH (0–20 %). Most of the PGL eluted with CHCl3 containing 2 % and 5 % CH3OH. The pure PGL preparation was obtained after normal-phase HPLC of the silica gel-purified material. Fractions off HPLC were screened for PGLs by TLC on aluminium-backed silica gel G plates (Merck) developed with CHCl3/CH3OH/H2O (90 : 10 : 1). The identity of the purified PGLs was confirmed using fast atom bombardment MS (results not shown) as reported elsewhere (Brennan et al., 1994). The purified PGLs were deacylated by procedures based on those described elsewhere (Hunter & Brennan, 1981). PGLs were hydrolysed with 10 % NaOH in toluene/CH3OH (1 : 2) in a sealed tube under N2 at 100 °C for 18 h. Lipids were extracted with CHCl3 from the water-diluted, acidified (pH 4) mixture. After two washes with water, the CHCl3 fraction was applied to a column of silicic acid/celite (2 : 1) (1×30 cm), which was irrigated first with CHCl3 to remove fatty acids and then with CHCl3 containing 5 % CH3OH to elute GPPOLs. The identity of the purified GPPOLs was confirmed by MS analysis as indicated below.
GPPOL palmitoylation assay.
Reactions for assessment of PapA5-dependent GPPOL palmitoylation contained 18 µM [14C]palmitoyl-CoA, 450 µM purified GPPOLs, 10 µM enzyme, 100 mM NaCl and 75 mM MES buffer (pH 6.2), and were incubated at 37 °C for 24 h. Negative control reactions contained the inactive PapA5 variant PapH124A (Onwueme et al., 2004) or an equivalent volume of enzyme buffer (100 mM NaCl, 75 mM MES, pH 6.2). After incubation, the reactions were quenched by addition of CHCl3/toluene/CH3OH (6 : 4 : 2) followed by vigorous mixing. The organic layers were recovered and analysed by TLC on aluminium-backed, 250 µm thick, silica gel plates (EM Science or Whatman). TLC plates were developed with CHCl3/CH3OH/H2O (90 : 10 : 1) and exposed to phosphor screens, and reaction products were quantified with a Storm 860 Imaging System (Molecular Dynamics). For dose–response experiments, reactions contained GPPOLs at the indicated concentrations, 18 µM [14C]palmitoyl-CoA, 0.5 µM PapA5, 100 mM NaCl and 75 mM MES, pH 6.5. After incubation (37 °C, 3 h), the reactions were quenched, extracted and analysed by radio-TLC as noted above. Dose–response data were fitted to the equation v = Vmax/[1+Km/S+S/Ki] to calculate apparent kinetic parameters and an apparent Ki value. Data were plotted and analysed using KaleidoGraph software. Recombinant PapA5 and PapH124A were purified as reported previously (Buglino et al., 2004; Onwueme et al., 2004).
MS analysis of GPPOL palmitoylation products.
GPPOL palmitoylation reactions for MS analysis of reaction products contained unlabelled palmitoyl-CoA (360 µM), GPPOL (360 µM) and enzyme (90 µM), or an equivalent volume of the enzyme buffer described above in negative control reactions. Reactions were incubated at 37 °C for 24 h and then quenched and extracted as above. The organic extracts were analysed by MALDI-TOF MS. The analyte was mixed with matrix solution (10 mg 2,5-dihydroxybenzoic acid ml−1 in CHCl3/CH3OH, 1 : 1) and the resulting mix was applied onto the MALDI sample plate. Spectra were acquired on a Voyager-DE PRO 6320 instrument (Applied Biosystems). The instrument was calibrated using bradykinin fragment 1-7 (monoistopic mass 757.4), human angiotensin II (monoisotopic mass 1046.5) and a synthetic peptide P14R (monoisotopic mass 1533.8). The distributions in the spectra of the reactions were calibrated based on the main GPPOL species found in the purified substrate {observed [(M+Na+)]+ m/z = 1123.79, calculated [(M+Na+)]+ m/z = 1123.75}. Data were processed with the Data Explorer software package (Applied Biosystems).
Results and Discussion
Construction of a papA5 deletion mutant of M. marinum
We have previously demonstrated that papA5 is required for production of PDIMs (Onwueme et al., 2004). In this study, we sought to determine whether papA5 was required for production of PGLs. Towards this end, we utilized M. marinum as a prototype representative of mycobacteria that produce both PGLs and PDIMs. M. marinum is an opportunistic human pathogen closely related to M. tuberculosis (Stinear et al., 2008) and offers superior experimental tractability compared with other PGL-producing mycobacteria.
We engineered strain M. marinum ΔpapA5, an unmarked, in-frame papA5 deletion mutant of M. marinum, and examined the capacity of this strain to produce PGLs and PDIMs as described below. The deletion was engineered using the papA5 deletion cassette-delivery suicide vector p2NIL-GOALc-ΔpapA5c (Fig. 2a) in a homologous recombination- and counterselection-based approach that replaced papA5 by a 4-codon remnant engineered into the papA5 deletion cassette of p2NIL-GOALc-ΔpapA5c. The deletion in M. marinum ΔpapA5 encompassed 412 central codons of papA5, and it was verified by PCR using two independent primer pairs, each producing diagnostic amplicons of different sizes depending on whether the genomic DNA used as template was wild-type (WT) or carried the papA5 deletion (Fig. 2b). The successful engineering of M. marinum ΔpapA5 set the stage for probing the involvement of papA5 in PGL production.
Fig. 2.

Construction of M. marinum ΔpapA5. (a) Scheme of papA5 deletion cassette (ΔpapA5c)-delivery suicide vector used for construction of M. marinum ΔpapA5. The papA5 deletion leaves behind a gene remnant consisting of the first two (black type) and last two (white type) amino acids of PapA5. The gene remnant in ΔpapA5c is flanked by WT sequence for homologous recombination with the chromosome. (b) Agarose gel electrophoresis showing PCR-based confirmation of the papA5 deletion in M. marinum ΔpapA5. Lanes: 1, M. marinum WT (2270 bp amplicon expected with primers papA5OF and papA5OR); 2, M. marinum ΔpapA5 (1035 bp amplicon expected with papA5OF and papA5OR); 3, M. marinum WT (1275 bp amplicon expected with primers papA5F and papA5R); 4, M. marinum ΔpapA5 (no amplicon expected with papA5F and papA5R); L, DNA ladder marker.
papA5 is required for production of both PGLs and PDIMs
We investigated the effect of the papA5 deletion in M. marinum ΔpapA5 on PGL and PDIM production using published radio-TLC methods (see Methods). In addition, a control strain for genetic complementation analysis was constructed (M. marinum ΔpapA5+pCP0-papA5mm, a plasmid expressing papA5mm), and the ability of this strain and that of M. marinum WT controls to produce PGLs and PDIMs was examined. We carried out metabolic labelling of mycobacterial cultures of these strains by feeding [14C]propionate to obtain labelled PGLs and PDIMs for radio-TLC analysis. Representative results of this radio-TLC analysis are shown in Fig. 3. The analysis of lipid samples from M. marinum WT and M. marinum WT+pCP0 (empty vector control) revealed the presence of both PGLs and PDIMs, as expected based on previous reports (Ferreras et al., 2008). More importantly, the analysis of samples from M. marinum ΔpapA5 and M. marinum ΔpapA5+pCP0 indicated that deletion of papA5 led not only to PDIM deficiency, a result expected based on the phenotype of M. tuberculosis ΔpapA5 (Onwueme et al., 2004), but also to PGL deficiency. Introduction of pCP0-papA5mm into M. marinum ΔpapA5 complemented the mutant. The episomal expression of papA5mm restored PGL and PDIM production in M. marinum ΔpapA5+pCP0-papA5mm to levels similar to those seen in M. marinum WT (Fig. 3). Overall, the results of the mutational analysis conclusively demonstrate that papA5 is required for production of both PGLs and PDIMs in M. marinum.
Fig. 3.
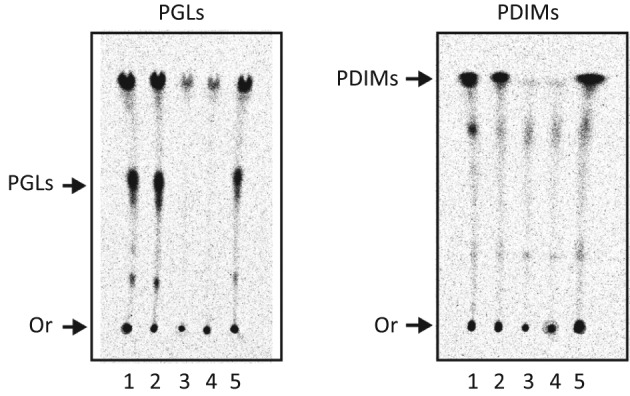
PapA5 is required for production of both PGLs and PDIMs. Radio-TLC analysis of 14C-labelled PGLs (with CHCl3/CH3OH, 95 : 5) and PDIMs (with petroleum ether/diethyl ether, 9 : 1). Lanes: 1, M. marinum WT; 2, M. marinum WT+pCP0; 3, M. marinum ΔpapA5; 4, M. marinum ΔpapA5+pCP0; 5, M. marinum ΔpapA5+pCP0-papA5mm. Or, origin. Liquid chromatography-MS analysis (conducted as recently reported; Chavadi et al., 2011b) demonstrated that the marginal signal in lanes 3 and 4 of the PDIM panel does not arise from PDIMs (not shown), but rather arises from other mycobacterial lipids that incorporate the label from [1-14C]propionate.
PapA5 can catalyse acylation of mycobacterial diol-containing aliphatic polyketides in vitro
Using commercially available surrogate substrates we have previously demonstrated the capacity of M. tuberculosis PapA5 to catalyse hydroxyl-ester formation chemistry (Onwueme et al., 2004). In these previous studies, surrogate alcohol and acyl-CoA thioester substrates replaced the suspected mycobacterial diol-containing aliphatic polyketide and multimethyl-branched fatty acyl thioester substrates, respectively, in the proposed esterification reactions catalysed by PapA5 (Buglino et al., 2004; Onwueme et al., 2004, 2005a). Herein, we set out to conduct a first examination of the ability of PapA5 to utilize mycobacterial diol-containing aliphatic polyketides as acyl acceptor substrates. To this end, we purified M. leprae PGL-derived GPPOLs to use as test substrates. M. leprae in infected Armadillo tissue produces PGLs in amounts that facilitate PGL isolation, and subsequent PGL hydrolytic deacylation and GPPOL purification (see Methods). The identity of the purified GPPOL preparation was verified by MS analysis. The mass spectrum of purified GPPOLs revealed the expected ion peak array, with a mass distribution pattern showing the 14 a.m.u. (-CH2- unit) periodicity arising from the characteristic carbon chain-length heterogeneity in the phenolphthiocerol component of PGLs (Onwueme et al., 2005a) (Fig. 4c). The main GPPOL species in the GPPOL preparation had an observed m/z of 1123.79. This m/z value is in agreement with the calculated m/z of 1123.75 for the sodium adduct [(M+Na+)]+ of the GPPOL species shown in Fig. 4(d). The availability of purified GPPOLs set the stage for a first examination of the competency of PapA5 to utilize a mycobacterial acyl acceptor substrate.
Fig. 4.

PapA5-catalysed palmitoylation of GPPOLs in vitro. (a) Radio-TLC analysis showing PapA5-dependent formation of GPPOL-monopalmitate (GPPOL-P) esters. The main GPPOL-P product is marked. Each TLC panel shows triplicate reactions (lanes 1–3). The TLC solvent system is indicated. Or, origin; SF, solvent front; PA, palmitic acid. (b) Effect of GPPOL concentration on the rate of PapA5-catalysed GPPOL-monopalmitate formation. Means±sem of triplicate reactions are shown. (c) MS analysis demonstrating PapA5-dependent palmitoylation of GPPOLs. The spectra show ion peak arrays with m/z values arising from the natural structural heterogeneity in the phenolphthiocerol component of PGLs. (d) Scheme of the proposed GPPOL palmitoylation reaction catalysed by PapA5. Only the main GPPOL species is displayed. The asterisk indicates that only one of two possible regioisomers is shown.
We utilized a radio-TLC-based acyltransferase assay to investigate the ability of recombinant M. tuberculosis PapA5 to acylate purified GPPOLs in vitro. Representative results of this analysis are shown in Fig. 4(a, b). In these experiments, [14C]palmitoyl-CoA was used as both a surrogate acyl donor substrate and a convenient radiotracer for visualization of [14C]palmitoyl-derived ester products by radio-TLC analysis. Palmitoyl-CoA was selected as the acyl donor for these experiments because it is the most effective surrogate acyl donor among nearly 20 commercially available acyl-CoA thioesters tested as substrates of PapA5 (Onwueme et al., 2004). TLC analysis of reactions with PapA5, purified GPPOLs and [14C]palmitoyl-CoA revealed the formation of two main labelled products (Fig. 4a). The product with the higher Rf is palmitate, which is known to be formed by non-specific hydrolysis of [14C]palmitoyl-CoA under our reaction conditions (Onwueme et al., 2004). The product with the lower Rf was formed only in reactions containing both PapA5 and substrates. Formation of the lower Rf product was not detected in reactions lacking PapA5 (Fig. 4a), in reactions without GPPOL (Onwueme et al., 2004), or in reactions with the PapA5 variant PapH124A in place of PapA5 (not shown). PapH124A has the catalytic residue His-124 replaced by Ala and lacks acyltransferase activity (Buglino et al., 2004; Onwueme et al., 2004). We reasoned that the lower Rf product was emerging from PapA5-catalysed palmitoylation of GPPOL, and indeed MS analysis supported the identity of the product as GPPOL-monopalmitate (see below). Notably, the TLC analysis revealed the presence of two less abundant products flanking the lower Rf product (hereafter referred to as GPPOL-monopalmitate). These secondary products are likely to arise from formation of GPPOL-monopalmitates with carbon chain-length heterogeneity in the GPPOL component (Fig. 4c). This view is in agreement with the results of the MS analysis described below. PapA5-dependent formation of GPPOL-monopalmitate was dependent on the concentration of GPPOLs in the reaction mixture, and an apparent substrate inhibition effect was observed at high levels of GPPOLs in the reaction (Fig. 4b). Apparent Vmax, Km and Ki values determined from these data were 5×10−4 min−1, 103 µM and 496 µM, respectively.
We probed the PapA5-dependent formation of GPPOL-palmitate esters by MS. Comparative analysis of mass spectra obtained from reactions containing PapA5 and the mass spectra of negative control reactions (no enzyme) validated the PapA5-dependent formation of GPPOL-monopalmitate (Fig. 4c). As expected, the mass spectra of both PapA5-containing and negative control reactions displayed an ion peak array with the mass distribution characteristic of the purified GPPOLs. Most importantly, an ion peak array centred at m/z = 1361.86 was detected only in reactions containing PapA5. This m/z value corresponds to the sodium adduct [(M+Na+)]+ of the main GPPOL species present in the purified GPPOL preparation plus the addition of 238.07 a.m.u. This mass gain is concordant with the expected acylation of the main GPPOL species with a palmitoyl chain to form GPPOL-monopalmitate (Fig. 4d). The additional ion peaks in the array had m/z values that matched those predicted for the sodium adducts of monopalmitate esters derived from secondary GPPOL species present in the GPPOL preparation. Formation of polyacylated GPPOLs was not detected by MS. PapA5 is expected to acylate both hydroxyl groups in the diol of the phenolphthiocerol chains during in vivo PGL biosynthesis. The in vitro observed monopalmitoylation is not surprising considering that the use of a non-physiological acyl donor substrate and the lack of potentially relevant factors (protein partners, possibility of membrane association) in the acylation assay might affect the processivity and kinetic parameters of PapA5.
The ability of PapA5 to acylate GPPOLs of different carbon chain length in vitro is in line with the proposed role of this enzyme in the biosynthesis of PGLs with carbon chain-length variability in the phenolphthiocerol moiety. Due to the inability of the PapA5 active site cavity to accommodate the bulky oligosaccharide unit of GPPOL (Buglino et al., 2004), and since PapA5 acylates alkanol and alkanediol surrogate substrates with a hydrophobic character but not sugars (Onwueme et al., 2004), we propose that the observed palmitoylation is likely to take place at one of the hydroxyl groups of the diol of phenolphthiocerol (Fig. 4d). Future studies will be required to validate this prediction, which is consistent with the hypothesized physiological acylation target of PapA5.
In conclusion, the results of both the in vivo mutational analysis and the in vitro biochemical studies presented herein are in line with a model in which PapA5 catalyses the acylation of diol-containing aliphatic polyketides with multimethyl branched fatty acids during biosynthesis of PGLs. The enzymic assay developed herein will provide a basis for more advanced studies of PapA5 activity, whereas the availability of M. marinum ΔpapA5 will facilitate future studies of biosynthetic intermediate accumulation. Overall, our studies advance the understanding of the biosynthesis of an important group of mycobacterial cell wall glycolipids and suggest that PapA5 might be an attractive target for the development of drugs that inhibit the biosynthesis of both PGLs and PDIMs. Such antivirulence drugs might be combined with conventional antimicrobial drugs in the treatment of selected mycobacterial infections.
Acknowledgements
This work was supported by NIH/NIAID grant R01AI069209 to L. E. N. Q. L. E. N. Q. acknowledges the endowment support from Carol and Larry Zicklin. We are grateful to Albert Morrishow (Weill Medical College) for assistance with MS analysis, Professor Christopher Lima (Memorial Sloan-Kettering Cancer Center) for providing PapA5, and Catherine Chan (Quadri laboratory) for help with plasmid constructions. K. C. O. was supported by NIH/MSTP grant GM07739, Minority Predoctoral Fellowship AI054326, and a United Negro College Fund-Merck Fellowship. D. C. was supported in part by NIH/NIAID contract N01 AI-25469.
Abbreviations:
- GPPOL
glycosyl-phenolphthiocerol
- PDIM
phthiocerol dimycocerosate
- PGL
phenolic glycolipid
- SOE
splicing by overlap extension
- WT
wild-type
Footnotes
A supplementary figure, showing the construction of p2NIL-GOALc-ΔpapA5c, is available with the online version of this paper.
References
- Alibaud L., Rombouts Y., Trivelli X., Burguière A., Cirillo S. L., Cirillo J. D., Dubremetz J. F., Guérardel Y., Lutfalla G., Kremer L. (2011). A Mycobacterium marinum TesA mutant defective for major cell wall-associated lipids is highly attenuated in Dictyostelium discoideum and zebrafish embryos. Mol Microbiol 80, 919–934. 10.1111/j.1365-2958.2011.07618.x [DOI] [PubMed] [Google Scholar]
- Astarie-Dequeker C., Le Guyader L., Malaga W., Seaphanh F. K., Chalut C., Lopez A., Guilhot C. (2009). Phthiocerol dimycocerosates of M. tuberculosis participate in macrophage invasion by inducing changes in the organization of plasma membrane lipids. PLoS Pathog 5, e1000289. 10.1371/journal.ppat.1000289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan P. J., Nikaido H. (1995). The envelope of mycobacteria. Annu Rev Biochem 64, 29–63. 10.1146/annurev.bi.64.070195.000333 [DOI] [PubMed] [Google Scholar]
- Brennan P. J., Chatterjee D., Fujiwara T., Cho S. N. (1994). Leprosy-specific neoglycoconjugates: synthesis and application to serodiagnosis of leprosy. Methods Enzymol 242, 27–37. 10.1016/0076-6879(94)42005-X [DOI] [PubMed] [Google Scholar]
- Brodin P., Poquet Y., Levillain F., Peguillet I., Larrouy-Maumus G., Gilleron M., Ewann F., Christophe T., Fenistein D. & other authors (2010). High content phenotypic cell-based visual screen identifies Mycobacterium tuberculosis acyltrehalose-containing glycolipids involved in phagosome remodeling. PLoS Pathog 6, e1001100. 10.1371/journal.ppat.1001100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buglino J., Onwueme K. C., Ferreras J. A., Quadri L. E., Lima C. D. (2004). Crystal structure of PapA5, a phthiocerol dimycocerosyl transferase from Mycobacterium tuberculosis. J Biol Chem 279, 30634–30642. 10.1074/jbc.M404011200 [DOI] [PubMed] [Google Scholar]
- Camacho L. R., Ensergueix D., Perez E., Gicquel B., Guilhot C. (1999). Identification of a virulence gene cluster of Mycobacterium tuberculosis by signature-tagged transposon mutagenesis. Mol Microbiol 34, 257–267. 10.1046/j.1365-2958.1999.01593.x [DOI] [PubMed] [Google Scholar]
- Chavadi S. S., Stirrett K. L., Edupuganti U. R., Vergnolle O., Sadhanandan G., Marchiano E., Martin C., Qiu W. G., Soll C. E., Quadri L. E. (2011a). Mutational and phylogenetic analyses of the mycobacterial mbt gene cluster. J Bacteriol 193, 5905–5913. 10.1128/JB.05811-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavadi S. S., Edupuganti U. R., Vergnolle O., Fatima I., Singh S. M., Soll C. E., Quadri L. E. (2011b). Inactivation of tesA reduces cell wall lipid production and increases drug susceptibility in mycobacteria. J Biol Chem 286, 24616–24625. 10.1074/jbc.M111.247601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins D. M., Skou B., White S., Bassett S., Collins L., For R., Hurr K., Hotter G., de Lisle G. W. (2005). Generation of attenuated Mycobacterium bovis strains by signature-tagged mutagenesis for discovery of novel vaccine candidates. Infect Immun 73, 2379–2386. 10.1128/IAI.73.4.2379-2386.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constant P., Perez E., Malaga W., Lanéelle M. A., Saurel O., Daffé M., Guilhot C. (2002). Role of the pks15/1 gene in the biosynthesis of phenolglycolipids in the Mycobacterium tuberculosis complex. Evidence that all strains synthesize glycosylated p-hydroxybenzoic methyl esters and that strains devoid of phenolglycolipids harbor a frameshift mutation in the pks15/1 gene. J Biol Chem 277, 38148–38158. 10.1074/jbc.M206538200 [DOI] [PubMed] [Google Scholar]
- Cox J. S., Chen B., McNeil M., Jacobs W. R., Jr (1999). Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature 402, 79–83. [DOI] [PubMed] [Google Scholar]
- Crick D. C., Quadri L. E., Brennan P. J. (2008). Biochemistry of the cell envelope of Mycobacterium tuberculosis. In Handbook of Tuberculosis: Molecular Biology and Biochemistry, pp. 1–19. Edited by Kaufmann S. H. E., Rubin R. Weinheim: Wiley-VCH. [Google Scholar]
- Dhungel S., Ranjit C., Sapkota B. R., Macdonald M. (2008). Role of PGL-I of M. leprae in TNF-alpha production by in vitro whole blood assay. Nepal Med Coll J 10, 1–3. [PubMed] [Google Scholar]
- Ferreras J. A., Ryu J. S., Di Lello F., Tan D. S., Quadri L. E. (2005). Small-molecule inhibition of siderophore biosynthesis in Mycobacterium tuberculosis and Yersinia pestis. Nat Chem Biol 1, 29–32. 10.1038/nchembio706 [DOI] [PubMed] [Google Scholar]
- Ferreras J. A., Stirrett K. L., Lu X., Ryu J. S., Soll C. E., Tan D. S., Quadri L. E. (2008). Mycobacterial phenolic glycolipid virulence factor biosynthesis: mechanism and small-molecule inhibition of polyketide chain initiation. Chem Biol 15, 51–61. 10.1016/j.chembiol.2007.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W., Soll C. E., Chavadi S. S., Zhang G., Warren J. D., Quadri L. E. (2009). Cooperation between a coenzyme A-independent stand-alone initiation module and an iterative type I polyketide synthase during synthesis of mycobacterial phenolic glycolipids. J Am Chem Soc 131, 16744–16750. 10.1021/ja904792q [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton R. M., Hunt H. D., Ho S. N., Pullen J. K., Pease L. R. (1989). Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77, 61–68. 10.1016/0378-1119(89)90359-4 [DOI] [PubMed] [Google Scholar]
- Hunter S. W., Brennan P. J. (1981). A novel phenolic glycolipid from Mycobacterium leprae possibly involved in immunogenicity and pathogenicity. J Bacteriol 147, 728–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murry J. P., Pandey A. K., Sassetti C. M., Rubin E. J. (2009). Phthiocerol dimycocerosate transport is required for resisting interferon-γ-independent immunity. J Infect Dis 200, 774–782. 10.1086/605128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng V., Zanazzi G., Timpl R., Talts J. F., Salzer J. L., Brennan P. J., Rambukkana A. (2000). Role of the cell wall phenolic glycolipid-1 in the peripheral nerve predilection of Mycobacterium leprae. Cell 103, 511–524. 10.1016/S0092-8674(00)00142-2 [DOI] [PubMed] [Google Scholar]
- Onwueme K. C., Ferreras J. A., Buglino J., Lima C. D., Quadri L. E. (2004). Mycobacterial polyketide-associated proteins are acyltransferases: proof of principle with Mycobacterium tuberculosis PapA5. Proc Natl Acad Sci U S A 101, 4608–4613. 10.1073/pnas.0306928101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onwueme K. C., Vos C. J., Zurita J., Ferreras J. A., Quadri L. E. (2005a). The dimycocerosate ester polyketide virulence factors of mycobacteria. Prog Lipid Res 44, 259–302. 10.1016/j.plipres.2005.07.001 [DOI] [PubMed] [Google Scholar]
- Onwueme K. C., Vos C. J., Zurita J., Soll C. E., Quadri L. E. (2005b). Identification of phthiodiolone ketoreductase, an enzyme required for production of mycobacterial diacyl phthiocerol virulence factors. J Bacteriol 187, 4760–4766. 10.1128/JB.187.14.4760-4766.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parish T., Stoker N. G. (1998). Mycobacteria Protocols. Totowa, NJ: Humana Press; 10.1385/0896034712 [DOI] [Google Scholar]
- Parish T., Stoker N. G. (2000). Use of a flexible cassette method to generate a double unmarked Mycobacterium tuberculosis tlyA plcABC mutant by gene replacement. Microbiology 146, 1969–1975. [DOI] [PubMed] [Google Scholar]
- Quadri L. E. N. (2007). Strategic paradigm shifts in the antimicrobial drug discovery process of the 21st century. Infect Disord Drug Targets 7, 230–237. 10.2174/187152607782110040 [DOI] [PubMed] [Google Scholar]
- Rambukkana A., Zanazzi G., Tapinos N., Salzer J. L. (2002). Contact-dependent demyelination by Mycobacterium leprae in the absence of immune cells. Science 296, 927–931. 10.1126/science.1067631 [DOI] [PubMed] [Google Scholar]
- Reed M. B., Domenech P., Manca C., Su H., Barczak A. K., Kreiswirth B. N., Kaplan G., Barry C. E., III (2004). A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature 431, 84–87. 10.1038/nature02837 [DOI] [PubMed] [Google Scholar]
- Reed M. B., Gagneux S., Deriemer K., Small P. M., Barry C. E., III (2007). The W-Beijing lineage of Mycobacterium tuberculosis overproduces triglycerides and has the DosR dormancy regulon constitutively upregulated. J Bacteriol 189, 2583–2589. 10.1128/JB.01670-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson N., Kolter T., Wolke M., Rybniker J., Hartmann P., Plum G. (2008). Mycobacterial phenolic glycolipid inhibits phagosome maturation and subverts the pro-inflammatory cytokine response. Traffic 9, 1936–1947. 10.1111/j.1600-0854.2008.00804.x [DOI] [PubMed] [Google Scholar]
- Ruley K. M., Ansede J. H., Pritchett C. L., Talaat A. M., Reimschuessel R., Trucksis M. (2004). Identification of Mycobacterium marinum virulence genes using signature-tagged mutagenesis and the goldfish model of mycobacterial pathogenesis. FEMS Microbiol Lett 232, 75–81. 10.1016/S0378-1097(04)00017-5 [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E. F., Maniatis T. (1989). Molecular Cloning: a Laboratory Manual, 2nd edn Cold Spring Harbor, NY: Cold Spring Harbor Laboratory. [Google Scholar]
- Sinsimer D., Huet G., Manca C., Tsenova L., Koo M. S., Kurepina N., Kana B., Mathema B., Marras S. A. & other authors (2008). The phenolic glycolipid of Mycobacterium tuberculosis differentially modulates the early host cytokine response but does not in itself confer hypervirulence. Infect Immun 76, 3027–3036. 10.1128/IAI.01663-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinear T. P., Seemann T., Harrison P. F., Jenkin G. A., Davies J. K., Johnson P. D., Abdellah Z., Arrowsmith C., Chillingworth T. & other authors (2008). Insights from the complete genome sequence of Mycobacterium marinum on the evolution of Mycobacterium tuberculosis. Genome Res 18, 729–741. 10.1101/gr.075069.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsenova L., Ellison E., Harbacheuski R., Moreira A. L., Kurepina N., Reed M. B., Mathema B., Barry C. E., III, Kaplan G. (2005). Virulence of selected Mycobacterium tuberculosis clinical isolates in the rabbit model of meningitis is dependent on phenolic glycolipid produced by the bacilli. J Infect Dis 192, 98–106. 10.1086/430614 [DOI] [PubMed] [Google Scholar]