

Abstract
A rhodium-catalyzed dehydrogenation protocol has been developed for conversion of 3,5-diarylcyclopentenones to the corresponding 2,4-diarylcyclopentadienones. Using this protocol, analogues of the cytotoxic agent chamaecypanone C have been synthesized via Diels-Alder cycloaddition between the cyclopentadienones and in situ-generated ortho-quinols. Biological evaluation of these analogues revealed a compound with higher activity as a microtubule inhibitor and cytotoxic agent in comparison with the parent structure.
INTRODUCTION
Cyclopentadienones are highly reactive and unstable species and in many cases are known to readily dimerize to release antiaromaticity. 1 As useful synthons, cyclopentadienone and derivatives have been utilized in both electrocyclic reactions2 and pericyclic additions.3 In these cases, “deantiaromatization” has been proposed to be a significant driving force. Moreover, cyclopentadienones have been used as key reagents to access biologically active natural products including manzamenone A (1, Figure 1)4 and chamaecypanone C (2).5,6
Figure 1.

Cyclopentadienone-derived natural products.
We have previously reported the synthesis of the bicyclo[2.2.2]octenone-containing natural product (+)-chamaecypanone C (2) utilizing a retro-Diels-Alder/Diels-Alder cascade of a 6-alkyl-6-hydroxycyclohexa-2,4-dienone (ortho-quinol) dimer as the key step wherein an in situ-generated 2,4-diarylcyclopentadienone was reacted with an o-quinol in a highly diastereoselective manner.5a The observed reactivity of the diarylcyclopentadienone reaction partner prompted further study of the key transformation. Furthermore, on the basis of the demonstrated tubulin inhibitory activity of chamaecypanone C,5a, 7 a general approach to analogues with modified structures was desirable for further biological studies, in particular for evaluation of structure-activity relationships (SAR). With these considerations in mind, we focused on the synthesis of cycloadducts derived from 2,4-cyclopentadienones with variable aromatic substitution (Scheme 1). Herein, we report our studies on development of a general protocol for preparation of 2,4-diarylcyclopentadienones en route to chamaecypanone C analogues as well as the biological evaluation of the target compounds as microtubule inhibitors.
Scheme 1.

Approach to chamaecypanone C analogues
RESULTS AND DISCUSSION
Syntheses of cyclopentadienones have been achieved using various protocols, including elimination of bromocyclopentenones 8 and hydroxycyclopentenones,4a, 9 condensations of propanones with α-diketones, 10 photochemical decarbonylation11 and SO2 extrusion,12 and carbonylation of metallacyclopentadienes. 13 A recent elegant methodology developed by the Wender group involved rhodium(I)-catalyzed [3+2] cycloaddition of cyclopropenones and alkynes. 14 In most cases, trisubstituted or tetrasubstituted cyclopentadienones are produced. Mono- or disubstituted cyclopentadienones are less commonly reported, likely due to their instability or tendency towards dimerization.8c In particular, there are very few examples of 2,4-disubstituted cyclopentadienones reported in the literature, 15 and no reports of 2,4-diarylcyclopentadienones or derived dimers to date.
As a 2,4-diarylcyclopentadienone is a key precursor to chamaecypanone C, the preparation of analogues with similar substitution patterns requires a protocol to access the corresponding cyclopentadienones. Considering the abundance of the corresponding cyclopentenone precursors, it should be possible to dehydrogenate these compounds to the target molecules. In our previous studies, 2,4-diarylcyclopentenone 3 was used as a substrate (Scheme 2) under DDQ-mediated oxidation conditions which generated 2,4-diarylcyclopentadienone 4 in situ. In the same pot, thermolytic retro-Diels-Alder reaction of dimer 5 afforded ortho-quinol 6 which subsequently reacted with intermediate 4 to afford chamaecypanone C methyl ether 7 in a highly diastereoselective manner. Upon treatment with excess boron tribromide, cycloadduct 7 was demethylated to afford (+)-chamaecypanone C (2). This synthetic sequence also represents usage of a dehydrogenative Diels-Alder cycloaddition in natural product synthesis.16
Scheme 2.

First generation synthesis of chamaecypanone C
While we were able to prepare di-para-methoxyphenyl substituted cyclopentenone 3 from cyclopentene (8) using a double-Heck cross-coupling (Scheme 3a), 17 allylic hydroxylation, and IBX oxidation sequence,5a we recognized that this route may not be optimal to prepare diarylcyclopentadienone precursors due to the potential limitation of aryl substitution and formation of isomerized alkene byproducts in the double Heck reactions, as well as usage of substoichiometric amounts of toxic selenium dioxide in the allylic oxidation step.
Scheme 3.

Synthetic routes to diarylcyclopentenones
Recently Xu, McLaughlin, and coworkers established a general and scalable method for preparation of 3,5-diarylcyclopentenones with a broad scope (Scheme 3, b).18 We proposed that these cyclopentenones may react similarly as the 2,4-diaryl compounds in the dehydrogenative Diels-Alder event. Accordingly, our goal was to identify suitable reaction conditions to produce cyclopentadienones from the readily accessible 3,5-diarylcyclopentenones.
Investigation of Aerobic Dehydrogenation
Although the installation of α,β-unsaturation to a ketone substrate using IBX19 or DDQ20 is well known, we first focused our attention on the evaluation of catalytic, oxidative conditions to avoid stoichiometric use of oxidants. 21 Utilizing 3,5-diphenylcyclopentenone (9, Table 1)18 as a model substrate, representative conditions for catalytic, aerobic dehydrogenation including Pd(TFA)2/bipyridine/O2 22 and Au(NP)/TiO2, 23 were investigated (Table 1, entries 1–2). Interestingly, instead of the desired cyclopentadienone, 4,6-diphenyl-α-pyrone 10 was identified as the major product with no diarylcyclopentadienone or derived product produced under the reaction conditions. 1H and 13C-NMR spectra of α-pyrone 10 were identical to literature data24 and the structure was further confirmed by X-ray crystallography.25 Two other cyclopentenones 11 (entry 3) and 12 (entry 4) were further evaluated under the gold nanoparticle-catalyzed oxidation conditions in which case the corresponding 4,6-diaryl-α-pyrones 13 and 14 were observed as major products.
Table 1.
Generation of α-pyrones from diarylcyclopentenones under aerobic dehydrogenation conditions.a
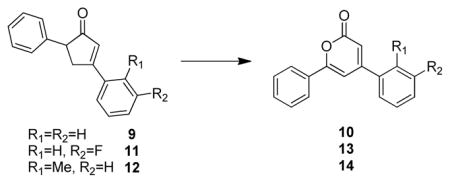
| ||||
|---|---|---|---|---|
| entry | cyclopentenone | Catalyst | time (h) | yieldb (%) |
| 1 | 9 | Pd(TFA)2/bipyridine | 12 | 36 |
| 2 | 9 | Au(NP)/TiO2 | 12 | 80 |
| 3 | 11 | Au(NP)/TiO2 | 12 | 46 |
| 4 | 12 | Au(NP)/TiO2 | 12 | 50c |
Reaction conditions: 5 mol% catalyst, O2, chlorobenzene, 100 °C;
isolated yield after silica gel column chromatography;
product inseparable from starting material, yield based on 1H-NMR integration.
Pd(TFA)2 = palladium(II) trifluoroacetate; Au(NP) = gold nanoparticles.
A plausible mechanism accounting for the generation of α-pyrone 10 is depicted in Figure 2. Under palladium or gold nanoparticle-catalyzed conditions, aerobic dehydrogenation of cyclopentenone 9 should afford cyclopentadienone 15 which is accompanied by the generation of hydrogen peroxide.21,26 Due to the instability of diarylcyclopentadienone 15, the compound may subsequently react with H2O2 to afford intermediates such as hydroperoxide 16, thereby releasing antiaromaticity.1 Baeyer-Villiger-type rearrangement of 16 should then generate α-pyrone 10.27 A literature reported example of aerobic oxidation of tetraphenylcyclopentadienone (tetracyclone) under thermolytic conditions to an α-pyrone 28 also supports the likely intermediacy of cyclopentadienone 15. 29 Moreover, treatment of cyclopentenone 9 with 2.0 equiv of H2O2 (dioxane, 80 °C, 16 h) led to no reaction which indicates that cyclopentadienone formation may be required prior to Baeyer-Villiger reaction.30
Figure 2.

Possible mechanism leading to α-pyrones.
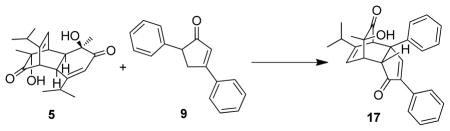
With the hope of intercepting the generated intermediate 15, a one-pot procedure was also investigated by heating a mixture of cyclopentenone 9 and the readily available dimer (±)-531 in the presence of a Au(NP)/TiO2 catalyst under an oxygen atmosphere. Unfortunately, the reaction afforded a complicated mixture, and no generation of the desired cycloadduct 17 was observed (Scheme 4).
Scheme 4.

In situ trapping experiment
Evaluation of Anaerobic Dehydrogenation Conditions
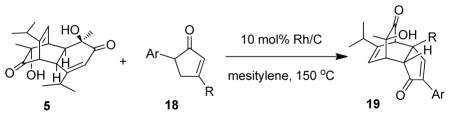
Taking into account the reactivity of 2,4-diarylcyclopentadienone to form byproducts, including α-pyrones under aerobic conditions, anaerobic dehydrogenation conditions were next investigated. Inspired by a reported dehydrogenation of arylcyclohexenones using palladium on carbon,32 several metal catalysts, including palladium(0), platinum(0), and rhodium(0), were evaluated for dehydrogenation of cyclopentenone 9 under an argon atmosphere at high temperatures. While in these cases no cyclopentadienones were observed or isolated, we discovered that in the presence of o-quinol dimer 5, Pd/C successfully catalyzed dehydrogenation of cyclopentenone substrates to afford the corresponding dienone intermediates which were reacted with an in situ generated o-quinol leading to the formation of the desired cycloadduct 17 (Table 2, entry 1). Although cyclopentenone 9 was completely consumed after the thermolysis in mesitylene for 6 h, a large amount of unreacted dimer 5 suggested the presence of other degradation pathways for 9. When Pt/C or Rh/Al2O3 were used as catalysts, production of a complex mixture of products including the phenol 3-hydroxy-α,4-dimethyl styrene33,34 (entry 2) or severe decomposition of starting materials (entry 3) was observed. After extensive experimentation, reactions with Rh/C as catalyst were found to cleanly afford the desired [4+2] cycloadduct 17 (entry 4). As the conversion of o-quinol dimer 5 was incomplete, whilst all cyclopentenone was consumed, we decided to increase the amount of easily accessed reaction partner 9. Accordingly, use of 5.0 equiv cyclopentenone 9 (2.5 equiv in respective to 1.0 equiv o-quinol monomer) and extending the reaction time led to improved conversion (entry 5) to afford product 17 in good yield (81%). The requirement for excess cyclopentenone may be due to the instability of generated cyclopentadienone under the reaction conditions.
Table 2.
Reaction optimization for dehydrogenation of diarylcyclopentenonesa
| |||
|---|---|---|---|
| entry | catalyst or oxidant | time (h) | % conversionb to cycloadduct 17 |
| 1 | Pd/C | 6 | 28 |
| 2 | Pt/C | 6 | <5 |
| 3 | Rh/Al2O3 | 6 | <5 |
| 4 | Rh/C | 16 | 56 |
| 5c | Rh/C | 18 | >95(81)d |
| 6e | DDQ | 1 | 33 |
Reaction conditions: 3.0 equiv cyclopentadienone 9 (1.5 equiv in respective to 1.0 equiv of o-quinol monomer generated), 10 mol% catalyst (based on cyclopentenone), mesitylene, 150 °C, argon atmosphere;
Conversion based on 1H-NMR integration of dimer 5 and product 17.
5.0 equiv of 9 (2.5 equiv with respect to 1.0 equiv of o-quinol monomer) used in the reaction.
Isolated yield in parenthesis.
5.0 equiv of 9, 6.0 equiv of DDQ, o-dichlorobenzene, 150 °C, argon atmosphere.
To further evaluate the efficiency of the Rh-catalyzed dehydrogenation in comparison to our previously developed protocol,5a to a mixture of dimer 5 and compound 9 (5.0 equiv) in o-dichlorobenzene was slowly added DDQ (6.0 equiv) at 150 °C over a period of one hour. Further reaction at the same temperature for an additional hour resulted in the generation of cycloadduct 17 in approximately 33% conversion (Table 2, entry 6) along with a significant amount of unreacted starting materials. Attempts to improve the conversion by extending the reaction time led to severe decomposition. These results suggested that DDQ-mediated oxidation was less effective in comparison to the Rh-catalyzed protocol for substrate 9
Utilizing the optimized conditions, a number of 2,4-diarylcyclopentadienone substrates were evaluated (Table 3). Substrates with electron rich aryl substituents reacted smoothly to afford cycloadducts in moderate to good yields (entries 1,2,4–6). In the case of 2-chlorophenyl-substituted compound 18c, a low conversion was observed due to its poor solubility leading to a low isolated yield of the desired product 19c (entry 3). Reaction with a substrate bearing a heterocyclic substituent also produced the [4+2] cycloadduct in moderate yield (entry 7). Styrenylcyclopentenone 18h also underwent dehydrogenation under Rh/C-catalyzed conditions. However, only a small amount of product 19h was isolated (entry 8), presumably due to the multiple reaction sites for the generated cyclopentadienone intermediate. In all cases, reactions to form [4+2] adducts proceeded in high diastereoselectivity which was in accordance with previous observations in our synthesis of chamaecypanone C.5a Endo-selectivity and facial-selectivity align well with the observed reactivity of o-quinols, 35 wherein the bulky aromatic substituents are oriented away from the sterically demanding quaternary center.
Table 3.
Diels-Alder cycloadditions of in situ generated o-quinols and 2,4-diarylcyclopentadienones
| ||||
|---|---|---|---|---|
| entry | precursor(equiv) | time(h) | cycloadduct | yielda (%) |
| 1 |
 18a |
18 |
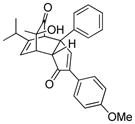 19a |
80 |
| 2 |
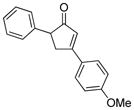 18b |
18 |
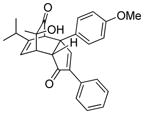 19b |
59 |
| 3 |
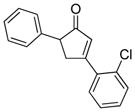 18c |
18 |
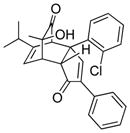 19c |
22 |
| 4 |
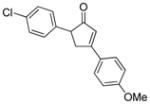 18d |
18 |
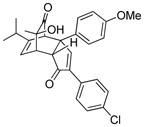 19d |
66 |
| 5 |
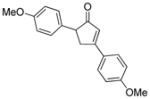 18e |
12 |
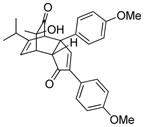 7 |
75 |
| 6 |
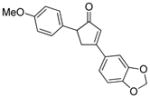 18f |
16 |
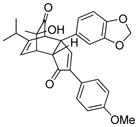 19f |
72 |
| 7 |
 18g |
18 |
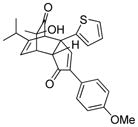 19g |
60 |
| 8 |
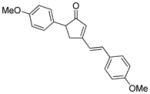 18h |
18 |
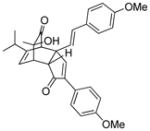 19h |
16b |
Reaction conditions: 10 mol% Rh/C (based on cyclopentenones), mesitylene, 150 °C, argon atmosphere.
Isolated yield after silica gel chromatography.
Full conversion observed based on cyclopentenone.
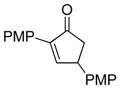
To evaluate the scope and limitations of the methodology, select o-quinol dimers32, 36 and masked-o-benzoquinone (MOB) dimers 37 were explored as 2,4-cyclohexadienone precursors. A number of cyclopentenone and indanone substrates were also investigated as substrates for Rh/C-catalyzed dehydrogenation. Dimers (±)-20 (Table 4, entry 1) and (±)-21 (entries 2–3) were found to have similar reactivities in comparison to dimer 5, and cycloadducts 22–24 were prepared in good yields from 3,5-diarylcyclopentenones 18e, 25, and 2,4-diarylcyclopentenone 3, respectively. In contrast, o-quinol dimers (±)-26 (Figure 3) and 27 and MOB dimer 28 did not afford any desired cycloadducts. In the failed cases, dienone-phenol rearrangement of the 2,4-cyclohexadienone intermediate was typically observed.38 The reaction using 5-arylcyclopentenone 29 (Table 4, entry 4) and dimer 5 under the rhodium-catalyzed conditions generated a mixture of two [4+2] adducts 30a and 30b in good combined yield. The two products were separated and characterized as isomers derived from reactivity of the unsubstituted cis-alkene. This result correlates well with the previously observed higher reactivity of disubstituted cis-alkenes in cycloaddition reactions with o-quinols.35 The lack of 4-aryl substitution on cyclopentadienone appears to have diminished the steric differentiation resulted from the aryl group and the tertiary alcohol, thereby leading to the formation of isomeric cycloadducts.
Table 4.
Dehydrogenative cycloadditions using alternative dimers and cyclopentenones
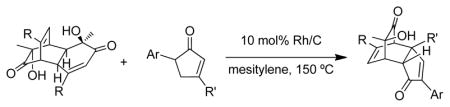
| |||||
|---|---|---|---|---|---|
| entry | dimer | cyclopentenone | time (h) | cycloadduct | yielda (%) |
| 1 | 20 (R = Me) |
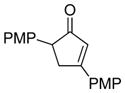 18e (3 equiv) |
18 |
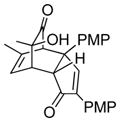 22 |
61 |
| 2 | 21 (R = tBu) |
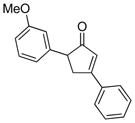 25 (5 equiv) |
18 |
 23 |
70 |
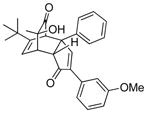
| 3 | 21 (R = tBu) |
 3 (5 equiv) |
12 |
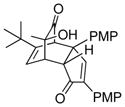 24 |
64 |
| 4 | 5 (R = iPr) |
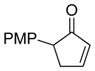 29 (5 equiv) |
12 |
 30a 30b |
45 29 |
Reaction conditions: 10 mol% Rh/C (based on cyclopentenones), mesitylene, 150 °C.
Isolated yield after silica gel chromatography.
PMP = para-Methoxyphenyl.
Figure 3.

Methodology limitations
When 3-arylcyclopentenone 3139 (Figure 3) was used as cyclopentadienone precursor, no conversion of either starting material was observed, which suggests that 5-aryl substitution is required for successful dehydrogenation. This assumption was further confirmed by the fact that cyclopentenone 32 was also inert to the rhodium-catalyzed dehydrogenation conditions. Further attempted experiments using indanones 33 and 34 did not yield any desired cycloadducts and no indenone formation was observed.
To prepare chamaecypanone C analogues with unprotected hydroxyl groups, previously prepared cycloadducts with methyl ethers were submitted to BBr3-mediated demethylation conditions and the corresponding hydroxyl products were isolated in moderate to good yields (Figure 4).
Figure 4.

Preparation of chamaecypanone C analogues with unprotected phenols. a Isolated yield in parenthesis.
Biological Studies
We utilized the developed synthetic route to explore the structure activity space of (+)-chamaecypanone C as a microtubule inhibitor and cytotoxic agent.5a,7 Racemic chamaecypanone C analogues were first evaluated in a tubulin assembly assay. The diphenyl analogue 17, chlorinated analogues 19c, and all methyl protected analogues were found to be inactive,25 while structures 35 to 38 (Figure 4) showed good activity. Compounds 39 and 40 were also found to be inactive. Taking note of the previous finding that the (+)-enantiomer of chamaecypanone C is active, while (−)-enantiomer is not, we therefore synthesized the enantiopure versions of active compounds (+)-35 to (+)-38 and tested them in both the tubulin assay and in the NCI 60-cell screen along with (+)-chamaecypanone C (2). As summarized in Table 5, these compounds showed comparable activity to that of the parent with the chaemacypanone C analogue (+)-38 (entry 4) which was found to have a slightly greater activity as an inhibitor of tubulin assembly and as a cytotoxic agent in the NCI-60 (mean GI50=180 nM) relative to the natural product (+)-2 (entry 5).
Table 5.
Tubulin and Growth Inhibitory Activity of Enantiopure Chamaecypanone Analogues
| Entry | Compound | Inhibition of Tubulin Assembly IC50±SD (μM) |
% Inhibition of Colchicine Binding ± SD 5 μM inhibitor |
NCI-60 Mean GI50 (μM) |
|---|---|---|---|---|
| 1 | (+)-35 | 2.8±0.4 | 30±0.8 | 0.55 |
| 2 | (+)-36 | 3.2±0.4 | 27±0.8 | 0.56 |
| 3 | (+)-37 | 2.4±0.3 | 32±0.5 | 0.91 |
| 4 | (+)-38 | 1.5±0.01 | 50±2.0 | 0.18 |
| 5 | (+)-2 | 2.2±0.3 | 30±4.0 | 0.28 (n=2) |
These results suggested that the 4-para-hydroxylphenyl group (Figure 4) is crucial for interactions with tubulin and showed that reducing the isopropyl appendage (cf. (+)-2) to a methyl group ((+)-38) slightly improved potency in both growth and tubulin assembly inhibition, while a tert-Bu group at C-11 (compound 41) completely eliminated activity.25 While these differences are modest, it is clear that minor modifications of the parent structure are tolerated, while larger changes are not. The 4′ phenolic group appears to be required as a hydrogen bond donor, as the 4′-desoxy analogue 39 and the dimethoxy analogue 19e were found to be inactive.25 The 4″-OH was found to be less critical, as both the thiophene analogue (+)-37 (Table 5, entry 3) and the 4″-desoxy analogue (+)-35 (entry 1) were active.
CONCLUSION
We have developed a rhodium-catalyzed, anaerobic dehydrogenation protocol to convert 3,5-diarylcyclopentenones to the corresponding 2,4-diarylcyclopentadienones. A series of in situ-generated, highly reactive cyclopentadienones were successfully utilized to prepare a series of chamaecypanone C analogues via [4+2] cycloaddition with in situ-generated ortho-quinols. Our investigation also led to the discovery of a gold nanoparticle-catalyzed transformation of 3,5-diarylcyclopentenones to diaryl-α-pyrones. Initial biological studies of chamaecypanone C analogues uncovered a chamaecypanone C derivative (+)-38 with higher activity as a microtubule inhibitor and cytotoxic agent in comparison with the parent structure. Further biological studies of chamaecypanone C derivatives, as well as the chemistry of the chamaecypanone C core structure, are ongoing and will be reported in due course.
Supplementary Material
Acknowledgments
Financial support from the NIH (GM-073855, J.A.P., Jr.), and the NIGMS CMLD Initiative (P50 GM067041, J.A.P., Jr.) is gratefully acknowledged. T.Q. thanks Vertex Pharmaceuticals, Inc. for a graduate fellowship. This research was also supported in part by the Intramural Research Program of NIH, National Cancer Institute, Center for Cancer Research and by the Developmental Therapeutics Program, Division of Cancer Diagnosis and Treatment. We thank Drs. Jeffrey Bacon (Boston University) and Charles Campana (Bruker AXS) for X-ray crystal structure analyses and Drs. Yingju Xu and Mark McLaughlin (Department of Process Chemistry, Merck Research Laboratories, Merck & Co., Inc., Rahway) for their kind donation of diarylcyclopentenones.
Footnotes
Supporting Information Available Experimental procedures and characterization data for all new compounds, including X-ray structure analysis of compound 10, X-ray crystallographic data (CIF), and detailed biological methods and results (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ogliaruso MA, Romanelli MG, Becker EI. Chem Rev. 1965;65:261. [Google Scholar]
- 2.Harmata M, Zheng P, Schreiner PR, Navarro-Vázquez A. Angew Chem, Int Ed. 2006;45:1966. doi: 10.1002/anie.200503812. [DOI] [PubMed] [Google Scholar]
- 3.Harmata M, Gomes MG. Eur J Org Chem. 2006:2273. [Google Scholar]
- 4.Al-Busafi S, Doncaster JR, Drew MGB, Regan AC, Whitehead RC. J Chem Soc, Perkin Trans 1. 2002:476.Isolation, see: Tsukamoto S, Takeuchi S, Ishibashi M, Kobayashi J. J Org Chem. 1992;57:5255.
- 5.Dong S, Hamel E, Bai R, Covell DG, Beutler JA, Porco JA., Jr Angew Chem, Int Ed. 2009;48:1494. doi: 10.1002/anie.200805486.For isolation of chamaecypanone C, see: Chien SC, Chang JY, Kuo CC, Hsieh CC, Yang NS, Kuo YH. Tetrahedron Lett. 2007;48:1567.
- 6.For other representative examples of utilizing cyclopentadienones as building blocks in natural product synthesis, see: Baraldi PG, Barco A, Benetti S, Pollini GP, Polo E, Simoni D. J Chem Soc, Chem Commun. 1984:1049.Schuda PF, Phillips JL, Morgan TM. J Org Chem. 1986;51:2742.Takano S, Sato T, Inomata K, Ogasawara K. J Chem Soc, Chem Commun. 1991:462.
- 7.Hsieh C, Kuo Y, Kuo C, Chen L, Cheung CA, Chao T, Lin C, Pan W, Chien S, Chen T, Lung C, Chang J. Biochem Pharmacol. 2010;79:1261. doi: 10.1016/j.bcp.2009.12.017. [DOI] [PubMed] [Google Scholar]
- 8.DePuy CH, Lyons CF. Chem Ind. 1961:429.DePuy CH, Isaks M, Eilers KL, Morris GF. J Org Chem. 1964;29:3503.For a recent report, see: Harmata M, Barnes CL, Brackley J, Bohnert G, Kirchhoefer P, Kürti L, Rashatasakhon P. J Org Chem. 2001;66:5232. doi: 10.1021/jo015671+.
- 9.Fuchs B, Pasternak M, Pazhenchevsky B. J Org Chem. 1981;46:2017. [Google Scholar]
- 10.(a) Allen CFH, VanAllan JA. J Am Chem Soc. 1950;72:5165. [Google Scholar]; (b) Van Ornum SG, Li J, Kubiak GG, Cook JM. J Chem Soc, Perkin Trans 1. 1997:3471. [Google Scholar]; (c) Walsh CJ, Mandal BK. J Org Chem. 1999;64:6102. [Google Scholar]; (d) Moore JE, York M, Harrity JPA. Synlett. 2005:860. [Google Scholar]
- 11.DeSelms RC, Schleigh WR. Synthesis. 1973:614. [Google Scholar]
- 12.Ried W, Bopp DCH. Angew Chem, Int Ed. 1977;16:653. [Google Scholar]
- 13.For a review of reactions of metallacyclodienes with leading references, see: Müller E. Synthesis. 1974:761–774.
- 14.Wender PA, Paxton TJ, Williams TJ. J Am Chem Soc. 2006;128:14814. doi: 10.1021/ja065868p. [DOI] [PubMed] [Google Scholar]
- 15.Bandyopadhyay C, Sur KR, Patra R. J Chem Res-S. 2002:414–415. [Google Scholar]
- 16.For a recent report of dehydrogenative Diels-Alder cycloadditions, see: Stang EM, White MC. J Am Chem Soc. 2011;133:14892. doi: 10.1021/ja2059704.
- 17.Prashad M, Tomesch JC, Wareing JR, Smith HC, Cheon SH. Tetrahedron Lett. 1989;30:2877. [Google Scholar]
- 18.Xu Y, McLaughling M, Chen C, Reamer RA, Dormer PG, Davies IW. J Org Chem. 2009;74:5100. doi: 10.1021/jo900696k. [DOI] [PubMed] [Google Scholar]
- 19.Nicolaou KC, Zhong Y, Baran PS. J Am Chem Soc. 2000;122:7596. [Google Scholar]
- 20.(a) Walker D, Hiebert JD. Chem Rev. 1967;67:153. doi: 10.1021/cr60246a002. [DOI] [PubMed] [Google Scholar]; (b) Buckle DR. In: Encyclopedia of Reagents for Organic Synthesis. 3. Paquette LA, editor. John Wiley & Sons; Chichester, UK: 1995. p. 1699. [Google Scholar]
- 21.For recent examples of Pd-catalyzed α,β-dehydrogenation of carbonyl compounds, see: Diao T, Stahl SS. J Am Chem Soc. 2011;133:14566. doi: 10.1021/ja206575j.Diao T, Wadzinski TJ, Stahl SS. Chem Sci. 2012;3:887. doi: 10.1039/C1SC00724F.
- 22.Tokunaga M, Harada S, Iwasawa T, Obora Y, Tsuji Y. Tetrahedron Lett. 2007;48:6860. [Google Scholar]
- 23.Sun H, Su F, Ni J, Cao Y, He H, Fan K. Angew Chem, Int Ed. 2009;48:4390. doi: 10.1002/anie.200900802. [DOI] [PubMed] [Google Scholar]
- 24.Li GQ, Dai LX, You SL. Org Lett. 2009;11:1623. doi: 10.1021/ol9002898. [DOI] [PubMed] [Google Scholar]
- 25.See Supporting Information for detailed experimental data.
- 26.For gold nanoparticle-catalyzed aerobic oxidation involving hydrogen peroxide formation, see: Miyamura H, Shiramizu M, Matsubara R, Kobayashi S. Chem Lett. 2008;37:360. doi: 10.1002/anie.200802192.
- 27.For a recent example of heterogeneous catalytic Baeyer-Villiger oxidation of ketones, see: Jeong EY, Ansari MB, Park SE. ACS Catal. 2011;1:855.
- 28.Thiemann T, Iniesta J, Walton DJ. J Chem Res. 2008:173.For Baeyer Villiger-type rearrangement of tetracyclone to an α-pyrone with bis(trimethylsilyl) monoperoxysulfate, see: Adam W, Rodriguez WA. J Org Chem. 1979;44:4969.
-
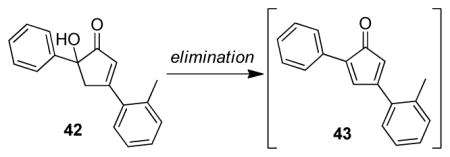
29.For Au(NP)-catalyzed oxidation of cyclopentenone 12, hydroxycyclopentenone 42 was isolated as an intermediate. Dehydration of this compound should lead to cyclopentadienone 43 (cf. refs 5 and 9a).
- 30.For a recent report of Sn (IV)-mediated Baeyer-Villiger reactions using H2O2 as oxidant at elevated temperature, see: Corma A, Iborra S, Mifsud M, Renz M, Susarte M. Adv Synth Catal. 2004;346:257.
- 31.Dong S, Zhu J, Porco JA., Jr J Am Chem Soc. 2008;130:2738. doi: 10.1021/ja711018z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horning EC, Horning MG. J Am Chem Soc. 1947;69:1359. [Google Scholar]
- 33.Patwardhan SA, Gupta AS. Phytochemistry. 1983;22:2080. [Google Scholar]
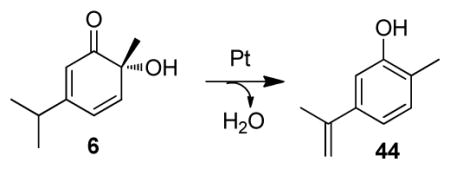
-
34.Phenol byproduct 44 was presumably derived from o-quinol intermediate 6 through an elimination process:
- 35.Dong S, Cahill KJ, Kang MI, Colburn NH, Henrich CJ, Wilson JA, Beutler JA, Johnson RP, Porco JA., Jr J Org Chem. 2011;76:8944. doi: 10.1021/jo201658y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gagnepain J, Castet F, Quideau S. Angew Chem, Int Ed. 2007;46:1533. doi: 10.1002/anie.200604610. [DOI] [PubMed] [Google Scholar]
- 37.Liao CC, Peddinti RK. Acc Chem Res. 2002;35:856. doi: 10.1021/ar000194n. and references therein. [DOI] [PubMed] [Google Scholar]
- 38.For a recent review on the dienone-phenol rearrangement, see: Whiting DA. In: Comp Org Synth. Trost BM, Fleming I, editors. Vol. 3. Pergamon Press; Oxford: 1991. pp. 803–821.
- 39.Li JH, Wang DP, Xie YX. Synthesis. 2005:2193. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.