

Abstract

The ability to monitor intracellular events in real time is paramount to advancing fundamental biological and clinical science. We present the first demonstration of a direct interface of vertically aligned single-walled carbon nanotubes (VASWCNTs) with eukaryotic cells, RAW 264.7 mouse macrophage cell line. The cells were cultured on indium tin oxide with VASWCNTs. VASWCNTs entered the cells naturally without application of any external force and were shown to sense the intracellular presence of a redox active moiety, methylene blue. The technology developed provides an alluring platform to enable electrochemical study of an intracellular environment.
Keywords: Intracellular electrochemical sensing, single-walled carbon nanotubes, eukaryotic cells, vertically aligned carbon nanotubes, indium tin oxide
The development of nanostructured surfaces for biomedical and biotechnological applications is an area of rapidly growing interest.1 In particular, three-dimensional (3D) nanostructured surfaces, based on arrays of vertically aligned nanostructures on a solid support, are attracting much attention owing to their potential cellular applications, including extracellular1c,2 and intracellular sensing3 and direct intracellular delivery.4 During recent years, a number of high-aspect-ratio semiconductor nanostructures have been synthesized to interface with cells. For instance, surface modified vertical silicon nanowires have been shown to penetrate mammalian cells without affecting cell viability5 and deliver biomolecules such as proteins and DNA plasmids into living cells.4 Other examples include cell viability studies using vertical arrays of indium–arsenide (InAs) nanowires6 and development of intracellular pH sensors based on high-density zinc oxide (ZnO) nanorods.3 While these nanoscale interfaces are furthering our ability to monitor and manipulate cellular processes, there are still significant hurdles to overcome in order to fully exploit the technological advantages offered by 3D nanostructured surfaces.
A critical remaining challenge is the development of platforms based on vertically aligned nanostructure for intracellular electrochemical sensing. Problems associated with some of the commonly used fluorescent techniques and challenges to be solved in monitoring the dynamic intracellular environment of cells was recently highlighted by Spiller et al.7 Notably, development of novel approaches for label-free intracellular detection is of great interest, and in this context, electrochemical probing platforms are particularly appealing due to their capability of measuring quantities down to the zeptomole level (allowing detection of trace levels of electroactive species such as proteins) in complex turbid environments.8
By virtue of their efficient electron transport and superior electrical conductivity, carbon nanotubes (CNTs) emerge as front-runners in ultrasensitive, nanostructured electrochemical sensing platform design. Other key advantages that CNTs offer are excellent high aspect ratios, a necessity for any structures needing to span the plasma membrane, high mechanical strength, and superb thermal and chemical stability.9 Furthermore, due to their versatile surface chemistry, CNTs have been functionalized with a number of biomolecules, including DNA,10 proteins11 such as immunoglobulins and enzymes important when requiring analyte specificity, and electrocatalyst probes important for catalyzing biologically relevant electron transfer reactions. Dispersed, randomly oriented CNTs have already been implemented in a number of biomedical applications, from biological imaging12 to CNT-mediated delivery of biomacromolecules.13
In this work, we explore the potential of vertically aligned single-walled carbon nanotubes (VASWCNTs) on an indium tin oxide (ITO) surface for intracellular electrochemical sensing, demonstrating VASWCNTs ability of naturally being inserted within the cytoplasm of a cell and electrochemical probing the intracellular environment. With an excellent combination of high electrical conductivity and high optical transparency, ITO is an attractive platform on which to build VASWCNTs intracellular electrical probes. Our approach involves (Figure 1) four major steps. (1) Formation of NH2-terminated surfaces on ITO surface by electrochemical grafting of an in situ generated diazonium cation.14 (2) The SWCNTs are then vertically aligned and covalently bound to the NH2-terminated surfaces on ITO surface via carbodiimide chemistry. (3) To achieve natural uptake into cells, the VASWCNTs are wrapped with DNA through ionic binding. (4) The resulting DNA-wrapped VASWCTNs are taken up naturally by a mouse macrophage cell and used to sense the intracellular presence of a redox active moiety, methylene blue (MB).
Figure 1.

Diagrammatic representation of (1) the electrochemical grafting of an arylamine to the ITO surface following in situ diazotisation of p-phenylenediamine and (2) its DCC coupling to SWCNTs, yielding VASWCNTs on a ITO surface. (3) Upon wrapping with DNA, (4) the VASWCNTs are capable to be taken up naturally by a mouse macrophage cell and subsequently to be used for intracellular electrochemical communication with the redox active MB moiety.
Results and Discussion
Our initial experiments were focused on establishing for the first time if the in situ generation of an aryl diazonium cation via reduction of p-phenylenediamine with NaNO2 could be electrochemically coupled to an ITO surface via its reduction in situ, forming an arylamine tether layer similar to that previously observed on pyrolyzed photoresist film (PPF) electrodes.15 Two cyclic voltammograms (CVs) were performed under the following conditions, a start potential of 0.4 V, a switching potential of −0.6 V, and an end potential of 0.4 V being applied using the ITO electrode at a scan rate of 0.1 V s–1. For the second scan, the electrode was held at a potential of −0.6 V for 120 s to ensure all the cation had been reduced. The resulting grafting scans at ITO electrodes of in situ generated aryl diazonium cation can be seen in Figure 2A. On scan one an irreversible reduction peak was observed which can be attributed to the reduction of the diazonium cation at a potential of approximately −0.1 V at the ITO surface. Others15,16 have reported the mechanism of surface modification in which the reduction of the diazonium cation leads to the generation of an aryl radical. The aryl radical then attacks the electrode surface forming a covalent bond between the aryl diazonium and the surface. Thus, it appears that the reaction mechanism is the same as previously reported at gold and carbon surfaces.15,16 On the second scan, at the ITO electrode, no subsequent cathodic peak is observed for the reduction of the aryl diazonium cation because the film becomes self-limiting. This is based on the process of multilayer formation by which an increase in thickness leads to a significant increase in the resistance, the potential at the outer surface of the diazonium film is not sufficient anymore to reduce more diazonium cation, and deposition ceases.16a,17
Figure 2.

(A) Two consecutive CVs were performed; on solutions of in situ generated diazonium cation. A starting potential of 0.4 V and a switching potential of −0.6 V at −0.1 V s–1; prior to the second scan the potential was held at −0.6 V for 120 s to confirm surface modification due to the blocking nature of the film. (B) Typical cyclic voltammograms obtained for solutions of 250 μM ferricyanide and 50 mM PBS at modified (in situ grafting of the diazonium cation) and unmodified electrodes.
To confirm surface modification, electrochemical behavior of the probe ferricyanide was performed and the resulting CVs can be seen in Figure 2B. CVs obtained for 250 μM ferricyanide at an unmodified ITO results in the observation of a quasi-reversible redox couple with a peak separation of 0.11 V. A cyclic voltammetric scan was then performed for ferricyanide using an arylamine modified ITO surface as the working electrode, and no redox couple was observed. The absence of a defined reversible redox couple can be attributed to the blocking nature of the arylamine film, as previously reported for other electrochemical mediators at diazonium salt modified surface.14,16a
High-resolution XPS spectra (Figure 3) were also acquired on the ITO electrodes before and after electrochemical grafting of the arylamine to the ITO surface. High-resolution scans of the N (1s) region show the presence of nitrogen on the arylamine modified ITO; importantly, no N (1s) peak was observed on bare ITO, thus confirming the grafting. The N (1s) spectrum of the arylamine modified ITO can be deconvoluted into two peaks with binding energies at 399.9 and 401.2 eV, which are attributed to amino groups (NH2) and protonated (NH3+) amino groups.18
Figure 3.

XPS spectra of the N (1s) peak region of ITO surface before and after the electrochemical grafting of the arylamine.
Following the electrochemical and XPS confirmation of the efficient electrochemical grafting, the viability of using chemical self-assembly of SWCNTs to vertically attach them to the ITO surface was demonstrated by atomic force microscopy (AFM). After performing electrochemical grafting, the SWCNTs were covalently tethered to the ITO surface via carbodiimide chemistry by forming amide bonds between the amino groups on the surface and the carboxylic acid groups on the SWCNTs. For comparison, control surfaces, i.e., bare ITO and electrografted arylamine modified ITO, were also analyzed by AFM. A clear difference was observed between the bare ITO surface (Figure 4A), arylamine modified ITO (Figure 4B) surface, and SWCNT modified ITO surface (Figure 4C). Importantly, the results show bundles of vertically aligned single-walled carbon nanotubes (VASWCNTs) with a maximum length of ∼60 nm (Figure 4C). Further evidence of surface modification of the corresponding tether layer and SWCNTs is given by comparing the roughness of each surface with root-mean-squared surface roughness (rms) values obtained of 2.73 ± 0.12, 3.50 ± 0.83, and 4.87 ± 0.92 nm for bare ITO, arylamine modified ITO, and with covalently coupled SWCNTs, respectively. Roughness values were obtained by sampling surface areas of 1 μm × 1 μm at three randomly selected locations on each of the three samples. A one-way ANOVA was performed to show that the mean values were significantly different (p < 0.000 000 1), demonstrating that with SWCNT modification there is significant increase in roughness as would be expected for a heterogeneous surface modified with varying lengths of SWCNT. Further evidence for surface modification is provided by cyclic voltammograms of 50 mM pH 7 PBS at the modified surfaces (Figure S1). As expected, immobilization of the VASWCNTs on the ITO surface increases the electrode area, resulting in higher charging currents for the SWCNT modified ITO surface, when compared with bare ITO and arylamine modified ITO surfaces.
Figure 4.
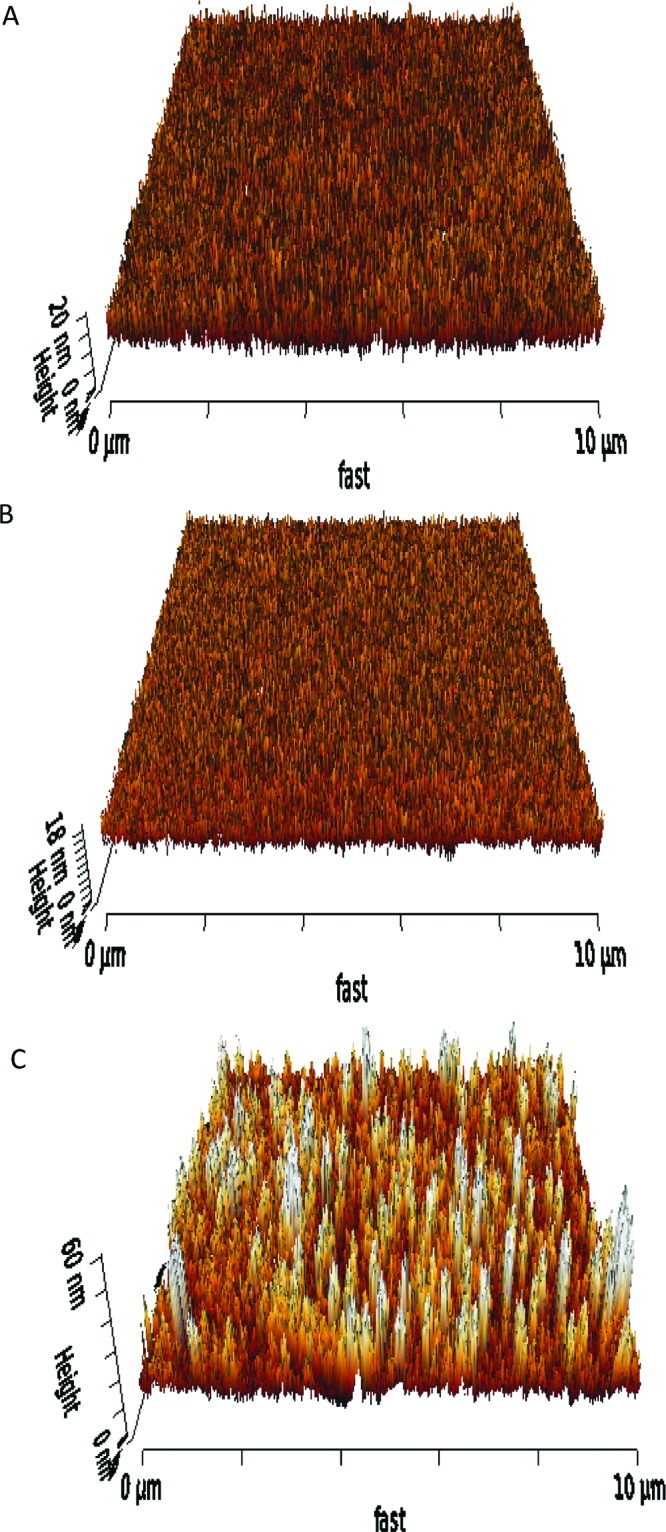
Typical AFM images of (A) bare ITO, (B) arylamine modified ITO, and (C) SWCNT modified ITO.
One of the aims of the research was to enable the efficient uptake of VASWCNTs into the intracellular space, while ensuring the SWCNTs were still attached to the underlying conducting ITO surface, to allow for electrochemical sensing inside the cells. To achieve this, SWCNTs were modified with DNA by denaturing dsDNA by boiling in water for 10 min and then placing the electrodes in the DNA solution and sonicating for 5 min which causes the produced ssDNA to form π-stacking interaction with the SWCNTs.19 In order to confirm the modification of SWCNTs with DNA, the electrochemical characterization of MB at the varying surfaces was performed. MB was used for this purpose at it intercalates with DNA, giving rise to different electrochemical characteristics when compared to its interaction at normal surfaces.20 These studies were also important to allow us to understand the electrochemical behavior of MB at the DNA modified surface as it was our intention to use intracellular MB to show proof of principle that intracellular signaling could be achieved using the fabricated surfaces.
MB has been reported to adsorb to SWCNTs through charge-transfer and hydrophobic interactions21 and and to interact with ssDNA through electrostatic interactions.20,22 Thus, a study was performed to assess the effect on the electron transfer kinetics of MB on the different surfaces. This was done by initially running a cyclic voltammogram from 0 to −0.4 V at 0.1 V s–1 in solutions of 500 μM MB (Figure S2). As the eventual monitoring of MB will be ascertained in solution using electrodes made in batches, it was important to ascertain their reproducibility. The mean cathodic peak current obtained for 500 μM MB at DNA-SWCNT modified ITO electrode was 22.73 μA with a coefficient of variation (SD/mean multiplied by 100) value of 11.1% obtained.
The electrodes were then washed in ethanol and water to remove none adsorbed MB prior to performing voltammograms in PBS, which was performed under the same operating conditions as mentioned previously but at varying scan rates with the following working electrodes: bare ITO, arylamine modified ITO, and SWCNT modified ITO, and the latter two surfaces were modified with DNA. For the ITO surfaces modified with the arylamine no or little adsorption of MB was observed. This can be attributed to the fact that at pH 7 the amino groups are partially protonated,14 meaning that they electrostatically repel the charged MB+ moieties inhibiting MB adsorption. Parts A(I), B(I), and C(I) of Figure 5 show typical cyclic voltammograms obtained in PBS at varying scan rates for bare ITO, SWCNT modified ITO, and SWCNT and DNA modified ITO surfaces, respectively, that were previously exposed to MB. As expected for a surface bound process, current was observed to be proportional to scan rate at bare ITO (Figure 5AII), SWCNT modified ITO (Figure 5BII), and SWCNT and DNA modified ITO (Figure 5CII) electrodes. If the process was under diffusion control, which would mean that the electroactive species was in solution, then current would be proportional to the square root of scan rate. Mean peak separation potentials (ΔEp) obtained at a scan rate of 0.1 V s–1 at bare ITO, SWCNT modified ITO, and SWCNT and DNA modified ITO were 26 ± 4, 50.7 ± 10.2, and 77.7 ± 2.08 mV, respectively. This data shows the kinetics for electron transfer to the electrode is quickest at the bare ITO electrode. The cyclic voltammograms obtained using the DNA modified SWCNT (Figure 5C) demonstrated that with increasing scan rates the ΔEp becomes larger, which is indicative of an electrochemical irreversible process. We can infer form this behavior that the DNA attenuates the rate of electron transfer of MB, and this is due to the electrons having to tunnel through the DNA.23 Additionally, this result also confirms the presence of DNA as this decrease in electron transfer rate over the scan range studied is significantly lower at surfaces modified with SWCNT only.
Figure 5.

Typical cyclic voltammograms obtained for solutions of pH 7. PBS at (AI) bare ITO, (BI) SWCNT modified ITO, and (CI) SWCNT and DNA modified ITO electrodes with absorbed MB at scan rates of 0.02, 0.05, 0.1, 0.2, and 0.4 V s–1. Plots of anodic and cathodic peak currents at different scan rates at (AII) ITO, (BII) SWCNT modified ITO, and (CII) SWCNT and DNA modified ITO.
In order to demonstrate that the VASWCNTs structures penetrate the plasma membrane, which is ∼5 nm thick, and have access to the cytoplasm, mouse macrophage cells were stained with MB. It is well established that MB can enter and is retained inside the cell,23 and most important, it is a redox active moiety that can be used as a fingerprint to identify if the nanostructures have accessed the cytoplasm.23 After performing experiments to establish the electrochemical characteristics of MB at the SWCNT and DNA modified electrode as described above, we performed transformation assays using Ca2+ to enable the cells to take up the SWCNTs. Evidence that SWCNT–DNA structures are positioned inside the cell is obtained by observing the characteristic MB redox peaks at approximately −0.233 and −0.128 V (Figure 6A, blue CV). Additionally, MB can be observed in the microscopic image in the inset of Figure 6. A control experiment was performed in an identical surface (i.e., SWCNT and DNA modified ITO) as above, but the cells were not incubated in MB. This control experiment did not display any redox peaks (Figure 6A, red CV), providing further support that the voltammetric peaks observed on the SWCNT and DNA modified ITO surfaces were due to the reduction and oxidation of MB within the cells.
Figure 6.

(A) CVs of MB stained cells on SWCNT and DNA modified ITO (blue curve) and SWCNT modified ITO with no DNA modification surfaces (green curve). CV (red curve) of cells not stained on a SWCNT and DNA modified ITO surface. Inset is a typical microscopic image of MB stained cells on the nanostructured electrodes.
It is also demonstrated that in order to observe the electrochemistry of MB the SWCNTs have to be modified with DNA. A control experiment, wherein SWCNTs modified ITO surfaces in the absence of DNA were subjected to the same transformation procedure as above, shows no redox chemistry occurring (Figure 6A, green CV). Thus, in order to transform the cells with surface-immobilized SWCNTs, it was essential that DNA was present. Recently, ssDNA modified SWCNTs have been shown, when internalized by an immune cell, not to induce an immune response.27 This demonstrates that ssDNA modified SWCNTs are biocompatible which is an important trait for any proposed intracellular sensing system utilizing a immune cell line such as used in current investigations.
With reasonable assumptions, it is possible to estimate the area and number of SWCNTs inside the cells that were attached to an area of 0.123 cm2, i.e., ITO electrode area exposed to the electrolyte. The magnitude of the peak current in the CV for SWCNT and DNA modified ITO (Figure S2, light blue curve) provides information regarding the electroactive area of the SWCNT–DNA system according to the Randles–Sevcik equation:
| 1 |
where ip is the peak current (2.66 × 10–5 A), n (equal to 2) is the number of electrons involved in the MB redox reaction, ν is the scan rate (0.10 V s–1), A is the electroactive area to be determined (cm2), D is the diffusion coefficient of MB in solution (4.0 × 10–6 cm2 s–1),24 and C is the bulk concentration of the MB solution (5.0 × 10–7 mol cm–3). The electroactive area, A, was calculated and was found to be 0.111 cm2. By knowing the relationship between peak current (2.66 × 10–5 A) and electroactive area (0.111 cm2) of the SWCNT–DNA system, it is now possible to calculate the electroactive area responsible for the peak current obtained for SWCNT based electrodes that the cells internalized (1.17 × 10–6 A, Figure 6, blue curve). Thus, the value of the electroactive area of SWCNT intracellular electrodes became 4.88 × 10–3 cm2. By considering that the maximum length of SWCNTs is up to 60 nm as demonstrated by the AFM images and the distance between the cells membrane and substrate is between 20 and 40 nm,25 we still have a length of SWCNT of up to 20–40 nm that can cross into the cytoplasm. Taking into account that the cell membrane is 4–5 nm,26 15–35 nm of the SWCNT can be inside the cell. If we assume that the diameter of all SWCNTs is 1 nm, a surface area per SWCNT between 4.79 × 10–13 cm2 (i.e., SWCNTs with an intracellular length of 15 nm) and 1.11 × 10–12 cm2 (i.e., SWCNTs with an intracellular length of 35 nm) can be in contact with cytoplasm. If we then divide the total electroactive area inside the cells (4.88 × 10–3 cm2) by the surface area per SWCNT (4.79 × 10–13–1.11 × 10–12 cm2), we can derive that approximately between 4.41 × 109 and 1.02 × 1010 SWCNTs have been taken up by cells that were attached to an ITO electrode area of 0.123 cm2.
Conclusions
In summary, we have shown that short VASWCNTs coupled to an arylamine tether layer on an ITO substrate were vertically aligned. These were further modified with DNA, which enables cells to naturally take up carbon nanotubes, and these remained covalently bound to the surface of the electrode via the arylamine tether layer. Once the SWCNTs had entered the cell, intracellular communication was established by sensing the electrochemical mediator methylene blue. Establishment of a generic entry mechanism for SWCNTs that remain covalently bound to a substrate is of fundamental importance and will facilitate future sensing platform that may be used for unprecedented spatial and temporal electrochemical intracellular biosensing.
Experimental Section
Chemicals
All chemicals were purchased from Sigma Aldrich unless otherwise stated. Phosphate buffered saline was prepared by mixing solutions of 50 mM K2HPO4 and 50 mM KH2PO4 to give the desired pH 7 and adding an appropriate amount of potassium chloride to give a final concentration of 0.1 M saline in 50 mM phosphate buffer. The PBS solution was prepared fresh each day. Methylene blue stock solution was prepared at a concentration of 10 mM in sterile PBS.
Electrode Preparation
Initially, ITO deposited on Corning low alkaline earth boro-aluminosililicate glass with indium tin oxide coated on one surface (resistivity = 20–25 Ω) purchased from Delta Technologies Limited was rinsed with HPLC grade ethanol and then ultrahigh purity H2O, exposed to UV light for an hour, and then rinsed in acetone and isopropyl alcohol. The strategy for preparation of functionalized SWCNTs covalently attached to ITO was based on using an electrografted tether layer which then acted as an anchor for the SWCNTs. ITO samples were modified in situ with p-phenylenediamine (AP) using a similar reported method for carbon surfaces.28 At room temperature, a 10 mM p-phenylenediamine solution was rapidly added to 1 M NaNO2 equivalent solution and allowed to react for 3 min. The reaction solution was then poured into the electrochemical cell, and the resultant diazonium cation was electrografted to the ITO scanning from 0.4 to −0.6 V followed by fixed potential deposition at −0.6 V for 2 min and finally a further scan from 0.4 to −0.6 V to confirm passivation (and hence grafting) of the electrode.
Uncut SWCNTs (NanoLab, Inc.) were acid-treated by adding 25 mg to 27 mL of a 3:1 mixture of concentrated H2SO4 and HNO3 and sonicating for 10 h. Following sonication, the contents were poured into 500 mL of distilled water and left to settle overnight. The SWCNTs were then filtered through a 0.22 μm hydrophilic PVDF filter (Millipore) under suction, with washing until the rinse water was close to pH 7. Once dried, the SWCNT mats were peeled off the filter disks. Suspensions of cut SWCNTs were prepared by sonication of dried SWCNTs mats in dimethyl sulfoxide (DMSO). SWCNTs were coupled to arylamine modified ITO samples by submerging modified ITO samples in a 0.2 mg mL–1 DMSO suspension of cut SWCNTs containing 0.5 mg mL–1 dicyclohexyl carbodiimide (DCC). The reactants were sonicated for 30 min and then heated to 65 °C for 24 h in a closed cell. After preparation, SWCNT modified electrodes were sonicated in acetone for 2 min and isopropyl alcohol for 10 s and finally rinsed in Milli-Q water. The electrodes were dried with argon gas between each washing step.
SWCNT were modified with DNA on the surface of the ITO electrodes by using an adapted method of that described by Zheng et al.19 First, DNA, obtained from salmon sperm, was denatured via placement of 2 mg mL–1 DNA into boiling water for 30 min. An aqueous solution of ssDNA (1 mg mL–1) was then prepared, and SWCNT modified ITO sample was immersed and sonicated for 5 min to allow π-stacking interactions to form between the DNA and SWCNTs.
Electrochemistry
All electrochemical studies were carried out with a Gamry 600 potentiostat and data acquisition software (Gamry electrochemistry software version 5.61a) and a three-electrode cell consisting of a saturated calomel reference electrode, Pt counter electrode, and then the working electrode of either bare ITO, ITO modified with an arylamine tether layer, an ITO modified with SWCNT, or ITO modified with SWCNT and ssDNA. The electrochemical area was controlled via use of a O-ring with a diameter of 4 mm.
Cyclic voltammetry was performed to confirm grafting of in situ produced diazonium cation by running scans of 250 μM solution of ferricyanide in 50 mM PBS (0.1 M KCl) from a starting potential of 1.0 V and a switching potential of −0.4 V and an end potential of 1.0 V at arylamine modified and unmodified surfaces. Cyclic voltammetric studies were performed to characterize the electrochemical behavior of MB at the various surfaces. The operating conditions for the cyclic voltammetry was a start potential of 0 V with a switching potential of −0.4 V at a scan rate of 0.1 V s–1. Controls of PBS were performed at each surface and 500 μM solution of methylene blue was utilized. For surface adsorption studies, the scan rate was varied between 0.02 and 0.4 V s–1. Prior to performing any scans on the electrodes exposed to MB, surfaces were rinsed in ethanol and water. Peak currents were obtained by subtracting the background using Linkfit software and measuring the cathodic peak current of three replicates.
AFM images were recorded using the Nanowizard II atomic force microscope (JPK instruments, Germany). The images were obtained with cantilever model RTESP part MPP-11100-11 (VEECO) operating in tapping mode at a frequency of 345–384 kHz; 10 μm × 10 μm scans were recorded at 1 μm s–1. Postprocessing of AFM images was performed on Version 3.6 Nanowizard image processing software and included digital leveling of the images and roughness calculations. Surface characterization was performed on three samples of each surface which included bare ITO, arylamine modified ITO, and SWCNT modified ITO.
X-ray Photoelectron Spectroscopy
X-ray photoelectron spectroscopy (XPS) spectra were obtained on the VG ESCAlab 250 instrument based at the Leeds EPSRC Nanoscience and Nanotechnology Research Equipment Facility (LENNF) at the University of Leeds, UK. XPS experiments were carried out using a monochromatic Al Kα X-ray source (1486.7 eV) and a takeoff angle of 90°. A high-resolution scan of N (1s) was recorded using a pass energy of 20 eV at a step size of 0.05 eV. Fitting of XPS peaks was performed using the Avantage V 2.2 processing software. Sensitivity factors used in this study were the following: N (1s), 1.73; C (1s), 0.298; O (1s), 0.711; In (3d 5/2), 13.32; In (3d 3/2), 9.22; Sn (3d 5/2), 14.8; Sn (3d 3/2), 10.25.
Cell Assays
Raw 264.7 cells (2.5 × 106 cells) were seeded in a 75 cm2 flask containing 12 mL of DMEM (10% FBS, 1 mL of penicillin/streptomycin, 2.4% glutamate, and 2.4% HEPES). The cells were grown for 3 days and reached an 80% confluence. The cells were then harvested and centrifuged at 3000 rpm for 3 min. The waste was decanted, and the cell pellet was resuspended in 1 mL of DMEM. Subsequently, the cells were counted, giving 13.0 × 106 cells/mL. This was further diluted, yielding a solution of 1 × 106 cells/mL. An aliquot of 10 mM stock solution of MB was added to cells, giving a final assay concentration of 25 μM. The cells were incubated for 30 min with MB and then washed three times in PBS in order to remove MB in solution. The cell pellet was then resuspended in DMEM to give final cell count of 1 × 106 cells/mL. 2 mL of DMEM and 2 mL of HBS solution were then aliquoted into a 6-well plate (12.5 μL of 2 × HBS 8.0 g of NaCl, 0.37 g of KCl, 201 mg of Na2HPO4·7H2O, 2.0 g of glucose, 5.0 g of HEPES/500 mL, pH 7.05), 65 μL of 1 M calcium chloride and 1 mL of the 1 × 106 cell suspension as added to a 6 well titer plate containing the ITO-SWCNT modified with DNA in the presence of calcium and incubated for 13 h. The electrodes were rinsed with DMEM, and electrochemical analysis was performed in PBS.
Acknowledgments
We thank the Leverhulme Trust grant number F/00 094/BD and Wellcome Trust Value-in-People Award for financial support. This research was in part supported through Birmingham Science City: Innovative Uses for Advanced Materials in the Modern World (West Midlands Centre for Advanced Materials Project 2). We thank Dr. John Loring for the use of Linkfit curve-fitting software.
Supporting Information Available
Figures S1 and S2. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- a Mendes P. M.; Yeung C. L.; Preece J. A. Nanoscale Res. Lett. 2007, 2, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mendes P. M. Chem. Soc. Rev. 2008, 37, 2512. [DOI] [PubMed] [Google Scholar]; c Rawson F. J.; Garrett D. J.; Leech D.; Downard A. J.; Baronian K. H. R. Biosens. Bioelectron. 2011, 26, 2383. [DOI] [PubMed] [Google Scholar]
- a Peng L.; You S.-J.; Wang J.-Y. Biosens. Bioelectron. 2010, 25, 1248. [DOI] [PubMed] [Google Scholar]; b Timur S.; Anik U.; Odaci D.; Gorton L. Electrochem. Commun. 2007, 9, 1810. [Google Scholar]
- Willander M.; Al-Hilli S.; Lee J. W. F. R. S. Micro- and Nanotechnol. Bioanal.: Methods Protocols 2009, 544, 187. [Google Scholar]
- Shalek A. K.; Robinson J. T.; Karp E. S.; Lee J. S.; Ahn D.-R.; Yoon M.-H.; Sutton A.; Jorgolli M.; Gertner R. S.; Gujral T. S.; MacBeath G.; Yang E. G.; Park H. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W.; Ng J. K.; Kunitake M. E.; Conklin B. R.; Yang P. J. Am. Chem. Soc. 2007, 129, 7228. [DOI] [PubMed] [Google Scholar]
- Berthing T.; Bonde S.; Sørensen C. B.; Utko P.; Nygård J.; Martinez K. L. Small 2011, 7, 640. [DOI] [PubMed] [Google Scholar]
- Spiller D. G.; Wood C. D.; Rand D. A.; White M. R. H. Nature 2010, 465, 736. [DOI] [PubMed] [Google Scholar]
- Bauer C. G.; Eremenko A. V.; EhrentreichForster E.; Bier F. F.; Makower A.; Halsall H. B.; Heineman W. R.; Scheller F. W. Anal. Chem. 1996, 68, 2453. [DOI] [PubMed] [Google Scholar]
- Yang W.; Ratinac K. R.; Ringer S. P.; Thordarson P.; Gooding J. J.; Braet F. Angew. Chem., Int. Ed. 2010, 49, 2114. [DOI] [PubMed] [Google Scholar]
- Lee C.-S.; Baker S. E.; Marcus M. S.; Yang W.; Eriksson M. A.; Hamers R. J. Nano Lett. 2004, 4, 1713. [Google Scholar]
- a Gooding J. J.; Wibowo R.; Liu; Yang W.; Losic D.; Orbons S.; Mearns F. J.; Shapter J. G.; Hibbert D. B. J. Am. Chem. Soc. 2003, 125, 9006. [DOI] [PubMed] [Google Scholar]; b Yu X.; Kim S. N.; Papadimitrakopoulos F.; Rusling J. F. Mol. Biosyst. 2005, 1, 70. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Yang K.; Lee S.-T. J. Mater. Chem. 2011, 21, 586. [Google Scholar]
- Kam N. W. S.; Liu Z. A.; Dai H. J. Angew. Chem., Int. Ed. 2006, 45, 577. [DOI] [PubMed] [Google Scholar]
- Garrett D. J.; Flavel B. S.; Shapter J. G.; Baronian K. H. R.; Downard A. J. Langmuir 2009, 26, 1848. [DOI] [PubMed] [Google Scholar]
- Brooksby P. A.; Downard A. J. Langmuir 2004, 20, 5038. [DOI] [PubMed] [Google Scholar]
- a Maldonado S.; Smith T. J.; Williams R. D.; Morin S.; Barton E.; Stevenson K. J. Langmuir 2006, 22, 2884. [DOI] [PubMed] [Google Scholar]; b Laurentius L.; Stoyanov S. R.; Gusarov S.; Kovalenko A.; Du R.; Lopinski G. P.; McDermott M. T. ACS Nano 2011, 5, 4219. [DOI] [PubMed] [Google Scholar]
- Lehr J.; Williamson B. E.; Downard A. J. J. Phys. Chem. C 2011, 115, 6629. [Google Scholar]
- Yeung C. L.; Iqbal P.; Allan M.; Lashkor M.; Preece J. A.; Mendes P. M.. Adv. Funct. Mater. 2010, not supplied.
- Zheng M.; Jagota A.; Semke E. D.; Diner B. A.; McLean R. S.; Lustig S. R.; Richardson R. E.; Tassi N. G. Nature Mater. 2003, 2, 338. [DOI] [PubMed] [Google Scholar]
- Kelley S. O.; Barton J. K.; Jackson N. M.; Hill M. G. Bioconjugate Chem. 1997, 8, 31. [DOI] [PubMed] [Google Scholar]
- Yan Y.; Zhang M.; Gong K.; Su L.; Guo Z.; Mao L. Chem. Mater. 2005, 17, 3457. [Google Scholar]
- Pan D.; Zuo X.; Wan Y.; Wang L.; Zhang J.; Song S.; Fan C. Sensors 2007, 7, 2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F.; Yang J.; Liu T.; Zhu X.; Li G. Anal. Chem. 2009, 81, 9168. [DOI] [PubMed] [Google Scholar]
- Bronzino J. D.The Biomedical Engineering Handbook, 2nd ed.; CRC Press: Boca Raton, FL. [Google Scholar]
- a Lo C. M.; Keese C. R.; Giaever I. Biophys. J. 1995, 69, 2800. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pierres A.; Benoliel A. M.; Bongrand P. Eur. Cell. Mater. 2002, 3, 31. [DOI] [PubMed] [Google Scholar]; c Estrada-Bernal A.; Gatlin J. C.; Sunpaweravong S.; Pfenninger K. H. J. Cell Sci. 2009, 122, 2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mitra K.; Ubarretxena-Belandia I.; Taguchi T.; Warren G.; Engelman D. M. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 4083. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Boal D.Mechanics of the Cell; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]
- Medepalli K.; Alphenaar B.; Raj A.; Sethu P. Nanomed. Nanotech. Biol. Med. 2011, 7, 983. [DOI] [PubMed] [Google Scholar]
- Boland S.; Barriere F.; Leech D. Langmuir 2008, 24, 6351. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.