

Abstract
The development of Aβ-PET imaging agents has allowed for detection of fibrillar Aβ deposition in vivo and marks a major advancement in understanding the role of Aβ in Alzheimer’s disease (AD). Imaging Aβ thus has many potential clinical benefits: early or perhaps preclinical detection of disease and accurately distinguishing AD from dementias of other non-Aβ causes in patients presenting with mild or atypical symptoms or confounding comorbidities (in which the distinction is difficult to make clinically). From a research perspective, imaging Aβ allows us to study relationships between amyloid pathology and changes in cognition, brain structure, and function across the continuum from normal aging to mild cognitive impairment (MCI) to AD; and to monitor the effectiveness of anti-Aβ drugs and relate them to neurodegeneration and clinical symptoms. Here, we will discuss the application of one of the most broadly studied and widely used Aβ imaging agents, Pittsburgh Compound-B (PiB).
I. Introduction
Alzheimer’s disease (AD) is the most common cause of dementia in the elderly and its prevalence is increasing at an alarming rate, with a worldwide prevalence estimated to quadruple over the next 50 years. AD is pathologically characterized by the presence of amyloid plaques, containing amyloid-β (Aβ), and neurofibrillary tangles (NFT), containing hyperphosphorylated tau, as well as significant loss of neurons and deficits in neurotransmitter systems. A growing consensus points to deposition of Aβ plaques as a central event in the pathogenesis of AD. This “amyloid cascade hypothesis” (Hardy & Allsop, 1991; Hardy & Higgins, 1992) states that overproduction of Aβ, or failure to clear this peptide, leads to AD primarily through amyloid deposition, which triggers the production of NFT, cell death and, ultimately, the clinical symptoms such as memory loss and cognitive impairment (Hardy et al., 1998). Further, the presence of Aβ in AD has been associated with synaptic loss (for review see Wilcox et al., 2011), which is significantly correlated with cognitive impairment in AD (DeKosky et al., 1996; Terry et al., 1991;). The single, most important piece of evidence for this “amyloid cascade hypothesis” of AD is the demonstration that at least five different mutations in the Aβ precursor protein (APP) gene on chromosome 21, all lying in or near the Aβ peptide region, cause early-onset AD (Hardy et al.,1998; Price & Sisodia, et al., 1998; Tanzi et al., 1996). Further genetic support for the amyloid cascade hypothesis comes from the finding that the most common form of early-onset, autosomal dominant, familial AD (eoFAD) (the chromosome 14 mutations) is caused by mutations in the presenilin-1 (PS1) gene which codes for a protein that is a component of the “γ-secretase” enzyme complex responsible for C-terminal cleavage of Aβ from APP (Xia et al., 2000).
II. Rationale for Studying Amyloid Deposition
Definitive diagnosis of AD relies on the demonstration of sufficient amounts of Aβ plaques and NFT in autopsy brains (Mirra et al., 1991). Imaging Aβ thus has many potential clinical benefits: early or perhaps pre-clinical detection of disease and accurately distinguishing AD from non-Aβ causes of dementia in patients with mild or atypical symptoms or confounding comorbidities (in which the distinction is difficult to make clinically). From a research perspective, imaging Aβ allows us to study relationships between amyloid, cognition, and brain structure, and function across the continuum from normal aging to AD; and to monitor the biological effects of anti-Aβ drugs and relate them to effects on neurodegeneration and cognition. Here, we will discuss the application of one of the most broadly studied and widely used agents, Pittsburgh Compound-B (PiB).
III. General Properties of the Aβ Imaging Tracer, PiB
PiB (also known as [11C]6-OH-BTA-1 or [N-methyl-11C]2-(4’-methylaminophenyl)-6-hydroxybenzothiazole (Mathis et al., 2003)) is a thioflavin-T (ThT) derivative, a small molecule known to bind amyloid proteins aggregated into a beta-pleated sheet structure (Levine 1995). Figure 1 demonstrates the steps in development of PiB from ThT. The first step removed the methyl group from the positively charged quaternary heterocyclic nitrogen of the benzothiazolium group of ThT, yielding a compound called 6-Me-BTA-2. This alteration produced increased brain entry of the compound and improved the Aβ binding affinity and highly decreased the NFT binding affinity relative to the parent compound ThT. The inhibition constant (Ki, a measure of binding affinity closely related to the Kd (Bennett & Yamamura, 1985)) of 6-Me-BTA-2 for fibrillar Aβ was nearly ten times lower than ThT, although it did not reach the desired binding affinity of <10 nM (Fig. 1). Additionally, the brain clearance of 6-Me-BTA-2 from normal brain was very poor and brain levels actually increased 2-fold over 30 min; therefore, two additional methyl groups were removed from 6-Me-BTA-2, creating a compound known as BTA-1, which showed significantly better Aβ affinity, brain entry, and clearance (Mathis et al., 2003). However, the 6-hydroxy derivative of BTA-1 (6-OH-BTA-1 or PiB) had a better Aβ affinity, with a Ki of 4.3 nM (surpassing the initial goal of 10 nM) and a better normal brain clearance, with a 2’:30’ ratio of 12 (normal brain clearance t1/2 ~7.9 min) and was used for further human studies (Fig. 1).
FIGURE 1.

Chemical structures, lipophilicity (logPoct), Aβ binding affinity (Ki), and brain entry [%Injected Dose Index (%IDI) or (%ID × g body weight)/g brain weight] and brain clearance (2’:30’ ratio) of thioflavin-T, PiB and intervening derivatives. Numbers in parentheses indicate targets for each parameter.
IV. Early Human PiB Studies
The first human positron-emission tomography (PET) imaging studies with PiB were a collaboration between the University of Pittsburgh and Uppsala and Karolinska Universities (Engler et al., 2002; Klunk et al., 2004). This study included 16 AD patients, six elderly age-matched controls, and three young controls, chosen because of the likelihood that most would be amyloid negative. The healthy control (HC) subjects showed rapid entry and clearance of PiB from all cortical and subcortical grey matter areas, including the cerebellum (Fig. 2). Nearly identical uptake and clearance of PiB was seen in the cerebellum of HC and AD groups (Fig. 2A), an area of the brain known to have few fibrillar Aβ deposits. Subcortical white matter showed relatively lower entry and slower clearance in both HC subjects and AD patients compared to grey matter areas (Fig. 2B). However, in AD patients, markedly increased PiB retention was observed in brain areas known to contain high levels of amyloid plaques when compared to HC subjects, including brain regions such as parietal and frontal cortices (Figs. 2C–E) (Arnold H et al., 1991; Thal, Rub, Orantes, & Braak, 2002).
FIGURE 2.

Standardized uptake values (SUV; 1.0 SUV = 0.10 %IDI) demonstrating brain entry and clearance of PiB in varying brain regions. For color version of this figure, the reader is referred to the online version of this book.
(from Klunk et al., 2004)
The pattern of PiB retention was quite different in AD patients compared to the HC subjects (Fig. 3). PiB retention in AD patients was generally most prominent in cortical areas and lower in white matter areas, in a manner most consistent with postmortem studies of Aβ plaques in the AD brain (Thal et al., 2002). PiB retention was broadly observed in frontal cortex in AD, but also was observed in precuneus/posterior cingulate, temporal, and parietal cortices. The occipital cortex and lateral temporal cortex were also significantly affected with a relative sparing of the mesial temporal areas. Significant striatal PiB retention was also observed, consistent with previous reports of extensive Aβ deposition in the striatum of AD patients (Braak & Braak, 1990; Brilliant et al., 1997; Suenaga et al., 1990; Wolf et al., 1999). PiB images from HC subjects showed little or no PiB retention in cortical areas, and the accumulation of PiB in white matter was the same in AD patients and HC subjects (Fig. 2B).
FIGURE 3.
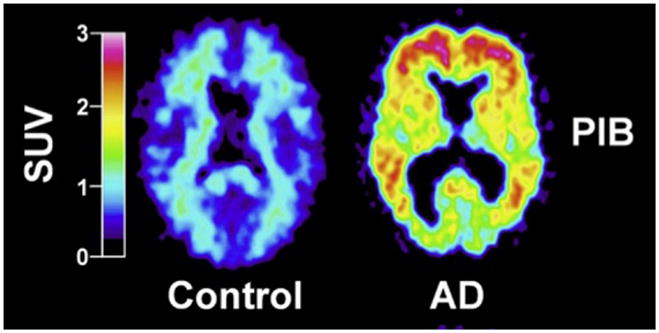
PIB standardized uptake value images demonstrate a marked difference between PIB retention in AD patients and HC subjects. For color version of this figure, the reader is referred to the online version of this book.
(from Klunk et al., 2004)
In the initial PiB-PET study three AD subjects displayed cortical PiB retention at the level of HC subjects – this is not a particularly surprising finding when one considers previous reports from postmortem studies that some people clinically diagnosed with AD do not have Aβ deposits at autopsy (Haroutunian et al., 1998; Price and Morris, 1999). Indeed, these three AD patients performed well on the mini-mental status exam and showed no significant cognitive deterioration over the 2–4 year follow-up period after the PiB study (i.e., MMSE remained 28–29) while the AD patients with significant PiB retention showed deterioration typical of clinical AD. Additionally, in the elderly HC group, the oldest subject (76 y/o) consistently showed the highest cortical PiB retention, consistent with postmortem studies identifying elderly HC subjects with significant amyloid deposits (Bennet et al., 2006). It was recognized very early that it would be critical to longitudinally follow PiB retention in these discordant subjects (i.e., clinical AD-absent PiB or HC-significant PiB) in order to gain insight into the natural history of Aβ deposition and the role it may (or may not) play in cognitive decline and clinical AD.
The initial PiB study was followed by a 2-year follow-up study which examined the clinical history of three PiB-negative [PiB(−)] AD patients and the PiB-positive [PiB(+)] HC subject (Engler et al., 2006). At 2-year follow-up, all three of the PiB-negative AD subjects were reclassified as mild cognitive impairment (MCI)—although it is not clear if this was by clinicians blinded to the PiB-PET results. The single PiB(+) HC subject showed no change in cognition or regional cerebral metabolic rate of glucose (rCMRglc), measured with Flurodeoxyglucose (FDG)-PET over the follow-up period, and little increase in PiB retention. These data suggest that the PET result was either false positive, if PiB retention followed a fairly rapid course, or true positive if PiB retention began long before clinical symptoms and followed a fairly lengthy course. These original studies provided a landmark description of the natural history of Aβ deposition in living subjects, and were later confirmed by additional studies using PiB in AD patients and cognitively normal subjects (Archer et al., 2006; Buckner et al., 2005; Edison et al., 2006; Fagan et al., 2006; Fagan et al., 2007; Jack et al., 2009; Kemppainen et al., 2006; Lopresti et al., 2005; Mintun et al., 2006; Nelissen et al., 2007; Price et al., 2005; Pike et al., 2007; Rabinovici et al., 2007; Rowe et al., 2007; Ziolko et al., 2006).
V. Amyloid Imaging and Apolipoprotein-E Genotype
Apolipoprotein E (ApoE) is a 299 amino-acid protein involved in lipid transport and metabolism in the periphery and in brain. ApoE plays a key role in neuronal maintenance and repair (for review see Mahley et al., PNAS 2006). The ApoE gene, found on chromosome 19, has three common isoforms: ε3 (allele frequency 65–70%), ε2 (5–10%), and ε4 (15–20%). The ε4 allele (ApoE4) is by far the strongest genetic risk factor for sporadic AD, associated with a 3-fold increased risk in heterozygotes and up to a 15-fold increased risk of AD in homozygotes (Farrer et al., 1997), while ApoE2 may be protective. ApoE4 has been implicated in multiple aspects of AD pathogenesis, including Aβ fibrillization and clearance (Mahley et al., 2006). Autopsy studies have demonstrated an increased likelihood of AD pathology in cognitively normal individuals who are ApoE4 carriers (Kok et al., 2009). Similarly, PiB-PET studies have found that ApoE4 genotype is associated with higher PiB retention in cognitively normal elderly in a dose-dependent manner (Reiman et al., 2009, Morris et al., 2010), and ApoE4 carriers are more than twice as likely to convert from PiB(−) to PiB(+) over time (Vlassenko et al., 2011). Conversely, ApoE2 has been associated with lower PiB retention in normal elderly (Morris et al., 2010). MCI patients who are ApoE4 carriers consistently show higher PiB retention than MCI noncarriers, though this is at least in part because the presence of ApoE4 increases the likelihood that MCI symptoms are due to underlying AD (Kemppainen et al., 2007; Rowe et al., 2007). Findings in AD patients have been mixed, with some studies demonstrating increased PiB retention in ApoE4 carriers cross-sectionally (Drezga et al., 2008) and longitudinally (Grimmer et al., 2010), while other studies did not find differences between ApoE4 carriers and noncarriers in AD (Klunk et al., 2004; Rowe et al., 2007; Rabinovici et al., 2010). Similarly, ambiguous results have been reported in the AD postmortem literature (Berg et al., 1998; Gomez-Isla et al., 1996). Amyloid imaging will be helpful in further elucidating the links between ApoE, Aβ, neurodegeneration, and cognition across the AD continuum.
VI. Amyloid Imaging in Normal Controls
Several studies have now demonstrated PiB retention in cognitively normal controls. Depending on the site, reports have ranged from a proportion of 10–30% of normal elderly subjects with significant PiB retention [i.e., PiB(+)] (Aizenstein et al., 2008; Jack et al., 2008; Kantarci et al., 2012; Klunk et al., 2004; Mintun et al., 2006; Mormino et al., 2009; Mormino et al., 2011; Pike et al., 2007; Reiman et al., 2009; Rowe et al., 2010; Villemagne et al., 2008). This wide range likely depends on factors such as the age of the cohort, proportion of subjects carrying the ApoE4 allele, definition of “cognitively normal,” and the threshold for defining amyloid-positivity. The relationship between increased PiB retention and cognition in the normal elderly has been difficult to define. It is apparent that among cognitively normal subjects, significant plaque load is not related to broad differences in cognitive performance between groups with and without significant PiB retention (Aizenstein et al., 2008; Jack et al., 2008; Mintun et al., 2006; Rowe et al., 2010). In other studies, an increase in PiB retention has been associated with poorer performance on episodic memory tests (Kantarci et al., 2012; Mormino et al., 2009; Pike et al., 2007; Villemagne et al., 2008). More consistently, PiB(+) cognitively normal individuals show, at a group level, “AD-like” changes in brain structure and network connectivity and activity (see PiB and MRI section, Section XIV: B, below). Most significantly, longitudinal studies have found that cognitively normal individuals with elevated PiB are at much higher risk for longitudinal cognitive decline and the emergence of clinically significant cognitive impairment than PiB(−) age and education matched subjects (Morris et al., 2010; Resnick et al., 2010; Storandt et al., 2009; Villemagne et al., 2008; Villemagne et al., 2011a). These data have led to the hypothesis that, at least in many older individuals, PiB-positivity is a marker for preclinical AD (Sperling et al., 2011).
VII. Amyloid Imaging in MCI
In early studies of MCI subjects, PiB appeared to show a bimodal distribution, with 60–75% of subjects showing a typical, AD-like pattern and burden of PiB retention, while the remaining subjects showed levels typical of PiB(−) controls (Jack et al., 2008; Lopresti et al., 2005; Price et al., 2005; Rowe et al. 2007). Variations in PiB retention have also been explored when examining MCI subjects based on MCI subtype; subjects with nonamnestic MCI were much less likely to be PiB(+) than subjects with amnestic MCI, further suggesting that PiB may be superior to FDG in distinguishing MCI subtypes (Lowe et al., 2009; Pike et al., 2007). These studies have suggested that the nonamnestic MCI subtype may include depression or incipient dementia where Aβ deposition is not a feature [e.g., frontotemporal or vascular dementia (VaD)], or they may prove to be part of the 5–10% who have stable MCI, or the 20% who revert to apparent normality (Busse et al., 2006; Gauthier et al., 2006).
Longitudinal studies have suggested that MCI subjects with high PiB retention are much more likely to convert to AD than subjects with low PiB retention. In a study by Forsberg and colleagues (Forsberg et al., 2007), all 7 MCI-to-AD converters were amyloid-positive at baseline and 9 of the 14 nonconverters were amyloid-negative. In addition, none of the baseline PiB(−) MCI subjects converted to AD. This effect has also been observed in several subsequent studies, with MCI subjects with increased PiB retention showing much more frequent conversion to AD (Koivunen et al., 2011; Villemagne et al., 2011a; Wolk et al., 2009). Therefore, amyloid PET is likely to have a prognostic role in the clinical evaluation of MCI, by identifying subjects who have underlying AD pathophysiology and are therefore at high risk for further clinical decline (Albert et al., 2011).
VIII. Amyloid Deposition in Early-Onset, Autosomal Dominant, Familial AD
Roughly 1% of all AD cases are caused by single gene mutations that are transmitted in an autosomal dominant pattern with nearly 100% penetrance. Familial AD has been linked to mutations in presenilin-1 (PS1, chromosome 14, the most commonly involved gene), amyloid precursor protein (APP, chromosome 21) or presenilin-2 (PS2, chromosome 1). All these mutations are thought to cause eoFAD by promoting the cleavage of APP to the proaggregatory Aβ1–42 peptide (Hardy et al., 1998). In order to explore the natural history of preclinical amyloid deposition in people at high risk for AD, individuals with eoFAD have been evaluated in several studies. In the first PiB-PET study, subjects with two different PS1 mutations were explored (Klunk et al., 2005). The PS1 mutation carriers, both symptomatic and asymptomatic, showed a strikingly similar, focal amyloid deposition that appeared to begin in the striatum (Fig. 4). This is in contrast to early deposition of amyloid in nonmutation carriers, typically in the frontal cortex and the precuneus/posterior cingulate region but not in striatum.
FIGURE 4.
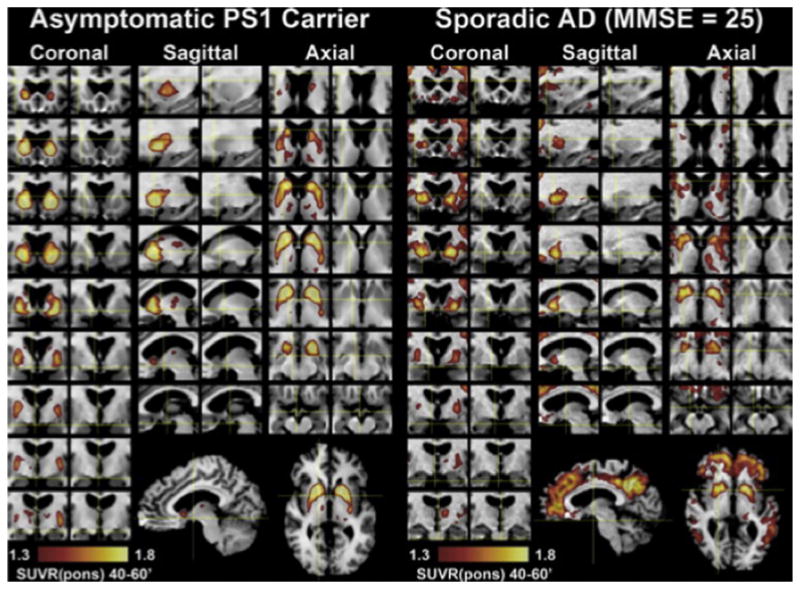
Detailed regional distribution of PiB retention in an asymptomatic PS1 carrier compared with a subject with sporadic AD. For color version of this figure, the reader is referred to the online version of this book.
(from Klunk et al., 2007)
These data have been extended to 49-year-old and 60-year-old siblings with autosomal dominant dementia and frequent cerebral amyloid angiopathy (CAA) and intracerebral hemorrhages due to an APP locus duplication (Remes et al., 2004; Rovelet-Lecrux et al., 2007). Similar to previous findings, PiB retention was highest in the striatum (up to 280% of the control mean) and the overall pattern of increased PiB retention was different from that seen in sporadic AD (Remes et al., 2007).
Theuns et al. (2006) reported widespread retention of PiB, typical of that observed in sporadic AD, in a 57-year-old patient (MMSE of 18) with a novel K724N mutation in the C-terminal intracytosolic fragment of APP. The subject showed no disproportionate PiB retention in the striatum. However, Villemagne et al. (2009), has demonstrated increased striatal PiB deposition in PS1 and APP mutation carriers. Further, Pittsburgh investigators have shown a similar striatal PiB retention pattern in older nondemented subjects with Down syndrome (Handen et al., in press), while Landt et al. (2011) showed a typical AD PiB retention pattern in one older subject with Down syndrome. These early-onset forms of AD all share overproduction of Aβ (particularly the 42 amino acid form) as a proposed mechanism of Aβ deposition (Younkin, 1997), whereas decreased clearance might be more important in late-onset AD (Whitaker et al., 2003). It may be that the cellular milieu of the striatum is particularly prone to amyloid deposition under conditions of overproduction.
It has been reported that two genetic forms of AD, the Arctic APP mutation and the Osaka APP mutation, were found to have little PiB retention in the brains of mutation carriers—in contrast to subjects with late-onset AD. Interestingly, these mutations have been associated with enhanced formation of Aβ oligomers without Aβ fibril formation (Nilsberth et al., 2001; Tomiyama et al., 2008). The lack of PiB-PET signal in both the Arctic and Osaka mutations suggest that oligomeric Aβ, rather than fibrillar Aβ, plays a significant role in the cause of dementia symptoms observed in patients carrying these genetic mutations (Shimada et al., 2011; Scholl et al., in press; Tomiyama et al., 2008).
IX. Frontotemporal Dementia
Frontotemporal dementia (FTD) refers to a family of neurodegenerative disorders that preferentially affect the frontal and anterior temporal lobes (Rabinovici & Miller, 2010). Clinically, FTD presents with progressive changes in behavior and social-emotional function (in the behavioral-variant) or with decline in language in the semantic and nonfluent/agrammatic variants of primary progressive aphasia (Gorno-Tempini et al., 2011; Rascovsky et al., 2011). Histologically, FTD clinical syndromes are associated with a group of pathologies collectively referred to as frontotemporal lobar degeneration (FTLD). Inclusions in FTLD consist of tau, TDP-43, or (rarely) fused in sarcoma (FUS) proteins, but, significantly, do not include Aβ deposits (Mackenzie et al., 2010). AD and FTD can overlap clinically and anatomically, and misclassification rates of 10–40% are cited even at expert centers when clinical diagnosis during life is compared to postmortem findings (Alladi et al., 2007; Forman et al., 2006).
PiB-PET could be helpful in distinguishing AD and FTD, since amyloid plaques are a core feature of AD but are not part of the FTLD pathologic spectrum. Further, patients with FTD typically develop symptoms before age 65 (Johnson et al., 2005), when the prevalence of AD and FTD is similar (Ratnavalli et al., 2002) and “age-related” amyloid deposits are less common (Morris et al., 2010). Several early case series demonstrated the utility of PiB-PET in distinguishing AD and FTD (Drzezga et al., 2008; Engler et al., 2007; Rabinovici et al., 2007; Rowe et al., 2007). In the largest series published to date, Rabinovici et al. tested the diagnostic performance of PiB-PET in distinguishing clinically diagnosed AD (N = 62) and FTLD (N = 45) patients (Rabinovici et al., 2011), and compared it to the performance of FDG-PET, which has an established diagnostic role in this scenario (Foster et al., 2007). PET scans were rated visually (blinded to clinical diagnosis) as PiB(+) or PiB(−) and as consistent with the FDG patterns of AD (temporoparietal-predominant hypometabolism) or FTLD (hypometabolism most severe in frontal or anterior temporal lobes). Scans were also classified quantitatively based on comparisons with normal controls. PiB visual reads were more sensitive for AD than FDG reads (89.5% vs. 77.5%) with similar specificity (83% vs. 84%). On quantitative classification, the sensitivity and specificity of PiB were essentially unchanged compared to visual reads, whereas FDG was slightly less sensitive (73%) but significantly more specific (98%). PiB outperformed FDG in a subset of 12 patients who underwent autopsy or carried a known pathogenic gene mutation, with an overall accuracy of 97% for PiB and 87% for FDG (see Section XIII for more details).
X. Dementia with Lewy Bodies and Parkinson’s Disease
Dementia with Lewy bodies (DLB) is the second most common degenerative cause of dementia after AD (McKeith et al., 1996). Clinically, DLB is characterized by the coincident onset of cognitive decline (often affecting executive and visuospatial function with relative sparing of memory) and motor features of Parkinson’s disease (PD) such as tremor, bradykinesia, rigidity, and postural instability (McKeith, 2006). Additional core features include visual hallucinations and fluctuations in cognition and arousal. DLB has significant clinical and pathological overlap with AD (McKeith, 2006). While pure DLB shows extensive deposition of α-synuclein protein in the form of Lewy bodies (Dickson, 2002), but no significant Aβ pathology, DLB with Aβ pathology (i.e., Lewy body variant of AD) is more frequently observed (Ballard et al., 2006). Evidence from in vitro binding and in vivo imaging studies suggests that PiB does not bind to α-synuclein deposits in detectable amounts (Bacskai et al., 2007; Burack et al., 2010; Fodero-Tavoletti et al., 2006; Klunk et al., 2003;), so PiB-PET can rule in or rule out the presence of significant Aβ pathology. Rowe et al. (2007) examined whether PiB retention can distinguish different types of dementia (AD, DLB, FTD), and found that cortical PiB retention was markedly elevated in every AD subject regardless of clinical severity (n = 17) but was generally lower and more variable in DLB (n = 10) and below detection in FTD (n = 6). In the DLB subjects, high neocortical PiB retention (especially in precuneus/posterior cingulate) correlated with shorter time between the onset of cognitive impairment and clinical manifestation of DLB, suggesting that Aβ pathology may accelerate DLB development. Additionally, studies support that PiB can distinguish DLB from other neurodegenerative syndromes with similar clinical and pathological phenotypes, such as multiple systems atrophy (Claassen et al., 2011). When compared to Parkinson’s disease dementia (PDD), another condition associated with extensive α-synuclein pathology, DLB subjects have significantly more Aβ deposition measured by PiB-PET (Claassen et al., 2011; Edison et al., 2008; Gomperts et al., 2008; Kalaitzakis et al., 2011; Maetzler et al., 2008), further supporting that Aβ deposition may have greater influence on the clinical development of DLB than PDD. However, vascular Aβ deposition is also common in PD and Aβ plaques are often found in PDD (Jellinger, 2003; Mastaglia et al., 2003). Johansson et al. (2007) reported that compared to HCs, cognitively intact PD patients do not show significantly increased PiB-PET retention, and PiB PET scan can be positive in more advanced PD patients. Indeed, higher PiB retention was reported in subjects with PDD (Foster et al., 2010; Kalaitzakis et al., 2011) and in two of three PiB-PET imaged PDD autopsy cases where PiB positivity was associated with the presence of frequent Aβ plaques (Fig. 5; Burack et al., 2010).
FIGURE 5.

PiB-PET images from Parkinson’s dementia cases with autopsy confirmed amyloid plaque pathology (A, B), a Parkinson’s case without amyloid plaque pathology (C), and a control participant (D). For color version of this figure, the reader is referred to the online version of this book.
(from Burack et al., 2010).
In conclusion, PiB imaging cannot distinguish DLB from AD given the high rate of Aβ co-pathology in DLB. This clinical distinction can be better accomplished by molecular imaging of the dopamine system, which is deficient in DLB but not AD (Koeppe et al., 2008). Further, a recent report has suggested that concomitant imaging of Aβ and markers of the presynaptic dopaminergic system in the same individuals aids in the differential diagnosis of DLB and AD (Villemagne et al., 2012). PiB imaging may have prognostic value, with a positive scan suggesting a more precipitous clinical course, though this needs to be more definitively demonstrated in longitudinal studies.
XI. Cerebral Amyloid Angiopathy (CAA)
An accumulating body of evidence from clinical, epidemiological, and autopsy studies suggest a relationship between cardiovascular disease (CVD) and Aβ pathology. Whether cerebral vascular pathology and Aβ deposition can influence each other, and the extent to which these changes affect cognition, is not clear. Recent autopsy studies and clinical imaging combining MRI and PiB-PET have contributed to our better understating of this potential relationship. CAA results from Aβ deposition in cerebral vessels’ wall. Several postmortem studies reported high incidence of CAA (up to 98%) in AD (for review see (Jellinger, 2002)). While CAA can be found in the absence of dementia it is often found in association with AD. This is particularly the case in ApoE4 carriers, where CAA is associated with a risk of blood vessel rupture and cerebral hemorrhages including strokes (Maia et al., 2007) which can contribute to VaD. CAA-associated strokes are most frequently located in the occipital lobe (Attems et al., 2007; Rosand et al., 2005;) which is less severely affected with plaques when compared to frontal and parietal (precuneus) cortices. Both plaques and CAA are detectable with PiB (Bacskai et al., 2007; Ikonomovic et al., 2008; Lockhart et al., 2007) and contribute to PiB retention in vivo.
Johnson et al. (2007) evaluated the sensitivity of PiB-PET to detect CAA in six nondemented subjects diagnosed with clinically probable CAA and compared them to patients with probable AD, and HCs. They found that all of the CAA and AD subjects were PiB(+). Global cortical PiB retention in the CAA group was significantly higher relative to HC subjects but was lower than in AD subjects. The occipital-to-global PiB ratio, however, was significantly greater in CAA than in AD subjects—consistent with the known predilection of CAA for the occipital lobe. Similarly, in a 42-year-old man with Iowa-type hereditary CAA, PiB retention was observed only in the occipital cortex, consistent with the pathology of this type of CAA (Greenberg et al., 2008). These findings have been replicated in additional CAA cohorts showing significantly increased occipital-to-global ratio of PiB retention (Ly et al., 2010).
XII. Atypical Presentations of AD
While episodic memory loss is considered the clinical hallmark of AD, ~15% of AD patients seen at academic centers have a nonamnestic presentation (Snowden et al., 2007). Two clinical syndromes in particular—posterior cortical atrophy (PCA), a progressive disorder of visuospatial function, and logopenic-variant primary progressive aphasia (lvPPA), a language disorder characterized by difficulties with naming, word retrieval, and repetition—have been strongly associated with AD pathophysiology (Alladi et al., 2007; Gorno-Tempini et al., 2004; Mesulam et al., 2008; Tang-Wai et al., 2004). These nonamnestic presentations have been incorporated into new AD diagnostic guidelines (McKhann et al., 2011).
Amyloid PET can be helpful in diagnosing AD in patients presenting with PCA and lvPPA during life, particularly since these syndromes are associated with early age-of-onset, and the alternative causative pathologies fall in the FTLD (non-Aβ) family. Indeed, a number of studies have demonstrated that patients diagnosed with PCA and lvPPA at expert centers are nearly always PiB(+) (de Souza et al., 2011; Formaglio et al., 2011; Leyton et al., 2011; Ng et al., 2007a; Rabinovici et al., 2008; Rabinovici et al., 2011; Rosenbloom et al., 2011). PiB may also be useful in diagnosing AD in patients with a dysexecutive-behavioral presentation (frontal-variant AD) (Johnson et al., 1999) and corticobasal syndrome, a disorder of sensorimotor integration, primary motor, and cognitive function (Lee et al., 2011), though data on these syndromes are limited to case reports (Laforce & Rabinovici, 2011). Interestingly, most group-level analyses have found that the distribution of amyloid in PCA and lvPPA is similar to the distribution in AD, though neurodegeneration patterns (as determined by MRI and FDG-PET) are distinct, with more occipital involvement in PCA and asymmetric left hemisphere degeneration in lvPPA (de Souza et al., 2011; Leyton et al., 2011; Rabinovici et al., 2008; Rosenbloom et al., 2011) (Fig. 6). These findings, along with the discordance between PiB and atrophy/hypometabolism patterns in typical AD ((Rabinovici et al., 2010), also see sections below, Section XIV), suggest that the burden and spatial distribution of fibrillar Aβ (as imaged by PiB) do not explain the clinical and anatomic heterogeneity of AD.
FIGURE 6.

Patterns of FDG and PIB binding in amnestic (AD), language (lvPPA) and visual (PCA) variants of AD compared with HCs. Shown are t-maps after correction for multiple comparisons (family-wise error correction at p < 0.05). Red in the FDG maps indicates significantly more hypometabolism in the patient groups compared with controls, whereas blue in the PIB maps indicates significantly more amyloid deposition in the patient groups. FDG patterns are distinct and correlate with the clinical deficits, while PIB binding is diffuse and indistinguishable across variants. For interpretation of the references to color in this figure legend, the reader is referred to the online version of this book.
XIII. Postmortem Validation of PiB-PET Imaging
From the earliest in vivo PiB-PET imaging studies it has been suggested that PiB retention reflects the extent of Aβ pathology in the brain (Klunk et al., 2004). However, strong, direct evidence in support of this idea became available only recently, after some of the PiB-PET imaged subjects came to autopsy. Autopsy studies of PiB-PET imaged brains allowed for the first time, that correlations can be examined between antemortem PiB retention levels and region-matched postmortem measures of fibrillar Aβ load and other neuropathology in the same brains. To date there has been more than a dozen of PiB-PET autopsy case reports in the literature (see Table I) that will facilitate elucidating the pathological substrates of PiB retention in brains of cognitively normal aged people and subjects with AD or other dementias.
TABLE I.
Overview of Studies Reporting PiB-PET Autopsy Cases
| Reference | a PiB (+/−) | b Clinical diagnosis (at time of PET scan) | Cognitive score (at time of PET scan) | PET-to-death interval (months) | c Cerebral amyloid angiopathy (severity) | c Cortical NP frequency | c Cortical DP frequency (load) | d CERAD/ NIA-RI diagnosis of AD | Braak stage for NFT |
|---|---|---|---|---|---|---|---|---|---|
| [1] | + | DLB | CDR = 1/MMSE = 25 | 3 | Severe | Sparse | Frequent | Possible/IL | IV |
| [2] | + | AD | MMSE = 1 | 10 | Sparse | Frequent | Frequent | Definite/HL | VI |
| [3] | − | Normal | CDR = 0 | 30 | Mild | Sparse | Focally frequent | Possible/LL | III |
| [4] | − | CJD | n/s | <1 | present (n/s) | None | None | n/s | n/s |
| [4] | − | CJD | n/s | <1 | present (n/s) | None | Sparse | n/s | n/s |
| [5] | + | PDD | CDR = 2/MMSE = 23 | < 15 | Mild | Sparse | Frequent | Possible/LL | III |
| [5] | + | PDD | CDR = 2/MMSE = 11 | <15 | None | Sparse | Frequent | Possible/LL | III |
| [5] | − | PDD | CDR = 1/MMSE = 24 | <15 | None | None | Sparse | n/s | I |
| [6] | + | DLB | MMSE = 10 | 18 | Mild | Moderate | Frequent | n/s/LL | III |
| [7] | + | AD | MMSE = 5 | 35 | present (n/s) | Frequent | Frequent | Definite/ HL | VI |
| [8] | + | Normal | CDR = 0 | 16 | present (n/s) | Sparse | High (>5%) | Normal/NO | IV |
| [8] | + | Dementia | CDR = 1 | 2 | present (n/s) | Moderate | High (>5%) | Probable/IL | III |
| [8] | − | Normal | CDR = 0 | 20 | None | None | Low (<5%) | Normal/NO | IV |
| [8] | − | Normal | CDR = 0 | 28 | None | Moderate | Low (<5%) | Possible/NO | III |
| [8] | − | Normal | CDR = 0 | 28 | None | Moderate | Low (<5%) | Possible/NO | IV |
| [8] | − | MCI | CDR = 0.5 | 13 | present (n/s) | Moderate | Low (<5%) | Possible/IL | III |
| [9] | − | DLB | MMSE = 10 | 17 | Moderate | Focally frequent | Focally frequent | eDefinite/LL | II |
PiB positivity (+) is defined by either local cutoffs defined by the authors or by cutoffs in standard use such as a DVR>1.4 (or BP>0.4) or an SUVR>1.5
Clinical diagnosis, AD (Alzheimer disease), CJD (Creutzfeldt–Jakob disease), DLB (dementia with Lewy bodies), MCI (mild cognitive impairment), PDD (Parkinson disease dementia). Highest regional values are shown for congophilic amyloid angiopathy and frequencies of neuritic plaques (NP) and diffuse plaques (DP)
CERAD = Consortium to establish a registry for Alzheimer’s disease (diagnoses of possible, probable, or definite AD); NIA-RI = The National Institute on Aging and Reagan Institute (LL = low likelihood of AD, IL = intermediate likelihood of AD, HL = high likelihood of AD, NO = not AD)
Diagnosis of definite AD was based on a single area of frequent neuritic plaques in the frontal cortex and strict application of the CERAD criteria.
NFT = neurofibrillary tangles
n/s = not specified
Modified from Ikonomovic et al. (2012).
The first PiB-PET autopsy case was described in 2007 by Bacskai and colleagues (Bacskai et al., 2007). This subject had a clinical diagnosis of DLB with mild impairment on the clinical dementia rating (CDR = 1) and mini-mental state examination (MMSE = 25) scales. A PiB-PET scan was performed 3 months prior to autopsy, and it showed positive PET retention when assessed using the reference Logan graphical analysis (Logan et al., 1996), with distribution volume ratios (DVR) ranging between 1.30 in the parietal cortex and 1.50 in the cingulate cortex. Postmortem neuropathology evaluation of the neocortex detected Lewy bodies in temporal and cingulate cortices, and moderate NFT in temporal, parietal, and occipital cortices, consistent with Braak stage IV (Braak & Braak, 1991). However, Aβ plaque pathology was surprisingly low in this case, with only rare neocortical cored plaques and numerous diffuse plaques observed using Aβ immunohistochemistry (6F/3D antibody). The low frequency of neuritic plaques and the NFT pathology in this case resulted in diagnosis of “possible AD” based on the Consortium to Establish a Registry of Alzheimer’s Disease (CERAD) (Mirra et al., 1991) and in an “intermediate likelihood of AD” based on the National Institute on Aging-Reagan Institute (NIA-RI) criteria (Consensus, 1997). Interestingly, both Aβ immunohistochemistry and PiB fluorescence in tissue sections revealed severe CAA. Biochemical analyses of soluble and insoluble Aβ concentrations in this PiB-PET positive case showed a preponderance of Aβ40 over Aβ42, supporting that vascular amyloid was the dominant form of Aβ pathology.
Several postmortem studies of AD cases without PiB-PET scan confirmed that CAA is a major pathologic substrate for PiB retention in the brain, and provided additional valuable information regarding PiB retention in dementia cases (see Section XI). Ikonomovic and colleagues (Ikonomovic et al., 2008) performed histological characterization of PiB retention using 6-CN-PiB, a highly fluorescent derivative of PiB, on postmortem tissue sections from multiple brain regions in 27 dementia cases from the University of Pittsburgh Alzheimer’s Disease Research Center (ADRC). PiB retention was most prominent in neocortical Aβ immunoreactive (6E10, 10D5, Aβ40, and Aβ42 antibodies) deposits in cerebral vasculature and in classic cored and neuritic plaques. PiB retention to neocortical and striatal diffuse plaques was far less prominent but still detectable, while diffuse Aβ plaques in the cerebellum were not detectable using 6-CN-PiB (Ikonomovic et al., 2008). A similar observation of PiB binding to CAA and classical and diffuse plaques was reported using [H-3]PiB autoradiography on brain tissue sections (Lockhart et al., 2007; Thompson et al., 2009). Lockhart and colleagues also reported that PiB binds to NFT (Lockhart et al., 2007); however, other studies did not support this idea and instead suggested that the extra-cellular (“ghost”) type of NFT is more likely to bind PiB due to the presence of Aβ fibrils in these extracellular tau aggregates (Ikonomovic et al., 2008; Fig. 7). Figure 7 illustrates selectivity of PiB retention to Aβ deposits in postmortem brain tissue sections; there is a very good correspondence between PiB retention and Aβ plaques while no binding of PiB to intracellular NFT is detectable. Regardless, at doses of PiB used for PET imaging it is unlikely that NFT could be detected in vivo.
FIGURE 7.
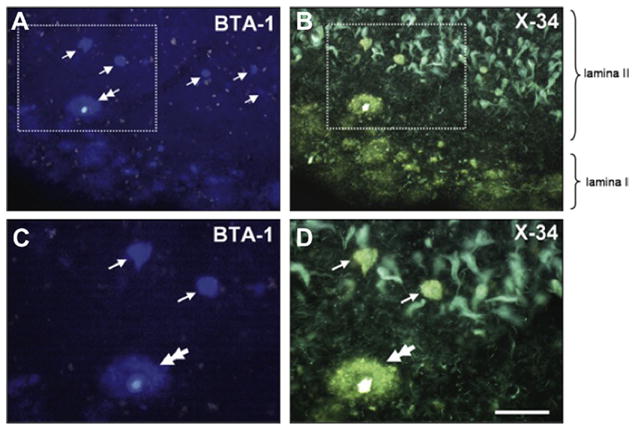
Double-histofluorescence staining of a single section of an AD entorhinal cortex, using BTA-1 (1 μM, A and C; U filter) and X-34 (100 μM, B and D; V filter) histofluorescence. Tissue autofluorescence (lipofuscin) is seen as bright bleed-through signal in A and C. Boxed areas in A and B delineate areas of higher magnification in C and D, respectively. Subpial diffuse Aβ plaques, and a single cored plaque (C and D, double-arrows) in lamina I are seen with both compounds. In lamina II, BTA-1 labels only a few isolated structures (A, arrows), while X-34 also labels abundant NFTs and neuropil threads (B). BTA-1 labeled structures inside layer II islands (C, arrows) with X-34 histofluorescence (D, arrows), similar to the neighboring cored plaque (D, double arrow); this makes them distinct from the surrounding NFTs that are seen with X-34 histofluorescence (B, D). Scale bar = 100 μm (A, B), 50 μm (C, D). For color version of this figure, the reader is referred to the online version of this book.
(from Ikonomovic et al. 2008)
It has also been of interest to determine if PiB retention reflects other types of intracellular protein aggregates such as α-synuclein in Lewy bodies (LB). Using [H-3]PiB binding, it was observed that PiB has very low binding affinity for α-synuclein fibrils, and no binding was detected in homogenates of DLB brains free of Aβ deposits (Fodero-Tavoletti et al., 2007). Collectively, these postmortem findings support that PiB retention is highly specific for fibrillar Aβ deposits, while binding to other types of neuropathology is negligible. The strong binding of PiB to CAA and classic cored plaques is due most likely to dense β-sheet structure of Aβ fibrils in these lesions. In contrast, it has been assumed that diffuse plaques lack fibrillar structure and therefore cannot bind PiB. The presence of detectable 6-CN-PiB histofluorescence (Ikonomovic, et al., 2008) and [H-3]PiB autoradiography signal (Lockhart et al. 2007) in diffuse plaques support that these lesions can retain PiB in vivo as a result of fibrillar Aβ present even in diffuse plaques. This is in agreement with reports of high PiB retention levels in familial AD (presenilin mutation) and variant AD subjects with large amounts of diffuse striatal plaques and cortical cotton wool plaques (Klunk et al., 2007; Koivunen et al., 2008). The absence of postmortem PiB labeling of diffuse plaques in the cerebellum (Ikonomovic et al., 2008) justifies using this region as a reference area for in vivo PiB retention analyses (Lopresti et al., 2005).
The first correlation analysis of region-matched antemortem PiB retention and postmortem measures of neuropathology was reported in a PiB-PET imaged typical AD subject with end-stage disease (Ikonomovic et al., 2008). The 64-year-old female subject examined in that study had a clinical diagnosis of probable AD and a positive PiB-PET scan 10 months prior to death. PiB retention was positive in all cortical regions (DVR range 1.59–2.38). Neuropathological diagnosis was “definite AD” by the CERAD criteria (Mirra et al., 1991) and Braak stage was V/VI (Braak & Braak, 1991). Frequent cortical plaques and mild CAA were Aβ immunoreactive (6E10 antibody) and positive for 6-CN-PiB fluorescence. Both Aβ immunoreactive and 6-CN-PiB positive plaque loads (% area) correlated strongly with region-matched DVR values determined antemortem in the same subject (Ikonomovic et al., 2008). Strong direct correlations were also observed between antemortem DVR values and region matched postmortem biochemistry measures of total Aβ42 and Aβ40 concentration or [H-3]PiB binding in frozen tissue homogenates from this case. Similar findings were reported by Kadir and colleagues (2011) who examined another case of typical end-stage AD; this 61-year-old female with severe dementia (MMSE = 5) was the first patient ever imaged using PiB-PET. She underwent PiB-PET imaging 35 months prior to death, and there was strong PiB-PET positivity in all regions examined. Neuropathology findings included frequent or widespread Aβ plaques detected using a battery of different Aβ antibodies (6E10, 4G8, 6F/3D, Aβ40, and Aβ42), neuropathology diagnosis was “definite AD” by the CERAD criteria (Mirra et al., 1991) and Braak stage for NFT was V/VI. Strong, direct correlations were detected between antemortem standardized uptake values (SUVs) and region-matched measures of Aβ plaque distribution, Aβ concentration, and [H-3]PiB binding (Kadir et al., 2011). Collectively, the results of these two studies of PiB autopsy brains from typical end-stage AD patients provide further support that in vivo PiB-PET retention reflects fibrillar Aβ burden. Other PiB brain autopsy studies examined additional cases with antemortem clinical diagnosis of DLB (Kantarci et al., 2010; Ikonomovic et al., 2012) and cases with PDD (Burack et al., 2010). These studies led to the conclusion that in patients with concomitant LB and Aβ pathology, it is the fibrillar Aβ burden, and not LB, which determines PiB retention in vivo (see Table 1).
The presence of even minimal Aβ deposits in a subject with a negative PiB-PET scan brings into question the sensitivity of this technique. Several postmortem studies reported various amounts of Aβ pathology in brains of PiB(−) subjects. Cairns et al. (2009) reported autopsy findings in a 91-year-old subject who had a negative PiB–PET scan (neocortical PiB retention ranged from 0.03 to 0.19) and normal cognition (CDR = 0) when evaluated 30 months prior to death. The subject later developed very mild dementia (CDR = 0.5) and underwent CSF analysis for Aβ/tau. Based on the neuropathology evaluation the case was diagnosed as “possible AD” by the CERAD criteria (Mirra et al., 1991) with a low likelihood that the mild dementia was caused by AD, based on the NIA-RI criteria (Consensus, 1997). There were sparse to focally frequent diffuse plaques, infrequent neuritic plaques, and mild CAA. Up to 5.4% area of neocortex was covered with Aβ immunoreactive plaques (10D5 antibody), an Aβ1–42 ELISA detected high levels of Aβ1–42 in cortical areas (range 687–1785 pmol/g wet tissue), and cortical [H-3]PiB binding ranged between 116–295 pmol/g. Interestingly, CSF was sampled 1 year after the PiB-PET scan was done, ~18 months prior to death, and it showed abnormal Aβ/tau levels, leading Cairns and colleagues to suggest that CSF profiling is more sensitive than PiB-PET in detecting fibrillar Aβ deposits in the brain (Cairns et al., 2009).
Ikonomovic et al (2012) reported autopsy findings in a PiB(−) subject with antemortem diagnoses of DLB and possible AD. PiB retention was low (DVR<1.2 in all cortical regions); however, postmortem neuropathology analysis 17 months later revealed mild to moderate and even focally frequent neocortical neuritic plaques which allowed for a diagnosis of “definite AD” by strict CERAD criteria (Mirra et al., 1991). Aβ immunoreactive plaque load was up to 1.8% of total plaque load but the majority of plaques were diffuse and they labeled weakly with PiB. While cortical Aβ1–40 concentration levels (up to 233 pmol/g) were similar to those in a typical PiB(+) AD case (Ikonomovic et al., 2008), Aβ1–42 concentrations were lower in all brain areas except the frontal cortex, where values (788 pmol/g) approached those measured in a typical PiB(+) AD case. However, [H-3]PiB binding in the frontal cortex and all other cortical regions from the PiB(−) case was low (60 pmol/g or less). The low ratios of PiB retention to Aβ measures in both histological and biochemical assays indicated very low fibrillar Aβ load in this PiB(−) brain (Ikonomovic et al., 2012). It is interesting that the amount of neuritic plaque pathology in this case was more substantial than in the PiB(−) case reported by Cairns et al. (Cairns et al., 2009), where “definite AD” diagnosis could result only by applying Khachaturian neuropathologic criteria (Khachaturian, 1985). Both cases were analyzed using the same methodology; however, the Cairns’ PiB(−) case had greater cortical Aβ-immunoreactive plaque load (up to 5.4 % area), Aβ1–42 concentration (687–1785 pmol/g wet tissue), and [H-3]PiB binding (116–295 pmol/g). The longer PET-to-death interval in the Cairns case (30 months) compared to the Ikonomovic case (17 months) may explain these differences. PiB(−) scans were also reported in two autopsy cases with a diagnosis of CJD (Villemagne et al., 2008) and in four autopsy cases with either mild (CDR = 0.5) or no cognitive impairment (Sojkova et al., 2011). While CJD cases in the study by Villemagne and colleagues had either absent or minimal Aβ plaques (Villemagne et al., 2008), several [C-11]PiB negative subjects examined by Sojkova and colleagues had moderate numbers of neocortical neuritic plaques (Sojkova et al., 2011).
The sensitivity of PiB-PET imaging is not well understood, and this technique may not be 100% sensitive for the presence of histologically detectable Aβ even if it were determined close to the time of the in vivo scan. On the other hand, so far there has been no report of an in vivo PiB(+) subject who failed to show Aβ deposits at autopsy, supporting good specificity of this technique. To-date, the most likely explanation for the few in vivo PiB(−) cases that have detectable postmortem Aβ is a combination of the following: (1) low amounts of Aβ that are below the in vivo threshold of the PiB-PET imaging technology and (2) a high percentage of nonfibrillar deposits of Aβ which are not easily detected with PiB-PET. There is some evidence that Aβ42 is more closely associated with in vivo PiB retention than Aβ40 (Ikonomovic et al., 2008, 2012). Additional analyses of large numbers of PiB(−) and PiB(+) cases, with short imaging-to-autopsy interval, are required to establish a threshold level of Aβ pathology necessary for in vivo PiB-PET detection.
XIV. Amyloid Imaging Compared to Other Biomarkers
A. PiB and FDG
Decreases in cerebral glucose metabolism, measured by FDG, show a characteristic regional pattern of posterior temporoparietal > frontal hypo-metabolism in AD (Foster et al., 2007; Friedland et al., 1983;Herholz, Carter, & Jones, 2007; Jagust et al., 2007). Similar changes have been reported in cognitively normal individuals at high risk for AD due to expression of the Apo-E4 alelle (Reiman et al., 1996; Small et al., 2000). Changes in cerebral metabolism also have been detected in MCI in many studies (Arnáiz et al., 2001; Chetelat et al., 2003a; Chetelat et al., 2003b; Del Sole et al., 2008; Garibotto et al., 2008; Li et al., 2008; Mevel et al., 2007; Mosconi et al., 2006; Mosconi et al., 2008 Perneczky et al., 2007). These early changes suggest FDG could play a predictive role in detecting which normal controls or MCI patients are most likely to convert to AD (Yuan et al., 2008). Indeed, several studies have shown that abnormalities in FDG PET predict progression from MCI to AD (Anchisi et al., 2005; Drzezga et al., 2005; Mosconi et al., 2004).
In the initial PiB-PET study, the largest and only significant difference in glucose metabolism (determined with FDG PET) between AD patients and control subjects was observed in parietal cortex. An inverse correlation between PiB retention and glucose metabolism was observed in most cortical areas, but this trend reached significance only in the parietal cortex. The lack of correlation between PiB and glucose metabolism in the frontal cortex suggests that Aβ deposition is not sufficient to locally reduce cerebral metabolism, suggesting that perhaps compensatory changes in neurotransmitter systems (i.e., DeKosky et al., 2002; Ikonomovic et al., 2007) in the frontal cortex delay FDG hypometabolism in frontal brain regions. Edison et al. (2006) investigated the association between PiB and FDG PET in AD. AD subjects showed significant increases in PiB retention in cingulate, frontal, temporal, parietal, and occipital cortical areas and levels of temporal and parietal rCMRglc were reduced by 20% in AD. Higher PiB retention correlated with lower rCMRglc in temporal and parietal cortices, but not in frontal areas. While these typical negative correlations were observed in AD, subjects with MCI often display positive correlations between PiB and FDG, reflecting increased brain reserve in those subjects who remain at the MCI level of cognitive impairment further into the process of Aβ deposition (Cohen et al., 2009).
Forsberg et al. explored MCI subjects with PiB and FDG PET, as well as assessment of cognitive function and CSF sampling. The MCI subjects that later converted to AD showed significantly higher PiB retention compared to nonconverting MCI patients. However, there was no significant difference in rCMRglc between MCI patients and HCs in any cortical brain region, suggesting PiB may better predict clinical conversion than FDG-PET. However, Furst et al. (2010) demonstrated that cognitive performance in AD correlated strongly with FDG but not at all with PiB, and did not demonstrate any significant correlations between PIB and FDG
Ng et al. (2007) compared a visual assessment to a quantitative assessment of PiB and FDG PET data for detection of AD compared to cognitively intact controls. Visual agreement between readers was excellent for PiB (kappa = 0.90) and good for FDG (kappa = 0.56). Based on the clinical diagnosis, Ng et al. found PiB was more accurate than FDG both on visual reading (accuracy, 90% vs. 70%) and ROC analysis (95% vs. 83%). The authors concluded that the visual analysis of PiB images appears more accurate than visual reading of FDG for identification of AD and had accuracy similar to quantitative analysis of a 90 min dynamic scan. Similar results were found in the Rabinovici et al. (2011) differential diagnosis study described above; inter-rater agreement was significantly higher for PiB (kappa = 0.96) than FDG (kappa = 0.72), as was agreement between visual and quantitative classifications (average kappa = 0.90 for PiB, 0.66 for FDG). The authors concluded that PiB was the superior qualitative technique in that visual assessment was both more accurate and more precise. While PiB and FDG demonstrate high (94%) agreement in differentiating AD from normal controls, agreement is lower in classifying MCI subjects (54%) (Li et al., 2008). Li et al. argues that “combining the two modalities improves the diagnostic accuracy for MCI.” In addition, when exploring the use of PiB and FDG among both AD and MCI subtypes it was demonstrated that while PiB and FDG displayed similar diagnostic accuracy, PiB was significantly better at separating MCI subtypes (Lowe et al., 2009). These findings are not surprising since the two tracers provide complementary information, with PiB quantifying molecular pathology, and FDG demonstrating neuronal dysfunction. The complementary nature of the two techniques are reflected in the new diagnostic guidelines for MCI and AD dementia, which require biomarker evidence of both Aβ deposition (CSF or amyloid PET) and neurodegeneration (hypometabolism on FDG-PET or atrophy on MRI) to diagnose AD pathophysiology with high-likelihood during life (McKhann AD criteria, Albert MCI criteria).
B. PiB and MRI
Many studies have demonstrated hippocampal atrophy in AD and MCI (Apostolova et al., 2006a; Becker et al., 2006; Grundman et al., 2002; Moretti et al., 2007; Morra et al., 2009). Furthermore, several studies have shown that the rate of hippocampal atrophy may identify those MCI patients soon to convert to clinical AD (Apostolova et al., 2006b; Apostolova et al., 2008; Chetelat et al., 2008; de Toledo-Morrell et al., 2004; Devanand et al., 2007; Grundman et al., 2002; Jack et al., 1999; Jack et al., 2000; van de Pol et al., 2007; Wang et al., 2009). When PiB-PET was correlated with volumetric MRI measurements in AD, a significant, positive correlation was observed between rates of whole brain atrophy and cortical PiB retention (Archer et al., 2006; Chetelat et al., 2010; Fotenos et al., 2008; Frisoni et al., 2009). In one study, PiB retention was shown to predict later decline in brain volume (Scheinin et al., 2009). However, in cognitively normal elderly, volume decline in the decade preceding PiB-PET is not correlated with cortical PiB retention (Driscoll et al., 2010). However, Chetelat et al. (2012), recently showed that cognitively unimpaired PiB(+) individuals have significantly higher rates of brain atrophy than their PiB(−) counterparts. Further, Jack et al. (2009) explored PiB and MRI across the AD continuum and observed a significant correlation between MMSE and ventricular atrophy, with only a weak correlation between PiB and ventricular size, suggesting a complementary use of PiB-PET and MRI in detection of MCI and AD, as reflected in the new diagnostic criteria (Jack et al., 2011).
C. PiB and Cerebrospinal Fluid (CSF) Aβ
Because neuritic Aβ plaques and NFT do not develop simultaneously in the brain, the availability of lesion-specific radioligands would facilitate evaluations of AD pathology in vivo. Histopathology studies demonstrated that PiB retention is specific for fibrillar Aβ pathology and that PiB binds negligibly or not at all to NFT and Lewy bodies (Fodero-Tavoletti et al., 2007; Ikonomovic et al., 2008; Lockhart et al., 2007; Thompson et al., 2009). Besides 2-(1-{6-[(2-[F-18] fluoroethyl) (methyl)amino]-2-naphthyl} ethylidene)malononitrile (FDDNP) PET which has been claimed to detect both Aβ plaques and NFT (Small et al., 2006), and some emerging tau-binding candidate radioligands such as [F-18]THK523 (Fodero-Tavoletti et al., 2011), none of the currently used imaging radiotracers allows for measurements of aggregated tau or phosphorylated tau (p-tau) pathology in brain tissues in living patients. Cerebrospinal fluid (CSF) analysis of Aβ42 and p-tau concentrations is an alternative, indirect method for quantifying both types of pathology in the brain; it has been reported to have high accuracy for identifying individuals with incipient AD (Mattsson et al., 2009) and for predicting the development and rate of cognitive decline (Buchhave et al., 2012; Fagan et al., 2007; Snider et al., 2009). CSF from AD patients contains higher concentrations of total and phosphorylated tau and lower levels of Aβ42 which correlate with the presence of post-mortem neurofibriallary and amyloid pathology respectively (Strozyk et al., 2003). However, the exact relationship between the amounts of fibrillar Aβ in brain parenchyma and soluble Aβ concentration in CSF is unclear. Based on a study in Tg2576 mice (Kawarabayashi et al., 2001) it has been assumed that lower CSF Aβ42 reflects deposition of fibrillar Aβ in brain tissues; however, no direct evidence from human studies is available to confirm this hypothesis and alternate hypotheses for lowered CSF Aβ such as impairments in clearance may apply better in humans.
Several clinical studies examined the relationship between Aβ changes in the brain and CSF by measuring in vivo PiB-PET retention and CSF Aβ42 concentration in the same subjects. A strong inverse correlation was observed between the two biomarkers, both in a mixed cohort of cognitively normal and demented subjects (Fagan et al., 2006) and in a homogeneous population of cognitively intact individuals (Fagan et al., 2009). While these associations were initially modeled as linear correlations, it has become increasingly recognized that the relationship between PiB retention and CSF Aβ42 is better modeled by a nonlinear approach. As expected, there was no correlation between PiB retention and CSF tau levels (Fagan et al., 2006). Similar associations between amyloid imaging and CSF Aβ were observed in cohorts of cognitively healthy (Storandt et al., 2012), MCI (Forsberg et al., 2007; Koivunen et al., 2008), and AD subjects (Grimmer et al., 2009). In a longitudinal study by Forsberg et al. (2007), all MCI subjects that converted to AD had high PiB retention, but <50% had pathological levels of Aβ42 in the CSF, suggesting that amyloid imaging may be more sensitive than CSF Aβ42 concentration in identifying MCI subjects who will develop AD (Forsberg et al., 2007). Observations by Koivunen et al. (Koivunen et al., 2008) lent further support to this idea; high PiB retention was detected in 87% of MCI patients while only 53% of MCI subjects had pathological levels of CSF Aβ42. The reason why some PiB(+) MCI subjects have normal Aβ42 concentration in the CSF is unknown. Grimmer and colleagues also reported an inverse correlation between overall [C-11]PiB retention in the brain and CSF Aβ42 levels in their cohort of AD subjects (Grimmer et al., 2009)—particularly in paraventricular regions, and more recently the same group reported that BACE1 activity in the CSF correlates with PiB-PET retention levels in the parahippocampal gyrus, thalamus, and pons (Grimmer et al., 2012).
In a cohort representing an entire spectrum of cognitive decline, Tolboom and colleagues (Tolboom et al., 2009a) compared CSF biomarkers to both PiB and [F-18]FDDNP. After adjusting for potential confounding variables, increased global or regional PiB retention was associated with low CSF Aβ42 (Tolboom et al., 2009). No association was observed between PiB and CSF tau, in agreement with some (Fagan et al., 2006; Forsberg et al., 2008) but not other (Storand et al., 2012) studies. Collectively, these studies support that PiB retention specifically reflects Aβ plaque pathology in the brain. In contrast, high [F-18]FDDNP retention was associated with high CSF tau, but no correlation was found with CSF Aβ42, suggesting that this radiotracer is more associated with NFT pathology in AD brains (Tolboom et al., 2009).
Cairns and colleagues studied a cognitively normal subject (CDR = 0) who had a negative PiB-PET scan; however, 12 months after the PET scan CSF analysis showed decreased Aβ42 and slightly increased tau and p-tau concentration, 18 months after the PET scan there were clinical signs of a very mild dementia (CDR = 0.5), and 30 months after the PET scan the subject died and neuropathology examination found evidence of primarily diffuse neocortical Aβ plaques (NIA-RI low likelihood AD) (Cairns et al., 2009). These observations may suggest that CSF Aβ42 may be a more sensitive bio-marker for detection of AD pathology when compared to PiB-PET. Additional studies in large numbers of subjects are needed to determine if amyloid imaging of fibrillar Aβ load or CSF Aβ concentration is a more sensitive biomarker and which one is better at predicting progression from MCI to AD.
C. PiB and Neuroinflammation
It is well known that inflammatory processes contribute to pathogenesis of AD. Activation of microglia appears to be an early reactive mechanism in response to amyloid deposition, and brain inflammation may even precede amyloid plaques and tangles in AD brain (for review see McGeer & McGeer, 2010). Studies in transgenic mice demonstrated that anti-inflammatory therapies are capable of reducing both microglia/cytokine reaction and Aβ load as determined by percent area and ELISA measurements (Lim et al., 2000). Thus, PET imaging of activated microglia, using radioligands that can specifically bind to peripheral benzodiazepine receptors expressed by these cells, is a valuable tool for evaluating the extent of inflammatory processes in living patients with chronic neurodegenerative disorders including AD (Venneti et al., 2009).
Several in vivo imaging studies examined both amyloid deposition and microglial activation using PET. Wiley and colleagues (2009) examined potential associations between amyloid pathology and microglial activation using PiB and (R)-PK11195 ([1-(2-chlorophenyl)-N-methyl-N- (1-methylpropyl)-3-isoquinoline carboxamide], a PET radiotracer for imaging peripheral benzodiazepine binding sites), respectively, in six mild-moderate AD, six MCI, and five cognitively normal subjects. There was no association between increased (R)-PK11195 uptake and positive PiB PET retention, suggesting that microglia activation occurs only during specific stages of amyloid deposition, and (R)-PK11195 may lack sensitivity to detect such changes. Similarly, in a study of amnestic MCI, Okello and colleagues (2009) found that not all of their PiB positive subjects had increased uptake of (R)-PK11195. Therefore, using this specific radioligand for measuring activated microglia in vivo, inflammatory process can be detected only in a subset of patients with increased amyloid burden. The same group examined 13 AD subjects and reported concomitant increases in (R)-PK11195 and PiB signal in multiple brain areas from AD brains. Interestingly, increased [C-11](R)-PK11195 uptake, but not PiB retention, correlated with impaired cognition in this AD cohort (Edison et al., 2008).
Collectively, these studies indicate that imaging brain inflammation is a valuable approach in evaluating AD pathology in vivo; however, more sensitive radioligands need to be developed. Furney et al., (2011) reported that compared to in vivo brain structural imaging alone, a combination of MRI imaging and inflammation (cytokine) biomarkers is a better predictor of a conversion from MCI to AD. PiB-PET imaging is particularly useful for monitoring changes in amyloid load in response to anti-amyloid therapies. While Aβ immunization appears to be effective in reducing amyloid pathology in AD patients, this intervention has been observed to activate microglia reaction in the brain and it can result in severe side effects (Boche et al., 2010). Therefore, combining in vivo PiB-PET imaging with biomarkers of inflammation will be of a particular importance when evaluation AD patients undergoing such therapies.
XV. Amyloid Imaging in AD Drug Development
Amyloid imaging will likely have two complimentary roles in clinical trials of future AD therapies. At the level of subject selection, amyloid PET will help ensure that patients enrolled in AD treatment trials truly have underlying Aβ deposits. This should increase the efficiency of AD-specific trials at the MCI phase (by eliminating the 25–40% of patients with non-AD causes of MCI who are unlikely to respond to the biological intervention) (Lorenzi et al., 2010), and ultimately by enabling primary prevention trials at the pre-clinical stage (Bateman et al., 2011; Reiman et al., 2011). Second, amyloid PET may be useful for demonstrating a biological effect of anti-Aβ therapies in early stages of drug development. Two studies thus far have illustrated this potential application of amyloid imaging. In a phase 2 trial of bapineuzumab, a humanized monoclonal antibody targeting Aβ, 19 patients receiving active treatment, and 8 receiving placebo underwent PiB-PET at baseline and following 18 months of treatment (up to six infusions) (Rinne et al., 2010). Mean cortical PiB SUVr values increased by an average of 16.9% from baseline in the placebo group, but decreased by an average of 8.5% from baseline in the active treatment group, resulting in an observed treatment effect of ~25%. Similar results were reported in a trial of gantenerumab, another human anti-Aβ monoclonal antibody, where patients receiving the drug at 60 mg (N = 6), 200 mg (N = 6), or placebo (N = 4) underwent PiB-PET at baseline and posttreatment (up to 7 monthly infusions) (Ostrowitzki et al., 2012). Mean PiB SUVr posttreatment was on average +11.0% of baseline in the placebo group, +2.1% in the low-dose treatment group, and −9.4% in the high-dose treatment group. While small and laden with caveats, these studies illustrate proof-of-concept for a very important translational application of amyloid PET. Ultimately, lower fibrillar Aβ burden will need to be linked to improved cognitive and functional outcomes for amyloid PET to be adopted as a true surrogate outcome measure in AD drug development.
XVI. F-18 Compounds
PiB is the most widely studied amyloid imaging agent and the first Aβ selective radiotracer to differentiate AD patients from HCs by in vivo PET imaging (Klunk et al., 2004). However, the short radioactive half-life of carbon-11 (about 20 min) limits the use of PiB only to those PET imaging centers with onsite capability to synthesize this radiotracer. Fluorine-18 (F-18) labeled PET tracers are longer lived (about 110 min) so they can be distributed to distant PET imaging sites. Several new [F-18]-labeled amyloid ligands have been developed recently for in vivo imaging of Aβ pathology. These radioligands include [F-18]flutemetamol, [F-18]AV-45 (florbetapir), [F-18]AV-1 (florbetaben), [F-18]AZD4694, and [F-18]FDDNP, and currently several are under development for use as clinically approved Aβ-imaging radiopharmaceuticals.
A. [F-18]Flutemetamol
[F-18]Flutemetamol is a 3’-fluoro analog of PiB (3’-F-PiB) currently being examined in Phase III FDA clinical trials. Being structurally similar to PiB, [F-18]flutemetamol was expected to demonstrate comparable brain uptake and clearance. Indeed, initial PET imaging studies show that compared to PiB [F-18]flutemetamol has similar retention characteristics although somewhat more pronounced retention in white matter. A phase I clinical study of eight mild AD patients (MMSE 20–26) and eight HCs reported that [F-18]flutemetamol regional standardized uptake value ratios (SUVRs) were significantly higher in the neocortex and striatum of AD patients, while the values measured in white matter, cerebellum, and pons were not different from HCs (Nelissen et al., 2009). ). In a multicenter phase II trial of [F-18]flutemetamol, Vanderberghe and colleagues studied 27 early AD, 20 amnestic MCI, 15 controls >55 years of age, and 10 controls <55 years of age, and reported 93.1% sensitivity and 93.3% specificity for AD (Vanderberghe et al., 2010). The same study reported a strong correlation (0.89–0.92) between [F-18]flutemetamol and PiB regional SUVRs in 20 AD and 20 MCI subjects (Vanderberghe et al., 2010). These data indicate that [F-18]flutemetamol is comparable to [C-11]PiB in its ability to detect brain fibrillar Aβ pathology in living subjects. In further support of this, Wolk et al. (2011) provided histopathological evidence in seven subjects who had a frontal cortical biopsy (as part of a clinical work-up for suspected normal pressure hydrocephalus) and later underwent [F-18]flutemetamol PET imaging, similar to previous reports of brain biopsy using PiB (Leinonen et al., 2008). They reported that a higher [F-18]flutemetamol uptake in frontal cortex correlated with amyloid plaque load determined using Aβ immunohistochemistry or thioflavin S staining in the frontal biopsy samples, further supporting that [F-18]flutemetamol is sensitive in detecting fibrillar Aβ plaques in vivo (Wolk et al., 2011).
B. [F-18]Florbetapir
[F-18]Florbetapir {(E)-4-(2-(6-(2-(2-(2-[F-18]-fluoroethoxy)ethoxy)eth-oxy)pyridin-3-yl)vinyl)-N-methyl benzenamine; [F-18]AV-45; or amyvid} has proven to be effective in imaging Aβ fibrillar pathology in vivo (reviewed by Lister-James et al., 2011). Preclinical studies characterized postmortem binding of [F-18]florbetapir to Aβ plaques and demonstrated prominent in vitro labeling in brain tissue sections from AD patients but not in sections from control brains (Choi et al., 2009; Lin et al., 2010). A clinical trial performed on 18 mild-moderate AD patients (mean MMSE = 19.3) and 16 HCs showed that cortical regions had a higher [F-18]florbetapir retention, while white matter and cerebellar retention were not different between AD and control subjects (Wong et al., 2010). An analysis of multicenter PET data from 210 participants, pooled from four registered phase I and II trials of [F-18]florbetapir imaging, reported that positive PET scans indicative of fibrillar Aβ pathology were observed in 85% of 68 subjects with clinically probable AD, 47% of the 60 MCI subjects, and 28% of the 82 cognitively HCs >55 years old (Fleisher et al., 2011). [F-18]florbetapir PET scans were negative in all young subjects aged <50 years (n = 74) and there was a good correlation between [F-18]florbetapir retention in vivo and postmortem measures of Aβ immunoreactive and neuritic plaques in a cohort of 29 terminally ill patients with mixed diagnoses who were evaluated with [F-18] florbetapir PET and later came to autopsy (Clark et al., 2011). However, there was a substantial variability in ratings of PET scans by independent readers in that study. Neuropathological confirmation of increased [F-18] florbetapir uptake in areas of neocortex, striatum, and thalamus which contained heavy loads of fibrillar Aβ deposits was also reported in a [F-18]florbetapir imaged patient with Down syndrome and AD (Sabbagh et al., 2011). New preliminary data demonstrate high sensitivity (92%) and specificity (91%) using quantitative assessment of global cortical SUVr to differentiate AD subjects from HCs, and indicate that [F-18]florbetapir PET is suitable biomarker for routine clinical use (Camus et al., 2012).
C. [F-18]Florbetaben
[F-18]Florbetaben {(E)-4-(2-(4-(2-(2-(2-[F-18]fluoroethoxy)ethoxy)eth-oxy)phenyl)-vinyl)-N-methyl-benzenamine; [F-18]AV-1 or BAY-94-9172} is another [F-18]-labeled radioligand that is one atom chemically different from [F-18]florbetapir and in early PET brain scan clinical studies proved to be able to discriminate a group of 15 AD patients with significantly higher neocortical retention from 15 HCs and 5 FTLD cases (Rowe et al., 2008). A large multicenter phase II study of [F-18]florbetaben was conducted in 81 clinical probable AD patients and 69 HC subjects, and it showed 80% sensitivity and 91% specificity for distinguishing the AD group from controls (Barthel et al., 2011a). An exploratory, open-label, nonrandomized, single-center phase 0 study of [F-18]florbetaben PET imaging in 10 clinically probable AD and 10 HCs reported 90% sensitivity and 90% specificity (Barthel et al., 2011b). A recent review of three clinical studies involving 109 subjects with clinical diagnoses of AD, MCI, and various non-AD dementias (FTLD, VaD, DLB, PD) who were imaged with [F-18]florbetaben revealed that AD patients had significantly higher gray matter retention values (SUVRs), indicating higher Aβ burden, compared to other disease groups (Villemagne et al., 2011b). Florbetaben findings in DLB, PD, and MCI were similar to those previously described for PiB.
D. [F-18]FDDNP
[F-18]FDDNP (2-(1-{6-[(2-[F-18]fluoroethyl)(methyl)amino]-2-naphthyl} ethylidene)malononitrile) is a lipophilic tracer which binds in histological and autoradiography assays not only to aggregated Aβ in plaques but also to NFT (Agdeppa et al., 2001). PET imaging studies demonstrated that regional increases in [F-18]FDDNP uptake correlate with greater brain atrophy (i.e., lower MRI volumes) and reduced brain glucose metabolism (lower FDG-PET) in brain areas containing both Aβ plaques and NFT (Shoghi-Jadid et al., 2002; Small et al., 2006). Several subsequent in vivo imaging studies compared [F-18]FDDNP to PiB retention in cognitively impaired subjects and HCs. Using both radiotracers, Shin et al. imaged 10 clinical AD and 10 HCs, and demonstrated that [F-18]FDDNP and PiB retention patterns were similar in the neocortical regions; however, in the mesial temporal lobe structures, known to contain large amounts of neurofibrillary pathology in AD (Braak & Braak, 1991), [F-18]FDDNP binding was strongest while PiB retention was minimal (Shin et al., 2008). Tolboom and colleagues examined 14 clinical probable AD, 11 amnestic MCI, and 13 HCs with both PiB and [F-18]FDDNP PET scans performed on the same day for most of the subjects (Tolboom et al., 2009). Global cortical uptake values of PiB and [F-18]FDDNP correlated directly but there were different regional binding patterns and PiB was better in detecting differences among clinical groups; although with both tracers, AD and MCI groups had higher global cortical uptake when compared to control values, only PiB showed no overlap between AD and control groups. These observations suggested that PiB and [F-18]FDDNP detect different but related pathology in the brain (Tolboom et al., 2009), in agreement with the idea that [F-18]FDDNP is valuable in detecting NFT pathology in addition to aggregated Aβ (Shin et al., 2011).
Preclinical characterization of the novel fluorinated PET radioligand candidates AZD2184 and AZD4694 demonstrated their high specificity for Aβ plaques in brain tissue sections from AD cases and transgenic APP mice (Johnson et al., 2009; Juréus et al., 2010). Full reports of the properties of these two radiotracers in detecting and assessing Aβ plaque deposits in PET human imaging studies have not been published to date.
Further studies in large numbers of subjects representing different clinical categories are required to characterize the existing radiotracers and develop new radiotracers for imaging the distribution and quantity of AD lesions in living subjects. Single or multiple tracer imaging studies using [F-18]-labeled PET radioligands will be extremely important and will complement clinical neurocognitive testing, making possible earlier and more sensitive detection of AD pathology as well as for monitoring disease progression and effects of new drug treatments.
XVII. Detection of the Earliest Signs of Amyloid Deposition
Since the initial PiB-PET studies, the focus of many research studies has shifted away from the robust signal seen in symptomatic AD and toward detection of the earliest signs of fibrillar Aβ pathology in cognitively normal individuals (see above, Section VI). This shift toward initial detection has generated a need for reliable methods that can distinguish brains free of fibrillar Aβ from brains that have early-stage fibrillar Aβ deposition. It is important that such methods can be standardized and applied across many centers.
It should be noted that PiB retention is a continuous measure and need not necessarily be dichotomized into PiB(+) and PiB(-). Many studies have used PiB retention as a continuous variable, correlating PiB retention to a variety of cognitive or biochemical measures ( Bourgeat et al., 2010; Forsberg et al., 2010; Furst et al., 2010; Mormino et al., 2009; Pike et al. 2007; Rentz et al., 2010; Resnick et al., 2010). This approach may be preferred for some applications; however, in other applications it is necessary to dichotomize subjects into PiB(+) and PiB(−). This may be most important in the cognitively normal subjects when attempting to disentangle the effects of normal aging from the effects of preclinical AD (Sperling et al., 2011).
A variety of ad hoc objective approaches have been presented to define an amyloid-positive cutoff using amyloid imaging. These methods include using one or two standard deviations above the mean of the control data (Edison et al., 2008; Kemppainen et al., 2007; Klunk et al., 2004 Okello et al., 2009); inspection of quantitative PET data for natural breakpoints in the distribution of tracer retention in combinations of young controls, elderly controls and/or AD patients (Edison et al., 2008; Gomperts et al., 2008; Hedden et al., 2009; Jack et al., 2008; Maetzler et al., 2009; Mintun et al., 2006; Mormino et al., 2011; Morris et al., 2010; Rowe et al., 2007; Roe et al., 2008); the low end of the range of tracer retention in clinically (Sperling et al.,2009) or pathologically (Fleisher et al., 2011) defined AD patients; receiver operating characteristic (ROC) analyses of PET data from control and AD subjects (Devanand et al., 2007; Mormino et al., 2009; Ng et al., 2007; Pike et al., 2007); visual reads (Engler et al., 2007; Gomperts et al., 2008; Johnson et al., 2007; Ng et al., 2007; Rabinovici et al., 2007; Suotunen et al., 2010; Tolboom et al., 2009); and cluster analysis methods using both PiB(+) and PiB(−) elderly control subjects (Bourgeat et al., 2010). Each approach has advantages and shortcomings. Most of these approaches involve subjective choices such as the number of standard deviations above the control mean, the exact location of the natural breakpoints and the interpretation of the visual read. Others, like ROC analysis or using the low end of the AD range, rely on the composition of the AD group, which can vary widely depending on the nature of the particular control or AD population utilized. Methods that rely on analysis of the entire control group can result in cutoffs that are unduly affected by the amyloid-positive high outliers in the control group (e.g., control mean + standard deviations). While many of these methods yield similar results, further study will be required to identify a widely applicable and standardized method to identify both the earliest signs of Aβ deposition and Aβ deposition that is clinically meaningful, or “AD-like.”
XVIII. Limitations, Validity, and Unresolved Questions
While amyloid imaging represents a major advance in AD research, the field is still young and there are a number of unresolved questions and limitations. The dynamic range, threshold and ceiling effects, binding interactions as well as the relative selectivity of amyloid tracers for different tertiary structures of Aβ deposits remain works in progress. Roughly 10–20% of clinically diagnosed AD patients are amyloid-negative (Fleisher et al., 2011; Rabinovici et al., 2011; Rowe et al., 2010; Vandenberghe et al., 2010; Villemagne et al., 2011b), and while some of these may have been clinically misdiagnosed, a case report of deficient in vitro PiB retention to an otherwise typical AD postmortem brain (Rosen et al., 2010) suggests that there are factors other than low Aβ burden that can lead to a negative in vivo study. Methodologically, the fundamental factors impacting white matter binding are incompletely understood (Fodero-Tavoletti et al., 2009). Furthermore, the relative benefit and optimal methodology for implementing partial volume correction are actively being debated. Partial volume effects are important to consider when quantifying binding in the atrophic brain, where low counts in enlarged CSF spaces can dilute signal from gray matter. This is a particularly relevant issue for quantifying amyloid in longitudinal studies, when progressive brain atrophy can be expected (Jack et al., 2009). Partial volume effects from white matter may be an issue for F-18 tracers, for which the dynamic range in white matter is similar to or even exceeds the dynamic range of gray matter (Baker et al., 2012). While most studies have employed cerebellar gray matter as the reference region for normalizing counts across subjects, some argue for inclusion of white matter (to account for the variability of white matter binding across subjects) (Clark et al., 2011) or even for a combined cerebellum-pons region that would be less susceptible to mis-registration errors when defined on a structural MRI (Koeppe, 2012).
In terms of translational applications, the relative advantages of qualitative visual versus quantitative classification are still being weighed. Visual interpretations may be easier to implement on a broad scale in the clinical arena. Quantitative methodologies are more objective, but also more prone to misclassification due to partial volume effects or errors in automated processing. The optimal threshold for defining a scan as positive (visually or quantitatively) is a moving target, as discussed in detail above, and will likely differ depending on whether the goal is early detection (more liberal threshold) or ruling-in AD as the cause of cognitive impairment (more conservative threshold). Whether and how the threshold should be adjusted for patient variables such as age, sex, education, and ApoE genotype is an open question. It is still not clear whether dichotomizing scans as positive or negative will be sufficient for clinical purposes, or whether there is additional information to be attained from the degree and spatial distribution of tracer binding. Only limited studies have directly compared the relative merit of amyloid PET to CSF biomarkers, MRI, FDG, or clinical measures in common clinical scenarios. Further, the data that are available about the clinical utility of amyloid imaging are almost entirely derived from highly selected research cohorts, and it is not yet clear how the technique will perform in typical clinical populations. Finally, even in scenarios where amyloid imaging will very likely yield helpful diagnostic and prognostic information (e.g., MCI, atypical dementia in a young patient), it is not at all clear that third party payers will cover the cost of PET unless a clear benefit in clinical outcome can be demonstrated.
XIX. Conclusion
PiB-PET and Aβ imaging mark a major advancement in the study of the pathology and treatment of AD. One facet of Aβ deposition that has become clear from PiB-PET studies is how early in the spectrum of AD the full burden of amyloid plaques begins to develop. Therefore, a major challenge of amyloid imaging is and will be how to determine the earliest signs of amyloid accumulation, its association with cognitive impairments and, ultimately, whether or not this early amyloid deposition will invariably lead to clinical dementia in a high percentage of individuals. This will likely require the field to focus on cognitively normal elderly and detection of the earliest signs of amyloid deposition, in order to determine the clinical significance of presymptomatic pathology. As anti-amyloid therapies are developed, it will be critical to effectively identify the earliest changes in amyloid deposition and the clinical significance of such changes. Further, as has been reflected in the new diagnostic criteria for AD, MCI, and “preclinical AD,” the use of amyloid imaging, alone or in conjunction with other biomarkers, will likely be critical to the identification of subjects at risk for AD and future decline.
Acknowledgments
Supported by The National Institutes of Health, National Institute on Aging: K01 AG037562; K23 AG031861; RO1 AG18402, AG014449, AG034570; R37 AG025516; P01 AG025204; P50 AG005133 and the John Douglas French Alzheimer’s Foundation.
List of Abbreviations
- [F-18]
fluorine-18
- [F-18]FDDNP
2-(1-{6-[(2-[F-18] fluoroethyl) (methyl)amino]-2-naphthyl}ethylidene)malononitrile
- [F-18]florbetaben
(E)-4-(2-(4-(2-(2-(2-[F-18]fluoroethoxy)ethoxy) ethoxy)phenyl)-vinyl)-N-methyl-benzenamine; [F-18]AV-1 or BAY-94-9172
- [F-18]florbetapir
(E)-4-(2-(6-(2-(2-(2-[F-18]-fluoroethoxy)ethoxy) ethoxy)pyridin-3-yl)vinyl)-N-methyl benzenamine; [F-18]AV-45; or amyvid
- [F-18]flutemetamol
2-{3-[18F]fluoro-4-(methylamino)phenyl}-6-hydroxybenzothiazole; or 3’-Fluoro-PiB
- [F-18]THK523
2-(4-aminophenyl)-6-(2-fluoroethoxy)quinoline)
- [H-3]
hydrogen-3 (Tritium)
- 6-CN-PiB
6-cyano-PiB
- AD
Alzheimer’s Disease
- ApoE
apolipoprotein E
- APP
Aβ precursor protein
- Aβ
amyloid-β
- BACE1
beta-secretase 1
- CAA
cerebral amyloid angiopathy
- CDR
clinical dementia rating
- CERAD
Consortium to Establish a Registry of Alzheimer’s Disease
- CJD
Creutzfeldt–Jakob disease
- CSF
cerebrospinal fluid
- CVD
cardiovascular disease
- DLB
dementia with Lewy bodies
- DVR
distribution volume ratios
- ELISA
enzyme-linked immunosorbent assay
- eoFAD
early-onset familial Alzheimer’s disease
- FDG
fludeoxyglucose
- FDG-PET
FDG-positron-emission tomography
- FTD
frontotemporal dementia
- FTLD
ftrontotemporal lobar degeneration
- FUS
fused-in sarcoma
- HC
healthy control
- Kd
dissociation constant
- Ki
inhibition constant
- LB
Lewy bodies
- lvPPA
logopenic-variant primary progressive aphasia
- MCI
mild cognitive impairment
- MMSE
mini-mental status exam
- MRI
magnetic resonance imaging
- NFT
neurofibrillary tangles
- NIA-RI
National Institute on Aging and Reagan Institute
- PCA
posterior cortical atrophy
- PD
Parkinson’s disease
- PDD
Parkinson’s disease dementia
- PET
positron-emission tomography
- PiB
Pittsburgh compound-B ( [N-methyl-11C]2-(4’-methylaminophenyl)-6-hydroxybenzothiazole; or [11C]6-OH-BTA-1)
- PiB(−)
PiB-negative
- PiB(+)
PiB-positive
- PiB-PET
PiB-positron-emission tomography
- PS1
presenilin-1
- p-tau
phosphorylated tau
- rCMRglc
regional cerebral metabolic rate of glucose
- ROC
receiver operating characteristic
- SUV
standardized uptake value
- SUVr
standardized uptake value ratio
- TDP-43
TAR DNA-binding protein 43
- ThT
thioflavin-T
- VaD
vascular dementia
Footnotes
Conflicts of Interest: Dr. Rabinovici has received research support from Avid Radio-pharmaceuticals and has consulted for Eli Lily and Novartis Diagnostics. Dr. Jagust has consulted for General Electric (GE) Healthcare, Bayer Healthcare, Janssen Alzheimer Immunotherapy, Synarc, Genentech, and TauRx. GE Healthcare holds a license agreement with the University of Pittsburgh based on the technology described in this manuscript. Drs. Klunk and Mathis are co-inventors of PiB and, as such, have a financial interest in this license agreement and serve as consultants for GE Healthcare. Dr. Mathis also has consulting agreements with Janssen AI, Pfizer, and Genzyme. Dr. Ikonomovic has received research support and consulted for GE Healthcare.
References
- Agdeppa ED, Kepe V, Liu J, Flores-Torres S, Satyamurthy N, Petric A, Cole GM, Small GW, Huang SC, Barrio JR. Binding characteristics of radiofluorinated 6-dialkylamino-2-naphthylethylidene derivatives as positron emission tomography imaging probes for beta-amyloid plaques in Alzheimer’s disease. Journal of Neuroscience. 2001;21:RC189. doi: 10.1523/JNEUROSCI.21-24-j0004.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aizenstein HJ, Nebes RD, Saxton JA, Price JC, Mathis CA, Tsopelas ND, Ziolko SK, James JA, Snitz BE, Houck PR, Bi W, Cohen AD, Lopresti BJ, DeKosky ST, Halligan EM, Klunk WE. Frequent amyloid deposition without significant cognitive impairment among the elderly. Archives of Neurology. 2008;65:1509–1517. doi: 10.1001/archneur.65.11.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, Snyder PJ, Carrillo MC, Thies B, Phelps CH. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & Dementia. 2011;7(3):270–279. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alladi S, Xuereb J, Bak T, Nestor P, Knibb J, Patterson K, et al. Focal cortical presentations of Alzheimer’s disease. Brain. 2007;130:2636–2645. doi: 10.1093/brain/awm213. [DOI] [PubMed] [Google Scholar]
- Anchisi D, Borroni B, Franceschi M, Kerrouche N, Kalbe E, Beuthien-Beumann B, Cappa S, Lenz O, Ludecke S, Marcone A, Mielke R, Ortelli P, Padovani A, Pelati O, Pupi A, Scarpini E, Weisenbach S, Herholz K, Salmon E, Holthoff V, Sorbi S, Fazio F, Perani D. Heterogeneity of brain glucose metabolism in mild cognitive impairment and clinical progression to Alzheimer disease. Archives of Neurology. 2005;62(11):1728–1733. doi: 10.1001/archneur.62.11.1728. [DOI] [PubMed] [Google Scholar]
- Apostolova LG, Dinov ID, Dutton RA, Hayashi KM, Toga AW, Cummings JL, Thompson PM. 3D comparison of hippocampal atrophy in amnestic mild cognitive impairment and Alzheimer’s disease. Brain. 2006a;129(Pt 11):2867–2873. doi: 10.1093/brain/awl274. [DOI] [PubMed] [Google Scholar]
- Apostolova LG, Dutton RA, Dinov ID, Hayashi KM, Toga AW, Cummings JL, Thompson PM. Conversion of mild cognitive impairment to Alzheimer disease predicted by hippocampal atrophy maps. Archives of Neurology. 2006b;63(5):693–699. doi: 10.1001/archneur.63.5.693. [DOI] [PubMed] [Google Scholar]
- Apostolova LG, Mosconi L, Thompson PM, Green AE, Hwang KS, Ramirez A, Mistur R, Tsui WH, de Leon MJ. Subregional hippocampal atrophy predicts Alzheimer’s dementia in the cognitively normal. Neurobiology of Aging. 2008;31(7):1077–1088. doi: 10.1016/j.neurobiolaging.2008.08.008. Epub 2008 Sep 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer HA, Edison P, Brooks DJ, Barnes J, Frost C, Yeatman T, Fox NC, Rossor MN. Amyloid load and cerebral atrophy in Alzheimer’s disease: An 11C-PIB positron emission tomography study. Annals of Neurology. 2006;60:145–147. doi: 10.1002/ana.20889. [DOI] [PubMed] [Google Scholar]
- Arnáiz E, Jelic V, Almkvist O, Wahlund LO, Winblad B, Valind S, Nordberg A. Impaired cerebral glucose metabolism and cognitive functioning predict deterioration in mild cognitive impairment. Neuroreport. 2001;12(4):851–855. doi: 10.1097/00001756-200103260-00045. [DOI] [PubMed] [Google Scholar]
- Arnold SE, Hyman BT, Flory J, Damasio AR, Van Hoesen GW. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cerebral Cortex. 1991;1:103–116. doi: 10.1093/cercor/1.1.103. [DOI] [PubMed] [Google Scholar]
- Attems J, Quass M, Jellinger KA, Lintner F. Topographical distribution of cerebral amyloid angiopathy and its effect on cognitive decline are influenced by Alzheimer disease pathology. Journal of the Neurological Sciences. 2007;257:49–55. doi: 10.1016/j.jns.2007.01.013. [DOI] [PubMed] [Google Scholar]
- Bacskai BJ, Frosch MP, Freeman SH, Raymond SB, Augustinack JC, Johnson KA, Irizarry MC, Klunk WE, Mathis CA, Dekosky ST, Greenberg SM, Hyman BT, Growdon JH. Molecular imaging with Pittsburgh Compound B confirmed at autopsy: A case report. Archives of Neurology. 2007;64:431–434. doi: 10.1001/archneur.64.3.431. [DOI] [PubMed] [Google Scholar]
- Baker S, Landau S, Jagust W. Effect of white matter binding on florbetapir and PIB image classification. Miami, FL: Human Amyloid Imaging; 2012. [Google Scholar]
- Ballard C, Ziabreva I, Perry R, Larsen JP, O’Brien J, McKeith I, Perry E, Aarsland D. Differences in neuropathologic characteristics across the Lewy body dementia spectrum. Neurology. 2006;67(11):1931–1934. doi: 10.1212/01.wnl.0000249130.63615.cc. [DOI] [PubMed] [Google Scholar]
- Barthel H, Gertz HJ, Dresel S, Peters O, Bartenstein P, Buerger K, et al. Cerebral amyloid-β PET with florbetaben (18F) in patients with Alzheimer’s disease and healthy controls: A multicentre phase 2 diagnostic study. Lancet Neurology. 2011a;10:424–435. doi: 10.1016/S1474-4422(11)70077-1. [DOI] [PubMed] [Google Scholar]
- Barthel H, Luthardt J, Becker G, Patt M, Hammerstein E, Hartwig K, Eggers B, Sattler B, Schildan A, Hesse S, Meyer PM, Wolf H, Zimmermann T, Reischl J, Rohde B, Gertz HJ, Reininger C, Sabri O. Individualized quantification of brain β-amyloid burden: Results of a proof of mechanism phase 0 florbetaben PET trial in patients with Alzheimer’s disease and healthy controls. European Journal of Nuclear Medicine and Molecular Imaging. 2011b;38(9):1702–1714. doi: 10.1007/s00259-011-1821-1. [DOI] [PubMed] [Google Scholar]
- Bateman RJ, Aisen PS, De Strooper B, Fox NC, Lemere CA, Ringman JM, et al. Autosomal-dominant Alzheimer’s disease: A review and proposal for the prevention of Alzheimer’s disease. Alzheimer’s Research & Therapy. 2011;3:1. doi: 10.1186/alzrt59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker JT, Davis SW, Hayashi KM, Meltzer CC, Toga AW, Lopez OL, Thompson PM Imaging Methods Analysis in Geriatrics Research Group. Three-dimensional patterns of hippocampal atrophy in mild cognitive impairment. Archives of Neurology. 2006;63(1):97–101. doi: 10.1001/archneur.63.1.97. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Arvanitakis Z, Kelly JF, Aggarwal NT, Shah RC, Wilson RS. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66(12):1837–1844. doi: 10.1212/01.wnl.0000219668.47116.e6. [DOI] [PubMed] [Google Scholar]
- Bennett JP, Yamamura HI. Neurotransmitter, hormone, or drug receptor binding methods. In: Yamamura HI, Enna SJ, Kuhar MJ, editors. Neurotransmitter receptor binding. New York: Raven Press; 1985. pp. 61–89. [Google Scholar]
- Berg L, McKeel DWJ, Miller JP, Storandt M, Rubin EH, Morris JC, Baty J, Coats M, Norton J, Goate AM, Price JL, Gearing M, Mirra SS, Saunders AM. Clinicopathologic studies in cognitively healthy aging and Alzheimer’s disease: Relation of histologic markers to dementia severity, age, sex, and apolipoprotein E genotype. Archives of Neurology. 1998;55:326–335. doi: 10.1001/archneur.55.3.326. [DOI] [PubMed] [Google Scholar]
- Blennow K. CSF biomarkers for mild cognitive impairment. Journal of Internal Medicine. 2004;256(3):224–234. doi: 10.1111/j.1365-2796.2004.01368.x. [DOI] [PubMed] [Google Scholar]
- Blennow K, Hampel H. CSF markers for incipient Alzheimer’s disease. Lancet Neurology. 2003;2(10):605–613. doi: 10.1016/s1474-4422(03)00530-1. [DOI] [PubMed] [Google Scholar]
- Bombois S, Maurage CA, Gompel M, Deramecourt V, Mackowiak-Cordoliani MA, Black RS, Lavielle R, Delacourte A, Pasquier F. Absence of beta-amyloid deposits after immunization in Alzheimer disease with Lewy body dementia. Archives of Neurology. 2007;64:583–587. doi: 10.1001/archneur.64.4.583. [DOI] [PubMed] [Google Scholar]
- Boche D, Denham N, Holmes C, Nicoll JA. Neuropathology after active Abeta42 immunotherapy: Implications for Alzheimer’s disease pathogenesis. Acta Neuropathologica. 2010 Sep;120(3):369–384. doi: 10.1007/s00401-010-0719-5. Epub 2010 Jul 15. [DOI] [PubMed] [Google Scholar]
- Bourgeat P, Chetelat G, Villemagne VL, Fripp J, Raniga P, Pike K, Acosta O, Szoeke C, Ourselin S, Ames D, Ellis KA, Martins RN, Masters CL, Rowe CC, Salvado O. Beta-amyloid burden in the temporal neocortex is related to hippocampal atrophy in elderly subjects without dementia. Neurology. 2010;(74):121–127. doi: 10.1212/WNL.0b013e3181c918b5. [DOI] [PubMed] [Google Scholar]
- Boxer AL, Rabinovici GD, Kepe V, Goldman J, Furst AJ, Huang SC, Baker SL, O’Neil JP, Chui H, Geschwind MD, Small GW, Barrio JR, Jagust W, Miller BL. Amyloid imaging in distinguishing atypical prion disease from Alzheimer disease. Neurology. 2007;69:283–290. doi: 10.1212/01.wnl.0000265815.38958.b6. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Alzheimer’s disease: Striatal amyloid deposits and neurofibrillary changes. Journal of Neuropathology & Experimental Neurology. 1990;49:215–224. [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathologica (Berlin) 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiology of Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Brilliant MJ, Elble RJ, Ghobrial M, Struble RG. The distribution of amyloid beta protein deposition in the corpus striatum of patients with Alzheimer’s disease. Neuropathology and Applied Neurobiology. 1997;23:322–325. [PubMed] [Google Scholar]
- Buchhave P, Minthon L, Zetterberg H, Wallin AK, Blennow K, Hansson O. Cerebrospinal fluid levels of β-Amyloid 1–42, but not of Tau, are fully changed already 5–10 years before the onset of Alzheimer dementia. Archives of General Psychiatry. 2012;69(1):98–106. doi: 10.1001/archgenpsychiatry.2011.155. [DOI] [PubMed] [Google Scholar]
- Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, Sheline YI, Klunk WE, Mathis CA, Morris JC, Mintun MA. Molecular, structural, and functional characterization of Alzheimer’s disease: Evidence for a relationship between default activity, amyloid, and memory. Journal of Neuroscience. 2005;25:7709–7717. doi: 10.1523/JNEUROSCI.2177-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burack MA, Hartlein J, Flores HP, Taylor-Reinwald L, Perlmutter JS, Cairns NJ. In vivo amyloid imaging in autopsy-confirmed Parkinson disease with dementia. Neurology. 2010;74(1):77–84. doi: 10.1212/WNL.0b013e3181c7da8e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busse A, Hensel A, Guhne U, Angermeyer MC, Riedel-Heller SG. Mild cognitive impairment: Long-term course of four clinical subtypes. Neurology. 2006;67:2176–2185. doi: 10.1212/01.wnl.0000249117.23318.e1. [DOI] [PubMed] [Google Scholar]
- Cairns NJ, Ikonomovic MD, Benzinger T, Storandt M, Fagan AM, Shah AR, Reinwald LT, Carter D, Felton A, Holtzman DM, Mintun MA, Klunk WE, Morris JC. Absence of Pittsburgh compound B detec tion of cerebral amyloid beta in a patient with clinical, cognitive, and cerebrospinal fluid markers of Alzheimer disease: A case report. Archives of Neurology. 2009;66(12):1557–1562. doi: 10.1001/archneurol.2009.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camus V, Payoux P, Barré L, Desgranges B, Voisin T, Tauber C, La Joie R, Tafani M, Hommet C, Chételat G, Mondon K, de La Sayette V, Cottier JP, Beaufils E, Ribeiro MJ, Gissot V, Vierron E, Vercouillie J, Vellas B, Eustache F, Guilloteau D. Using PET with (18)F-AV-45 (florbetapir) to quantify brain amyloid load in a clinical environment. European Journal of Nuclear Medicine and Molecular Imaging. 2012 doi: 10.1007/s00259-011-2021-8. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetelat G, Villemagne VL, Villain N, Jones G, Ellis KA, Ames D, Masters CL, Rowe CC, Martins RN AIBL Research Group. Accelerated cortical atrophy in cognitively normal elderly with high beta-amyloid deposition. Neurology. 2012;78(7):477–484. doi: 10.1212/WNL.0b013e318246d67a. [DOI] [PubMed] [Google Scholar]
- Chételat G, Desgranges B, de la Sayette V, Viader F, Eustache F, Baron JC. Mild cognitive impairment: Can FDG-PET predict who is to rapidly convert to Alzheimer’s disease? Neurology. 2003;60(8):1374–1377. doi: 10.1212/01.wnl.0000055847.17752.e6. [DOI] [PubMed] [Google Scholar]
- Chételat G, Desgranges B, de la Sayette V, Viader F, Berkouk K, Landeau B, Lalevée C, Le Doze F, Dupuy B, Hannequin D, Baron JC, Eustache F. Dissociating atrophy and hypometabolism impact on episodic memory in mild cognitive impairment. Brain. 2003;126(Pt 9):1955–1967. doi: 10.1093/brain/awg196. [DOI] [PubMed] [Google Scholar]
- Chételat G, Fouquet M, Kalpouzos G, Denghien I, De la Sayette V, Viader F, Mézenge F, Landeau B, Baron JC, Eustache F, Desgranges B. Three-dimensional surface mapping of hippocampal atrophy progression from MCI to AD and over normal aging as assessed using voxel-based morphometry. Neuropsychologia. 2008;46(6):1721–1731. doi: 10.1016/j.neuropsychologia.2007.11.037. [DOI] [PubMed] [Google Scholar]
- Chételat G, Villemagne VL, Bourgeat P, Pike KE, Jones G, Ames D, Ellis KA, Szoeke C, Martins RN, O’Keefe GJ, Salvado O, Masters CL, Rowe CC Australian Imaging Biomarkers Lifestyle Research Group. Relationship between atrophy and beta-amyloid deposition in Alzheimer disease. Annals of Neurology. 2010;67(3):317–324. doi: 10.1002/ana.21955. [DOI] [PubMed] [Google Scholar]
- Choi SR, Golding G, Zhuang Z, Zhang W, Lim N, Hefti F, Benedum TE, Kilbourn MR, Skovronsky D, Kung HF. Preclinical properties of 18F-AV-45: A PET agent for Abeta plaques in the brain. Journal of Nuclear Medicine. 2009;50(11):1887–1894. doi: 10.2967/jnumed.109.065284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claassen DO, Lowe VJ, Peller PJ, Petersen RC, Josephs KA. Amyloid and glucose imaging in dementia with Lewy bodies and multiple systems atrophy. Parkinsonism & Related Disorders. 2011;17(3):160–165. doi: 10.1016/j.parkreldis.2010.12.006. [DOI] [PubMed] [Google Scholar]
- Clark CM, Schneider JA, Bedell BJ, Beach TG, Bilker WB, Mintun MA, et al. Use of florbetapir-PET for imaging beta-amyloid pathology. The Journal of the American Medical Association. 2011;305:275–283. doi: 10.1001/jama.2010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AD, Price JC, Weissfeld LA, James J, Rosario BL, Bi W, Nebes RD, Saxton JA, Snitz BE, Aizenstein HA, Wolk DA, DeKosky ST, Mathis CA, Klunk WE. Basal cerebral metabolism may modulate the cognitive effects of Aβ in mild cognitive impairment: An example of brain reserve. Journal of Neuroscience. 2009;29:14770–14778. doi: 10.1523/JNEUROSCI.3669-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway KA, Jr, Harper JD, Lansbury PT. Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry. 2000;39:2552–2563. doi: 10.1021/bi991447r. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Mathis CM, Price JC, Ikonomovic MD, Hamilton RL, Abrahamson EE, Paljug WR, Debnath ML, Hope CE, Isanski BA, Tsopelas ND, Lopresti BJ, Ziolko S, Bi W, Klunk WE. Correlation of regional in vivo Pittsburgh compound-B (PIB) retention with in vitro PIB, Aβ levels, and amyloid plaque density: Validation of PIB-PET in postmortem human brain. Alzheimer’s & Dementia. 2007;3:S105. [Google Scholar]
- DeKosky ST, Ikonomovic MD, Styren SD, Beckett L, Wisniewski S, Cochran EJ, Kordower JH, Mufson EJ, Bennett DA. Upregulation of choline acetyl-transferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Annals of Neurology. 2002;51(2):145–155. doi: 10.1002/ana.10069. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Scheff SW, Styren SD. Structural correlates of cognition in dementia: Quantification and assessment of synapse change. Neurodegeneration. 1996;5(4):417–421. doi: 10.1006/neur.1996.0056. [DOI] [PubMed] [Google Scholar]
- Del Sole A, Clerici F, Chiti A, Lecchi M, Mariani C, Maggiore L, Mosconi L, Lucignani G. Individual cerebral metabolic deficits in Alzheimer’s disease and amnestic mild cognitive impairment: An FDG PET study. European Journal of Nuclear Medicine and Molecular Imaging. 2008;35(7):1357–1366. doi: 10.1007/s00259-008-0773-6. [DOI] [PubMed] [Google Scholar]
- de Souza LC, Corlier F, Habert MO, Uspenskaya O, Maroy R, Lamari F, et al. Similar amyloid-beta burden in posterior cortical atrophy and Alzheimer’s disease. Brain. 2011;134:2036–2043. doi: 10.1093/brain/awr130. [DOI] [PubMed] [Google Scholar]
- de Toledo-Morrell L, Stoub TR, Bulgakova M, Wilson RS, Bennett DA, Leurgans S, Wuu J, Turner DA. MRI-derived entorhinal volume is a good predictor of conversion from MCI to AD. Neurobiology of Aging. 2004;25(9):1197–1203. doi: 10.1016/j.neurobiolaging.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Devanand DP, Pradhaban G, Liu X, Khandji A, De Santi S, Segal S, Rusinek H, Pelton GH, Honig LS, Mayeux R, Stern Y, Tabert MH, de Leon MJ. Hippocampal and entorhinal atrophy in mild cognitive impairment: Prediction of Alzheimer disease. Neurology. 2007;68(11):828–836. doi: 10.1212/01.wnl.0000256697.20968.d7. [DOI] [PubMed] [Google Scholar]
- Dickson DW. Dementia with Lewy bodies: Neuropathology. Journal of Geriatric Psychiatry and Neurology. 2002;15:210–216. doi: 10.1177/089198870201500406. [DOI] [PubMed] [Google Scholar]
- Dierksen GA, Skehan ME, Khan MA, Jeng J, Nandigam RN, Becker JA, Kumar A, Neal KL, Betensky RA, Frosch MP, Rosand J, Johnson KA, Viswanathan A, Salat DH, Greenberg SM. Spatial relation between microbleeds and amyloid deposits in amyloid angiopathy. Annals of Neurology. 2010;68(4):545–548. doi: 10.1002/ana.22099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll I, Zhou Y, An Y, Sojkova J, Davatzikos C, Kraut MA, Ye W, Ferrucci L, Mathis CA, Klunk WE, Wong DF, Resnick SM. Lack of association between 11C-PiB and longitudinal brain atrophy in non-demented older individuals. Neurobiology of Aging. 2010;32(12):2123–2130. doi: 10.1016/j.neurobiolaging.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drzezga A, Grimmer T, Henriksen G, Stangier I, Perneczky R, Diehl-Schmid J, Mathis CA, Klunk WE, Price J, Dekosky S, Wester HJ, Schwaiger M, Kurz A. Imaging of amyloid plaques and cerebral glucose metabolism in semantic dementia and Alzheimer’s disease. Neuroimage. 2008;39:619–633. doi: 10.1016/j.neuroimage.2007.09.020. [DOI] [PubMed] [Google Scholar]
- Drzezga A, Grimmer T, Riemenschneider M, Lautenschlager N, Siebner H, Alexopoulus P, Minoshima S, Schwaiger M, Kurz A. Prediction of individual clinical outcome in MCI by means of genetic assessment and (18)F-FDG PET. Journal of Nuclear Medicine. 2005;46(10):1625–1632. [PubMed] [Google Scholar]
- Edison P, Archer HA, Hinz R, Hammers A, Pavese N, Tai YF, Hotton G, Cutler D, Fox N, Kennedy A, Rossor M, Brooks DJ. Amyloid, hypometabolism, and cognition in Alzheimer disease. An [11C]PIB and [18F]FDG PET study. Neurology. 2006;68:501–508. doi: 10.1212/01.wnl.0000244749.20056.d4. [DOI] [PubMed] [Google Scholar]
- Edison P, Rowe CC, Rinne JO, Ng S, Ahmed I, Kemppainen N, Villemagne VL, O’Keefe G, Nagren K, Chaudhury KR, Masters CL, Brooks DJ. Amyloid load in Parkinson’s disease dementia and Lewy body dementia measured with [11C]PIB positron emission tomography. Journal of Neurology, Neurosurgery, and Psychiatry. 2008;79:1331–1338. doi: 10.1136/jnnp.2007.127878. [DOI] [PubMed] [Google Scholar]
- Edison P, Archer HA, Gerhard A, Hinz R, Pavese N, Turkheimer FE, Hammers A, Tai YF, Fox N, Kennedy A, Rossor M, Brooks DJ. Microglia, amyloid, and cognition in Alzheimer’s disease: An [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiology of Disease. 2008;32(3):412–419. doi: 10.1016/j.nbd.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Engler H, Santillo AF, Wang SX, Lindau M, Savitcheva I, Nordberg A, Lannfelt L, Langstrom B, Kilander L. In vivo amyloid imaging with PET in frontotemporal dementia. European Journal of Nuclear Medicine and Molecular Imaging. 2007;35:100–106. doi: 10.1007/s00259-007-0523-1. [DOI] [PubMed] [Google Scholar]
- Engler H, Forsberg A, Almkvist O, Blomquist G, Larsson E, Savitcheva I, Wall A, Ringheim A, Langstrom B, Nordberg A. Two-year follow-up of amyloid deposition in patients with Alzheimer’s disease. Brain. 2006;129:2856–2866. doi: 10.1093/brain/awl178. [DOI] [PubMed] [Google Scholar]
- Engler H, Nordberg A, Blomqvist G, Bergström M, Estrada S, Barletta J, Sandell J, Antoni G, Långström B, Klunk WE, Debnath ML, Holt DP, Wang Y, Huang G-F, Mathis CA. First human study with a benzothiazole amyloid-imaging agent in Alzheimer’s disease and control subjects. Neurobiology of Aging. 2002;23(1S):S429. [Google Scholar]
- Fagan AM, Mintun MA, Shah AR, Aldea P, Roe CM, Mach RH, Marcus D, Morris JC, Holtzman DM. Cerebrospinal fluid tau and ptau181 increase with cortical amyloid deposition in cognitively normal individuals: Implications for future clinical trials of Alzheimer’s disease. EMBO Molecular Medicine. 2009;1:371–380. doi: 10.1002/emmm.200900048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Archives of Neurology. 2007;64:343–349. doi: 10.1001/archneur.64.3.noc60123. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, Larossa GN, Spinner ML, Klunk WE, Mathis CA, Dekosky ST, Morris JC, Holtzman DM. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta(42) in humans. Annals of Neurology. 2006;59:512–519. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. The Journal of the American Medical Association. 1997;278(16):1349–1356. [PubMed] [Google Scholar]
- Fleisher AS, Chen K, Liu X, Roontiva A, Thiyyagura P, Ayutyanont N, Joshi AD, Clark CM, Mintun MA, Pontecorvo MJ, Doraiswamy PM, Johnson KA, Skovronsky DM, Reiman EM. Using positron emission tomography and florbetapir F 18 to image cortical amyloid in patients with mild cognitive impairment or dementia due to Alzheimer disease. Archives of Neurology. 2011;68(11):1404–1411. doi: 10.1001/archneurol.2011.150. [DOI] [PubMed] [Google Scholar]
- Fodero-Tavoletti MT, Okamura N, Furumoto S, Mulligan RS, Connor AR, McLean CA, Cao D, Rigopoulos A, Cartwright GA, O’Keefe G, Gong S, Adlard PA, Barnham KJ, Rowe CC, Masters CL, Kudo Y, Cappai R, Yanai K, Villemagne VL. 18F-THK523: A novel in vivo tau imaging ligand for Alzheimer’s disease. Brain. 2011;134(Pt 4):1089–1100. doi: 10.1093/brain/awr038. [DOI] [PubMed] [Google Scholar]
- Fodero-Tavoletti MT, Rowe CC, McLean CA, Leone L, Li QX, Masters CL, et al. Characterization of PiB binding to white matter in Alzheimer disease and other dementias. Journal of Nuclear Medicine. 2009;50:198–204. doi: 10.2967/jnumed.108.057984. [DOI] [PubMed] [Google Scholar]
- Fodero-Tavoletti MT, Smith DP, McLean CA, Adlard PA, Barnham KJ, Foster LE, Leone L, Perez K, Cortés M, Culvenor JG, Li QX, Laughton KM, Rowe CC, Masters CL, Cappai R, Villemagne VL. In vitro characterization of Pittsburgh compound-B binding to Lewy bodies. Journal of Neuroscience. 2007;27(39):10365–10371. doi: 10.1523/JNEUROSCI.0630-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodero-Tavoletti M, Cappai R, Krause S, Lippoldt A, Foster L, Leone L, Smith D, McLean C, Rowe CC, Dyrks T, Masters CL, Villemagne VL. In vitro characterization of PIB binding to α-synuclein. Alzheimer’s & Dementia. 2006;2:S333–S334. [Google Scholar]
- Formaglio M, Costes N, Seguin J, Tholance Y, Le Bars D, Roullet-Solignac I, et al. In vivo demonstration of amyloid burden in posterior cortical atrophy: A case series with PET and CSF findings. Journal of Neurology. 2011;258:1841–1851. doi: 10.1007/s00415-011-6030-0. [DOI] [PubMed] [Google Scholar]
- Forman MS, Farmer J, Johnson JK, Clark CM, Arnold SE, Coslett HB, et al. Frontotemporal dementia: Clinicopathological correlations. Annals of Neurology. 2006;59:952–962. doi: 10.1002/ana.20873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg A, Almkvist O, Engler H, Wall A, Langstrom B, Nordberg A. High PIB retention in Alzheimer’s disease is an early event with complex relationship with CSF biomarkers and functional parameters. Current Alzheimer Research. 2010;7:56–66. doi: 10.2174/156720510790274446. [DOI] [PubMed] [Google Scholar]
- Forsberg A, Engler H, Almkvist O, Blomquist G, Hagman G, Wall A, Ringheim A, Langstrom B, Nordberg A. PET imaging of amyloid deposition in patients with mild cognitive impairment. Neurobiology of Aging. 2007;29(10):1456–1465. doi: 10.1016/j.neurobiolaging.2007.03.029. [DOI] [PubMed] [Google Scholar]
- Foster ER, Campbell MC, Burack MA, Hartlein J, Flores HP, Cairns NJ, Hershey T, Perlmutter JS. Amyloid imaging of Lewy body-associated disorders. Movement Disorders. 2010;25(15):2516–2523. doi: 10.1002/mds.23393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster NL, Heidebrink JL, Clark CM, Jagust WJ, Arnold SE, Barbas NR, et al. FDG-PET improves accuracy in distinguishing frontotemporal dementia and Alzheimer’s disease. Brain. 2007;130:2616–2635. doi: 10.1093/brain/awm177. [DOI] [PubMed] [Google Scholar]
- Fotenos AF, Mintun MA, Snyder AZ, Morris JC, Buckner RL. Brain volume decline in aging: Evidence for a relation between socioeconomic status, preclinical Alzheimer disease, and reserve. Archives of Neurology. 2008;65(1):113–120. doi: 10.1001/archneurol.2007.27. [DOI] [PubMed] [Google Scholar]
- Friedland RP, Budinger TF, Ganz E, Yano Y, Mathis CA, Koss B, Ober BA, Huesman RH, Derenzo SE. Regional cerebral metabolic alterations in dementia of the Alzheimer type: Positron emission tomography with [18F]fluorodeoxy-glucose. Journal of Computer Assisted Tomography. 1983;7(4):590–598. doi: 10.1097/00004728-198308000-00003. [DOI] [PubMed] [Google Scholar]
- Frisoni GB, Lorenzi M, Caroli A, Kemppainen N, Någren K, Rinne JO. In vivo mapping of amyloid toxicity in Alzheimer disease. Neurology. 2009;72(17):1504–1511. doi: 10.1212/WNL.0b013e3181a2e896. [DOI] [PubMed] [Google Scholar]
- Furney SJ, Kronenberg D, Simmons A, Güntert A, Dobson RJ, Proitsi P, Wahlund LO, Kloszewska I, Mecocci P, Soininen H, Tsolaki M, Vellas B, Spenger C, Lovestone S. Combinatorial markers of mild cognitive impairment conversion to Alzheimer’s disease–cytokines and MRI measures together predict disease progression. J Alzheimers Dis. 2011;3(Suppl 26):395–405. doi: 10.3233/JAD-2011-0044. [DOI] [PubMed] [Google Scholar]
- Furst AJ, Rabinovici GD, Rostomian AH, Steed T, Alkalay A, Racine C, Miller BL, Jagust WJ. Cognition, glucose metabolism and amyloid burden in Alzheimer’s disease. Neurobiology of Aging. 2010;33(2):215–225. doi: 10.1016/j.neurobiolaging.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garibotto V, Borroni B, Kalbe E, Herholz K, Salmon E, Holtoff V, Sorbi S, Cappa SF, Padovani A, Fazio F, Perani D. Education and occupation as proxies for reserve in aMCI converters and AD: FDG-PET evidence. Neurology. 2008;71(17):1342–1349. doi: 10.1212/01.wnl.0000327670.62378.c0. [DOI] [PubMed] [Google Scholar]
- Gauthier S, Reisberg B, Zaudig M, Petersen RC, Ritchie K, Broich K, Belleville S, Brodaty H, Bennett D, Chertkow H, Cummings JL, de Leon M, Feldman H, Ganguli M, Hampel H, Scheltens P, Tierney MC, Whitehouse P, Winblad B. Mild cognitive impairment. Lancet. 2006;367:1262–1270. doi: 10.1016/S0140-6736(06)68542-5. [DOI] [PubMed] [Google Scholar]
- Gomez-Isla T, West HL, Rebeck GW, Harr SD, Growdon JH, Locascio JJ, Perls TT, Lipsitz LA, Hyman BT. Clinical and pathological correlates of apolipoprotein E epsilon 4 in Alzheimer’s disease. Annals of Neurology. 1996;39(1):62–70. doi: 10.1002/ana.410390110. [DOI] [PubMed] [Google Scholar]
- Gomperts SN, Rentz DM, Moran E, Becker JA, Locascio JJ, Klunk WE, Mathis CA, Elmaleh DR, Shoup T, Fischman AJ, Hyman BT, Growdon JH, Johnson KA. Imaging amyloid deposition in Lewy body diseases. Neurology. 2008;71(12):903–910. doi: 10.1212/01.wnl.0000326146.60732.d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76:1006–1014. doi: 10.1212/WNL.0b013e31821103e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorno-Tempini ML, Dronkers NF, Rankin KP, Ogar JM, Phengrasamy L, Rosen HJ, et al. Cognition and anatomy in three variants of primary progressive aphasia. Annals of Neurology. 2004;55:335–346. doi: 10.1002/ana.10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg SM, Grabowski T, Gurol ME, Skehan ME, Nandigam RN, Becker JA, Garcia-Alloza M, Prada C, Frosch MP, Rosand J, Viswanathan A, Smith EE, Johnson KA. Detection of isolated cerebrovascular beta-amyloid with Pittsburgh compound B. Annals of Neurology. 2008;64(5):587–591. doi: 10.1002/ana.21528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimmer T, Alexopoulos P, Tsolakidou A, Guo LH, Henriksen G, Yousefi BH, Förstl H, Sorg C, Kurz A, Drzezga A, Perneczky R. Cerebrospinal fluid BACE1 activity and brain amyloid load in Alzheimer’s disease. Scientific World Journal. 2012;2012:712048. doi: 10.1100/2012/712048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimmer T, Tholen S, Yousefi BH, Alexopoulos P, Förschler A, Förstl H, Henriksen G, Klunk WE, Mathis CA, Perneczky R, Sorg C, Kurz A, Drzezga A. Progression of cerebral amyloid load is associated with the apolipoprotein E ε4 genotype in Alzheimer’s disease. Biological Psychiatry. 2010;68(10):879–884. doi: 10.1016/j.biopsych.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimmer T, Riemenschneider M, Forstl H, Henriksen G, Klunk WE, Mathis CA, Shiga T, Wester HJ, Kurz A, Drzezga A. Beta amyloid in Alzheimer’s disease: Increased deposition in brain is reflected in reduced concentration in cerebrospinal fluid. Biological Psychiatry. 2009;(65):927–934. doi: 10.1016/j.biopsych.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundman M, Sencakova D, Jack CR, Jr, Petersen RC, Kim HT, Schultz A, Weiner MF, DeCarli C, DeKosky ST, van Dyck C, Thomas RG, Thal LJ Alzheimer’s Disease Cooperative Study. Brain MRI hippocampal volume and prediction of clinical status in a mild cognitive impairment trial. Journal of Molecular Neuroscience. 2002;19(1–2):23–27. doi: 10.1007/s12031-002-0006-6. [DOI] [PubMed] [Google Scholar]
- Handen B, Cohen AD, Channamalappa U, Bulova P, Cannon SA, Cohen WI, Mathis CA, Price JC, Klunk WE. Imaging brain amyloid in non-demented young adults with Down syndrome using Pittsburgh Compound-B. Alzheimer’s & Dementia. doi: 10.1016/j.jalz.2011.09.229. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends in Pharmacological Sciences. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- Hardy J, Duff K, Hardy KG, Perez-Tur J, Hutton M. Genetic dissection of Alzheimer’s disease and related dementias: Amyloid and its relationship to tau. Nature Neuroscience. 1998;1:355–358. doi: 10.1038/1565. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer’s disease: The amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Haroutunian V, Perl D, Purohit D, Marin D, Khan K, Lantz M, Davis K. Regional distribution of neuritic plaques in the nondemented elderly and subjects with very mild Alzheimer’s disease. Archives of Neurology. 1998;55:1185–1191. doi: 10.1001/archneur.55.9.1185. [DOI] [PubMed] [Google Scholar]
- Hedden T, Van Dijk KR, Becker JA, Mehta A, Sperling RA, Johnson KA, Buckner RL. Disruption of functional connectivity in clinically normal older adults harboring amyloid burden. Journal of Neuroscience. 2009;29(40):12686–12694. doi: 10.1523/JNEUROSCI.3189-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herholz K, Carter SF, Jones M. Positron emission tomography imaging in dementia. The British Journal of Radiology. 2007;80:S160–S167. doi: 10.1259/bjr/97295129. [DOI] [PubMed] [Google Scholar]
- Ikonomovic MD, Abrahamson EE, Price JC, Hamilton RL, Mathis CA, Paljug WR, Debnath ML, Cohen AD, Mizukami K, Dekosky ST, Lopez OL, Klunk WE. Early AD pathology in a [C-11]PiB-negative case: A PiB-amyloid imaging, biochemical, and immunohistochemical study. Acta Neuropathologica. 2012;123(3):433–47. doi: 10.1007/s00401-012-0943-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomovic MD, Klunk WE, Abrahamson EE, Mathis CA, Price JC, Tsopelas ND, Lopresti BJ, Ziolko S, Bi W, Paljug WR, Debnath ML, Hope CE, Isanski BA, Hamilton RL, DeKosky ST. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain. 2008;131:1630–1645. doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomovic MD, Abrahamson EE, Isanski BA, Wuu J, Mufson EJ, DeKosky ST. Superior frontal cortex cholinergic axon density in mild cognitive impairment and early Alzheimer disease. Archives of Neurology. 2007;64(9):1312–1317. doi: 10.1001/archneur.64.9.1312. [DOI] [PubMed] [Google Scholar]
- Jack CR, Jr, Albert MS, Knopman DS, McKhann GM, Sperling RA, Carrillo MC, Thies B, Phelps CH. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & Dementia. 2011;7(3):257–262. doi: 10.1016/j.jalz.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, Knopman DS, Shiung MM, Gunter JL, Boeve BF, Kemp BJ, Weiner M, Petersen RC the Alzheimer’s Disease Neuroimaging I. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer’s disease: Implications for sequence of pathological events in Alzheimer’s disease. Brain. 2009;132(Pt 5):1355–1365. doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Lowe VJ, Senjem ML, Weigand SD, Kemp BJ, Shiung MM, Knopman DS, Boeve BF, Klunk WE, Mathis CA, Petersen RC. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer’s disease and amnestic mild cognitive impairment. Brain. 2008;131:665–680. doi: 10.1093/brain/awm336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Petersen RC, Xu Y, O’Brien PC, Smith GE, Ivnik RJ, Boeve BF, Tangalos EG, Kokmen E. Rates of hippocampal atrophy correlate with change in clinical status in aging and AD. Neurology. 2000;55(4):484–489. doi: 10.1212/wnl.55.4.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Petersen RC, Xu YC, O’Brien PC, Smith GE, Ivnik RJ, Boeve BF, Waring SC, Tangalos EG, Kokmen E. Prediction of AD with MRI-based hippocampal volume in mild cognitive impairment. Neurology. 1999;52(7):1397–1403. doi: 10.1212/wnl.52.7.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust W, Reed B, Mungas D, Ellis W, Decarli C. What does fluorodeoxyglucose PET imaging add to a clinical diagnosis of dementia? Neurology. 2007;69:871–877. doi: 10.1212/01.wnl.0000269790.05105.16. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. Alzheimer disease and cerebrovascular pathology: An update. Journal of Neural Transmission. 2002;109:813–836. doi: 10.1007/s007020200068. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. Prevalence of Alzheimer lesions in Parkinson’s disease. Movement Disorders. 2003;18:1207–1208. doi: 10.1002/mds.10513. [DOI] [PubMed] [Google Scholar]
- Johansson A, Savitcheva I, Forsberg A, Engler H, Langstrom B, Nordberg A, Askmark H. [(11)C]-PIB imaging in patients with Parkinson’s disease: Preliminary results. Parkinsonism & Related Disorders. 2007;14(4):345–347. doi: 10.1016/j.parkreldis.2007.07.010. [DOI] [PubMed] [Google Scholar]
- Johnson AE, Jeppsson F, Sandell J, Wensbo D, Neelissen JA, Juréus A, Ström P, Norman H, Farde L, Svensson SP. AZD2184: A radioligand for sensitive detection of beta-amyloid deposits. Journal of Neurochemistry. 2009;108(5):1177–1186. doi: 10.1111/j.1471-4159.2008.05861.x. [DOI] [PubMed] [Google Scholar]
- Johnson KA, Gregas M, Becker JA, Kinnecom C, Salat DH, Moran EK, Smith EE, Rosand J, Rentz DM, Klunk WE, Mathis CA, Price JC, Dekosky ST, Fischman AJ, Greenberg SM. Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Annals of Neurology. 2007;62(3):229–234. doi: 10.1002/ana.21164. [DOI] [PubMed] [Google Scholar]
- Johnson J, Head E, Kim R, Starr A, Cotman C. Clinical and pathological evidence for a frontal variant of Alzheimer disease. Archives of Neurology. 1999;56:1233–1239. doi: 10.1001/archneur.56.10.1233. [DOI] [PubMed] [Google Scholar]
- Johnson JK, Diehl J, Mendez MF, Neuhaus J, Shapira JS, Forman M, et al. Frontotemporal lobar degeneration: Demographic characteristics of 353 patients. Arch Neurol. 2005;62:925–930. doi: 10.1001/archneur.62.6.925. [DOI] [PubMed] [Google Scholar]
- Juréus A, Swahn BM, Sandell J, Jeppsson F, Johnson AE, Johnström P, Neelissen JA, Sunnemark D, Farde L, Svensson SP. Characterization of AZD4694, a novel fluorinated Abeta plaque neuroimaging PET radioligand. Journal of Neurochemistry. 2010 doi: 10.1111/j.1471-4159.2010.06812.x. [DOI] [PubMed] [Google Scholar]
- Kadir A, Marutle A, Gonzalez D, Schöll M, Almkvist O, Mousavi M, Mustafiz T, Darreh-Shori T, Nennesmo I, Nordberg A. Positron emission tomography imaging and clinical progression in relation to molecular pathology in the first Pittsburgh Compound B positron emission tomography patient with Alzheimer’s disease. Brain. 2011;134(Pt 1):301–317. doi: 10.1093/brain/awq349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaitzakis ME, Walls AJ, Pearce RK, Gentleman SM. Striatal Aβ peptide deposition mirrors dementia and differentiates DLB and PDD from other parkinsonian syndromes. Neurobiology of Disease. 2011;41(2):377–384. doi: 10.1016/j.nbd.2010.10.005. [DOI] [PubMed] [Google Scholar]
- Kantarci K, Lowe V, Przybelski SA, Weigand SD, Senjem ML, Ivnik RJ, Preboske GM, Roberts R, Geda YE, Boeve BF, Knopman DS, Petersen RC, Jack CR., Jr APOE modifies the association between Aβ load and cognition in cognitively normal older adults. Neurology. 2012;78(4):232–240. doi: 10.1212/WNL.0b013e31824365ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarci K, Yang C, Schneider JA, Senjem ML, Reyes DA, Lowe VJ, Barnes LL, Aggarwal NT, Bennett DA, Smith GE, Petersen RC, Jack CR, Boeve BF. Ante mortem amyloid imaging and B-amyloid pathology in a case with dementia with Lewy bodies. Neurobiology of Aging. 2010 Oct 18; doi: 10.1016/j.neurobiolaging.2010.08.007. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. Journal of Neuroscience. 2001;21(2):372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemppainen NM, Aalto S, Wilson IA, Nagren K, Helin S, Bruck A, Oikonen V, Kailajarvi M, Scheinin M, Viitanen M, Parkkola R, Rinne JO. Voxel-based analysis of PET amyloid ligand [11C]PIB uptake in Alzheimer disease. Neurology. 2006;67:1575–1580. doi: 10.1212/01.wnl.0000240117.55680.0a. [DOI] [PubMed] [Google Scholar]
- Kemppainen NM, Aalto S, Wilson IA, Nagren K, Helin S, Bruck A, Oikonen V, Kailajarvi M, Scheinin M, Viitanen M, Parkkola R, Rinne JO. PET amyloid ligand [11C]PIB uptake is increased in mild cognitive impairment. Neurology. 2007;68:1603–1606. doi: 10.1212/01.wnl.0000260969.94695.56. [DOI] [PubMed] [Google Scholar]
- Khachaturian ZS. Diagnosis of Alzheimer’s disease. Archives of Neurology. 1985;42:1097–1105. doi: 10.1001/archneur.1985.04060100083029. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Wang Y, Huang GF, Debnath ML, Holt DP, Shao L, Hamilton RL, Ikonomovic MD, DeKosky ST, Mathis CA. The binding of 2-(4’-methyl-aminophenyl)benzothiazole to postmortem brain homogenates is dominated by the amyloid component. Journal of Neuroscience. 2003;23:2086–2092. doi: 10.1523/JNEUROSCI.23-06-02086.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Lopresti BJ, Ikonomovic MD, Lefterov IM, Koldamova RP, Abrahamson EE, Debnath ML, Holt DP, Huang GF, Shao L, DeKosky ST, Price JC, Mathis CA. Binding of the positron emission tomography tracer Pittsburgh compound-B reflects the amount of amyloid-beta in Alzheimer’s disease brain but not in transgenic mouse brain. Journal of Neuroscience. 2005;25:10598–10606. doi: 10.1523/JNEUROSCI.2990-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergström M, Savitcheva I, Huang GF, Estrada S, Ausén B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Långström B. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Annals of Neurology. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Price JC, Mathis CA, Tsopelas ND, Lopresti BJ, Ziolko SK, Bi W, Hoge JA, Cohen AD, Ikonomovic MD, Saxton JA, Snitz BE, Pollen DA, Moonis M, Lippa CF, Swearer JM, Johnson KA, Rentz DM, Fischman AJ, Aizenstein HJ, DeKosky ST. Amyloid deposition begins in the striatum of presenilin-1 mutation carriers from two unrelated pedigrees. Journal of Neuroscience. 2007;27:6174–6184. doi: 10.1523/JNEUROSCI.0730-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeppe RA. Basic principles and controversies in PET amyloid imaging. Miami, FL: Human Amyloid Imaging; 2012. [Google Scholar]
- Koeppe RA, Gilman S, Junck L, Wernette K, Frey KA. Differentiating Alzheimer’s disease from dementia with Lewy bodies and Parkinson’s disease with (+)-[11C]dihydrotetrabenazine positron emission tomography. Alzheimer’s & Dementia. 2008;4(1 Suppl 1):S67–S76. doi: 10.1016/j.jalz.2007.11.016. [DOI] [PubMed] [Google Scholar]
- Koivunen J, Scheinin N, Virta JR, Aalto S, Vahlberg T, Någren K, Helin S, Parkkola R, Viitanen M, Rinne JO. Amyloid PET imaging in patients with mild cognitive impairment: A 2-year follow-up study. Neurology. 2011;76(12):1085–1090. doi: 10.1212/WNL.0b013e318212015e. [DOI] [PubMed] [Google Scholar]
- Koivunen J, Pirttilä T, Kemppainen N, Aalto S, Herukka SK, Jauhianen AM, Hänninen T, Hallikainen M, Någren K, Rinne JO, Soininen H. PET amyloid ligand [11C]PIB uptake and cerebrospinal fluid beta-amyloid in mild cognitive impairment. Dementia and Geriatric Cognitive Disorders. 2008;26(4):378–383. doi: 10.1159/000163927. [DOI] [PubMed] [Google Scholar]
- Kok E, Haikonen S, Luoto T, Huhtala H, Goebeler S, Haapasalo H, Karhunen PJ. Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Annals of Neurology. 2009;65(6):650–657. doi: 10.1002/ana.21696. [DOI] [PubMed] [Google Scholar]
- Laforce R, Jr, Rabinovici GD. Amyloid imaging in the differential diagnosis of dementia: Review and potential clinical applications. Alzheimer’s Research & Therapy. 2011;3:31. doi: 10.1186/alzrt93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt J, D’Abrera JC, Holland AJ, Aigbirhio FI, Fryer TD, Canales R, Hong YT, Menon DK, Baron JC, Zaman SH. Using positron emission tomography and Carbon 11-labeled Pittsburgh compound B to image brain fibrillar β-amyloid in adults with down syndrome: Safety, acceptability, and feasibility. Archives of Neurology. 2011;68(7):890–896. doi: 10.1001/archneurol.2011.36. [DOI] [PubMed] [Google Scholar]
- Lee SE, Rabinovici GD, Mayo MC, Wilson SM, Seeley WW, DeArmond SJ, et al. Clinicopathological correlations in corticobasal degeneration. Annals of Neurology. 2011;70:327–340. doi: 10.1002/ana.22424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinonen V, Alafuzoff I, Aalto S, Suotunen T, Savolainen S, Nagren K, Tapiola T, Pirttila T, Rinne J, Jaaskelainen JE, Soininen H, Rinne JO. Assessment of beta-amyloid in a frontal cortical brain biopsy specimen and by positron emission tomography with carbon 11-labeled Pittsburgh compound B. Archives of Neurology. 2008;65(10):1304–1309. doi: 10.1001/archneur.65.10.noc80013. [DOI] [PubMed] [Google Scholar]
- Levine H. Soluble multimeric Alzheimer beta(1–40) pre-amyloid complexes in dilute solution. Neurobiology of Aging. 1995;16(5):755–764. doi: 10.1016/0197-4580(95)00052-g. [DOI] [PubMed] [Google Scholar]
- Leyton CE, Villemagne VL, Savage S, Pike KE, Ballard KJ, Piguet O, et al. Subtypes of progressive aphasia: Application of the international consensus criteria and validation using {beta}-amyloid imaging. Brain. 2011;134:3030–3043. doi: 10.1093/brain/awr216. [DOI] [PubMed] [Google Scholar]
- Li Y, Rinne J, Mosconi L, Pirraglia E, Rusinek H, DeSanti S, Kemppainen N, Någren K, Kim B-C, Tsui W, de Leon M. Regional analysis of FDG and PIB-PET images in normal aging, mild cognitive impairment, and Alzheimer’s disease. European Journal of Nuclear Medicine and Molecular Imaging. 2008;35:2169–2181. doi: 10.1007/s00259-008-0833-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Ashe KH, Frautschy SA, Cole GM. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s disease. Journal of Neuroscience. 2000;20(15):5709–5714. doi: 10.1523/JNEUROSCI.20-15-05709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KJ, Hsu WC, Hsiao IT, Wey SP, Jin LW, Skovronsky D, Wai YY, Chang HP, Lo CW, Yao CH, Yen TC, Kung MP. Whole-body biodistribution and brain PET imaging with [18F]AV-45, a novel amyloid imaging agent – a pilot study. Nuclear Medicine and Biology. 2010;37(4):497–508. doi: 10.1016/j.nucmedbio.2010.02.003. [DOI] [PubMed] [Google Scholar]
- Lister-James J, Pontecorvo MJ, Clark C, Joshi AD, Mintun MA, Zhang W, Lim N, Zhuang Z, Golding G, Choi SR, Benedum TE, Kennedy P, Hefti F, Carpenter AP, Kung HF, Skovronsky DM. Florbetapir f-18: A histopathologically validated Beta-amyloid positron emission tomography imaging agent. Seminars in Nuclear Medicine. 2011;41(4):300–304. doi: 10.1053/j.semnuclmed.2011.03.001. [DOI] [PubMed] [Google Scholar]
- Lockhart A, Lamb JR, Osredkar T, Sue LI, Joyce JN, Ye L, Libri V, Leppert D, Beach TG. PIB is a non-specific imaging marker of amyloid-beta (Abeta) peptide-related cerebral amyloidosis. Brain. 2007;130(Pt 10):2607–2615. doi: 10.1093/brain/awm191. [DOI] [PubMed] [Google Scholar]
- Logan J, Fowler JS, Volkow ND, Wang GJ, Ding YS, Alexoff DL. Distribution volume ratios without blood sampling from graphical analysis of PET data. Journal of Cerebral Blood Flow and Metabolism. 1996;16(5):834–840. doi: 10.1097/00004647-199609000-00008. [DOI] [PubMed] [Google Scholar]
- Lopresti BJ, Klunk WE, Mathis CA, Hoge JA, Ziolko SK, Lu X, Meltzer CC, Schimmel K, Tsopelas ND, Dekosky ST, Price JC. Simplified quantification of Pittsburgh compound B amyloid imaging PET Studies: A comparative analysis. Journal of Nuclear Medicine. 2005;46:1959–1972. [PubMed] [Google Scholar]
- Lorenzi M, Donohue M, Paternicò M, Scarpazza C, Ostrowitzki S, Blin O, Irving E, Frisoni GB The Alzheimer’s Disease Neuroimaging Initiative. Enrichment through biomarkers in clinical trials of Alzheimer’s drugs in patients with mild cognitive impairment. Neurobiology of Aging. 2010;31(8):1443–1451. doi: 10.1016/j.neurobiolaging.2010.04.036. [DOI] [PubMed] [Google Scholar]
- Lowe VJ, Kemp BJ, Jack CR, Jr, Senjem M, Weigand S, Shiung M, Smith G, Knopman D, Boeve B, Mullan B, Petersen RC. Comparison of 18F-FDG and PiB PET in Cognitive Impairment. Journal of Nuclear Medicine. 2009;50(6):878–886. doi: 10.2967/jnumed.108.058529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly JV, Donnan GA, Villemagne VL, Zavala JA, Ma H, O’Keefe G, Gong SJ, Gunawan RM, Saunder T, Ackerman U, Tochon-Danguy H, Churilov L, Phan TG, Rowe CC. 11C-PIB binding is increased in patients with cerebral amyloid angiopathy-related hemorrhage. Neurology. 2010;74(6):487–493. doi: 10.1212/WNL.0b013e3181cef7e3. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathologica. 2010;119:1–4. doi: 10.1007/s00401-009-0612-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maetzler W, Liepelt I, Reimold M, Reischl G, Solbach C, Becker C, Schulte C, Leyhe T, Keller S, Melms A, Gasser T, Berg D. Cortical PIB binding in Lewy body disease is associated with Alzheimer-like characteristics. Neurobiology of Disease. 2009;34:107–112. doi: 10.1016/j.nbd.2008.12.008. [DOI] [PubMed] [Google Scholar]
- Maetzler W, Reimold M, Liepelt I, Solbach C, Leyhe T, Schweitzer K, Eschweiler GW, Mittelbronn M, Gaenslen A, Uebele M, Reischl G, Gasser T, Machulla HJ, Bares R, Berg D. [11C]PIB binding in Parkinson’s disease dementia. Neuroimage. 2008;39:1027–1033. doi: 10.1016/j.neuroimage.2007.09.072. [DOI] [PubMed] [Google Scholar]
- Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: A causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(15):5644–5651. doi: 10.1073/pnas.0600549103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maia LF, Mackenzie IR, Feldman HH. Clinical phenotypes of cerebral amyloid angiopathy. Journal of the Neurological Sciences. 2007;257:23–30. doi: 10.1016/j.jns.2007.01.054. [DOI] [PubMed] [Google Scholar]
- Masliah E, Hansen L, Adame A, Crews L, Bard F, Lee C, Seubert P, Games D, Kirby L, Schenk D. Ab vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology. 2005;64:129–131. doi: 10.1212/01.WNL.0000148590.39911.DF. [DOI] [PubMed] [Google Scholar]
- Mastaglia FL, Johnsen RD, Byrnes ML, Kakulas BA. Prevalence of amyloid-beta deposition in the cerebral cortex in Parkinson’s disease. Movement Disorders. 2003;18:81–86. doi: 10.1002/mds.10295. [DOI] [PubMed] [Google Scholar]
- Mathis CA, Lopresti B, Mason N, Price J, Flatt N, Bi W, Ziolko S, DeKosky S, Klunk W. Comparison of the amyloid imaging agents [F-18]3’-F-PIB and [C-11] PIB in Alzheimer’s disease and control subjects. Indian Journal of Nuclear Medicine. 2007;48:56. [Google Scholar]
- Mathis CA, Wang Y, Holt DP, Huang GF, Debnath ML, Klunk WE. Synthesis and evaluation of 11C-labeled 6-substituted 2-arylbenzothiazoles as amyloid imaging agents. Journal of Medicinal Chemistry. 2003;46:2740–2754. doi: 10.1021/jm030026b. [DOI] [PubMed] [Google Scholar]
- Mattsson N, Zetterberg H, Hansson O, Andreasen N, Parnetti L, Jonsson M, Herukka SK, van der Flier WM, Blankenstein MA, Ewers M, Rich K, Kaiser E, Verbeek M, Tsolaki M, Mulugeta E, Rosén E, Aarsland D, Visser PJ, Schröder J, Marcusson J, de Leon M, Hampel H, Scheltens P, Pirttilä T, Wallin A, Jönhagen ME, Minthon L, Winblad B, Blennow K. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. The Journal of the American Medical Association. 2009;302(4):385–393. doi: 10.1001/jama.2009.1064. [DOI] [PubMed] [Google Scholar]
- McGeer EG, McGeer PL. Neuroinflammation in Alzheimer’s disease and mild cognitive impairment: A field in its infancy. J Alzheimers Dis. 2010;19(1):355–361. doi: 10.3233/JAD-2010-1219. [DOI] [PubMed] [Google Scholar]
- McKeith IG. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): Report of the Consortium on DLB International Workshop. Journal of Alzheimer’s Disease. 2006;9:417–423. doi: 10.3233/jad-2006-9s347. [DOI] [PubMed] [Google Scholar]
- McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, Salmon DP, Lowe J, Mirra SS, Byrne EJ, Lennox G, Quinn NP, Edwardson JA, Ince PG, Bergeron C, Burns A, Miller BL, Lovestone S, Collerton D, Jansen EN, Ballard C, de Vos RA, Wilcock GK, Jellinger KA, Perry RH. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): Report of the consortium on DLB international workshop. Neurology. 1996;47(5):1113–1124. doi: 10.1212/wnl.47.5.1113. [DOI] [PubMed] [Google Scholar]
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, Scheltens P, Carrillo MC, Thies B, Weintraub S, Phelps CH. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & Dementia. 2011;7(3):263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA work group under the auspices of the Department of Health and Human Services Task Force on Alzheimer’s disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- Mesulam M, Wicklund A, Johnson N, Rogalski E, Leger GC, Rademaker A, et al. Alzheimer and frontotemporal pathology in subsets of primary progressive aphasia. Annals of Neurology. 2008;63:709–719. doi: 10.1002/ana.21388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mevel K, Desgranges B, Baron JC, Landeau B, De la Sayette V, Viader F, Eustache F, Chételat G. Detecting hippocampal hypometabolism in mild cognitive impairment using automatic voxel-based approaches. Neuroimage. 2007;37(1):18–25. doi: 10.1016/j.neuroimage.2007.04.048. Epub 2007 May 8. [DOI] [PubMed] [Google Scholar]
- Mintun MA, Larossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, Klunk WE, Mathis CA, DeKosky ST, Morris JC. [11C]PIB in a nondemented population: Potential antecedent marker of Alzheimer disease. Neurology. 2006;67:446–452. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- Moretti DV, Miniussi C, Frisoni GB, Geroldi C, Zanetti O, Binetti G, Rossini PM. Hippocampal atrophy and EEG markers in subjects with mild cognitive impairment. Clinical Neurophysiology. 2007;118(12):2716–2729. doi: 10.1016/j.clinph.2007.09.059. [DOI] [PubMed] [Google Scholar]
- Mormino EC, Brandel MG, Madison CM, Rabinovici GD, Marks S, Baker SL, Jagust WJ. Not quite PIB-positive, not quite PIB-negative: Slight PIB elevations in elderly normal control subjects are biologically relevant. Neuroimage. 2011;59(3):2362–2373. doi: 10.1016/j.neuroimage.2011.07.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mormino EC, Kluth JT, Madison CM, Rabinovici GD, Baker SL, Miller BL, Koeppe RA, Mathis CA, Weiner MW, Jagust WJ. Episodic memory loss is related to hippocampal-mediated beta-amyloid deposition in elderly subjects. Brain. 2009;132:1310–1323. doi: 10.1093/brain/awn320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morra JH, Tu Z, Apostolova LG, Green AE, Avedissian C, Madsen SK, Parikshak N, Hua X, Toga AW, Jack CR, Jr, Schuff N, Weiner MW, Thompson PM Alzheimer’s Disease Neuroimaging Initiative. Automated 3D mapping of hippocampal atrophy and its clinical correlates in 400 subjects with Alzheimer’s disease, mild cognitive impairment, and elderly controls. Human Brain Mapping. 2009;30(9):2766–2788. doi: 10.1002/hbm.20708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JC, Roe CM, Xiong C, Fagan AM, Goate AM, Holtzman DM, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Annals of Neurology. 2010;67:122–131. doi: 10.1002/ana.21843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JC, Price AL. Pathologic correlates of nondemented aging, mild cognitive impairment, and early-stage Alzheimer’s disease. Journal of Molecular Neuroscience. 2001;17:101–118. doi: 10.1385/jmn:17:2:101. [DOI] [PubMed] [Google Scholar]
- Morris JC, Storandt M, McKeel DW, Jr, Rubin EH, Price JL, Grant EA, Berg L. Cerebral amyloid deposition and diffuse plaques in “normal” aging: Evidence for presymptomatic and very mild Alzheimer’s disease. Neurology. 1996;46:707–719. doi: 10.1212/wnl.46.3.707. [DOI] [PubMed] [Google Scholar]
- Mosconi L, Perani D, Sorbi S, Herholz K, Nacmias B, Holthoff V, Salmon E, Baron JC, De Cristofaro MT, Padovani A, Borroni B, Franceschi M, Bracco L, Pupi A. MCI conversion to dementia and the APOE genotype: A prediction study with FDG-PET. Neurology. 2004;63(12):2332–2340. doi: 10.1212/01.wnl.0000147469.18313.3b. [DOI] [PubMed] [Google Scholar]
- Mosconi L, De Santi S, Li Y, Li J, Zhan J, Tsui WH, Boppana M, Pupi A, de Leon MJ. Visual rating of medial temporal lobe metabolism in mild cognitive impairment and Alzheimer’s disease using FDG-PET. European Journal of Nuclear Medicine and Molecular Imaging. 2006;33(2):210–221. doi: 10.1007/s00259-005-1956-z. [DOI] [PubMed] [Google Scholar]
- Mosconi L, De Santi S, Li J, Tsui WH, Li Y, Boppana M, Laska E, Rusinek H, de Leon MJ. Hippocampal hypometabolism predicts cognitive decline from normal aging. Neurobiology of Aging. 2008;29(5):676–692. doi: 10.1016/j.neurobiolaging.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motter R, Vigo-Pelfrey C, Kholodenko D, Barbour R, Johnson-Wood K, Galasko D, Chang L, Miller B, Clark C, Green R, et al. Reduction of beta-amyloid peptide42 in the cerebrospinal fluid of patients with Alzheimer’s disease. Annals of Neurology. 1995;38:643–648. doi: 10.1002/ana.410380413. [DOI] [PubMed] [Google Scholar]
- Mueller SG, Weiner MW, Thal LJ, Petersen RC, Jack CR, Jagust W, Trojanowski JQ, Toga AW, Beckett L. Ways toward an early diagnosis in Alzheimer’s disease: The Alzheimer’s Disease Neuroimaging Initiative (ADNI) Alzheimer’s & Dementia. 2005a;1:55–66. doi: 10.1016/j.jalz.2005.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller SG, Weiner MW, Thal LJ, Petersen RC, Jack C, Jagust W, Trojanowski JQ, Toga AW, Beckett L. The Alzheimer’s disease neuroimaging initiative. Neuroimaging Clinics of North America. 2005b;15:869–877. doi: 10.1016/j.nic.2005.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelissen N, Van Laere K, Thurfjell L, Owenius R, Vandenbulcke M, Koole M, Bormans G, Brooks DJ, Vandenberghe R. Phase 1 study of the Pittsburgh compound B derivative 18F-flutemetamol in healthy volunteers and patients with probable Alzheimer disease. Journal of Nuclear Medicine. 2009;50(8):1251–1259. doi: 10.2967/jnumed.109.063305. [DOI] [PubMed] [Google Scholar]
- Nelissen N, Vandenbulcke M, Fannes K, Verbruggen A, Peeters R, Dupont P, Van Laere K, Bormans G, Vandenberghe R. Abeta amyloid deposition in the language system and how the brain responds. Brain. 2007;130:2055–2069. doi: 10.1093/brain/awm133. [DOI] [PubMed] [Google Scholar]
- Newell KL, Hyman BT, Growdon JH, Hedley-Whyte ET. Application of the National Institute on Aging (NIA)-Reagan Institute criteria for the neuropathological diagnosis of Alzheimer disease. Journal of Neuropathology and Experimental Neurology. 1999;58:1147–1155. doi: 10.1097/00005072-199911000-00004. [DOI] [PubMed] [Google Scholar]
- Ng H, Cheng Y, Poon W. Alzheimer-type of pathological changes in Chinese. Journal of the Neurological Sciences. 1997;97(1):97–103. doi: 10.1016/s0022-510x(96)00253-5. [DOI] [PubMed] [Google Scholar]
- Ng SY, Villemagne VL, Masters CL, Rowe CC. Evaluating atypical dementia syndromes using positron emission tomography with carbon 11 labeled Pittsburgh compound B. Archives of Neurology. 2007a;64:1140–1144. doi: 10.1001/archneur.64.8.1140. [DOI] [PubMed] [Google Scholar]
- Ng S, Villemagne VL, Berlangieri S, Lee ST, Cherk M, Gong SJ, Ackermann U, Saunder T, Tochon-Danguy H, Jones G, Smith C, O’Keefe G, Masters CL, Rowe CC. Visual assessment versus quantitative assessment of 11C-PIB PET and 18F-FDG PET for detection of Alzheimer’s disease. Journal of Nuclear Medicine. 2007b;48:547–552. doi: 10.2967/jnumed.106.037762. [DOI] [PubMed] [Google Scholar]
- NIA/Reagan_Workgroup. Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. The National Institute on Aging, and Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer’s disease. Neurobiology of Aging. 1997;18:S1–S2. [PubMed] [Google Scholar]
- Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-b peptide: A case report. Nature Medicine. 2003;9:448–452. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, Stenh C, Luthman J, Teplow DB, Younkin SG, Näslund J, Lannfelt L. The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Abeta protofibril formation. Nature Neuroscience. 2001;4(9):887–893. doi: 10.1038/nn0901-887. [DOI] [PubMed] [Google Scholar]
- Nitsch RM, Rebeck GW, Deng M, Richardson UI, Tennis M, Schenk DB, Vigo-Pelfrey C, Lieberburg I, Wurtman RJ, Hyman BT, et al. Cerebrospinal fluid levels of amyloid beta-protein in Alzheimer’s disease: Inverse correlation with severity of dementia and effect of apolipoprotein E genotype. Annals of Neurology. 1995;37:512–518. doi: 10.1002/ana.410370414. [DOI] [PubMed] [Google Scholar]
- Nordberg A, Schöll M, Wall A, Thordadottir S, Ferreira D, Bogdanovic N, Långström B, Almkvist A, Graff C. Arctic APP mutation carriers show low PiB PET retention in the presence of pathological CSF biomarkers and reduced FDG uptake. Neurology. 2012 doi: 10.1212/WNL.0b013e31825fdf18. in press. [DOI] [PubMed] [Google Scholar]
- Ohm TG, Kirca M, Bohl J, Scharnagl H, Gross W, Marz W. Apolipoprotein E polymorphism influences not only cerebral senile plaque load but also Alzheimer-type neurofibrillary tangle formation. Neuroscience. 1995;66:583–587. doi: 10.1016/0306-4522(94)00596-w. [DOI] [PubMed] [Google Scholar]
- Okello A, Edison P, Archer HA, Turkheimer FE, Kennedy J, Bullock R, Walker Z, Kennedy A, Fox N, Rossor M, Brooks DJ. Microglial activation and amyloid deposition in mild cognitive impairment: A PET study. Neurology. 2009;72(1):56–62. doi: 10.1212/01.wnl.0000338622.27876.0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okello A, Koivunen J, Edison P, Archer HA, Turkheimer FE, Nagren K, Bullock R, Walker Z, Kennedy A, Fox NC, Rossor MN, Rinne JO, Brooks DJ. Conversion of amyloid positive and negative MCI to AD over 3 years: An 11C-PIB PET study. Neurology. 2009;73:754–760. doi: 10.1212/WNL.0b013e3181b23564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowitzki S, Deptula D, Thurfjell L, Barkhof F, Bohrmann B, Brooks DJ, et al. Mechanism of amyloid removal in patients with Alzheimer disease treated with Gantenerumab. Archives of Neurology. 2012;69(2):198–207. doi: 10.1001/archneurol.2011.1538. [DOI] [PubMed] [Google Scholar]
- Patton RL, Kalback WM, Esh CL, Kokjohn TA, Van Vickle GD, Luehrs DC, Kuo YM, Lopez J, Brune D, Ferrer I, Masliah E, Newel AJ, Beach TG, Castano EM, Roher AE. Amyloid-beta peptide remnants in AN-1792-immunized Alzheimer’s disease patients: A biochemical analysis. The American Journal of Pathology. 2006;169:1048–1063. doi: 10.2353/ajpath.2006.060269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perneczky R, Hartmann J, Grimmer T, Drzezga A, Kurz A. Cerebral metabolic correlates of the clinical dementia rating scale in mild cognitive impairment. Journal of Geriatric Psychiatry and Neurology. 2007;20(2):84–88. doi: 10.1177/0891988706297093. [DOI] [PubMed] [Google Scholar]
- Pike KEGS, Villemagne VL, Ng S, Moss SA, Maruff P, Mathis CA, Klunk WE, Masters CL, Rowe CC. β-Amyloid imaging and memory in nondemented individuals: Evidence for preclinical Alzheimer’s disease. Brain. 2007 doi: 10.1093/brain/awm238. (in press) [DOI] [PubMed] [Google Scholar]
- Price DL, Sisodia SS. Mutant genes in familial Alzheimer’s disease and transgenic models. Annual Review of Neuroscience. 1998;21:479–505. doi: 10.1146/annurev.neuro.21.1.479. [DOI] [PubMed] [Google Scholar]
- Price JC, Klunk WE, Lopresti BJ, Lu X, Hoge JA, Ziolko SK, Holt DP, Meltzer CC, Dekosky ST, Mathis CA. Kinetic modeling of amyloid binding in humans using PET imaging and Pittsburgh Compound-B. Journal of Cerebral Blood Flow and Metabolism. 2005;25:1528–1547. doi: 10.1038/sj.jcbfm.9600146. [DOI] [PubMed] [Google Scholar]
- Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Annals of Neurology. 1999;45:358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Rabinovici GD, Miller BL. Frontotemporal lobar degeneration: Epidemiology, pathophysiology, diagnosis and management. CNS Drugs. 2010;24:375–398. doi: 10.2165/11533100-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovici GD, Rosen HJ, Alkalay A, Kornak J, Furst AJ, Agarwal N, et al. Amyloid vs FDG-PET in the differential diagnosis of AD and FTLD. Neurology. 2011;77:2034–2042. doi: 10.1212/WNL.0b013e31823b9c5e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovici GD, Furst AJ, Alkalay A, Racine CA, O’Neil JP, Janabi M, et al. Increased metabolic vulnerability in early-onset Alzheimer’s disease is not related to amyloid burden. Brain. 2010;133:512–528. doi: 10.1093/brain/awp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovici GD, Jagust WJ, Furst AJ, Ogar JM, Racine CA, Mormino EC, et al. Abeta amyloid and glucose metabolism in three variants of primary progressive aphasia. Annals of Neurology. 2008;64:388–401. doi: 10.1002/ana.21451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovici GD, Furst AJ, O’Neil JP, Racine CA, Mormino EC, Baker SL, Chetty S, Patel P, Pagliaro TA, Klunk WE, Mathis CA, Rosen HJ, Miller BL, Jagust WJ. 11C-PIB PET imaging in Alzheimer disease and frontotemporal lobar degeneration. Neurology. 2007;68:1205–1212. doi: 10.1212/01.wnl.0000259035.98480.ed. [DOI] [PubMed] [Google Scholar]
- Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134:2456–2477. doi: 10.1093/brain/awr179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnavalli E, Brayne C, Dawson K, Hodges JR. The prevalence of frontotemporal dementia. Neurology. 2002;58:1615–1621. doi: 10.1212/wnl.58.11.1615. [DOI] [PubMed] [Google Scholar]
- Reiman EM, Langbaum JB, Fleisher AS, Caselli RJ, Chen K, Ayutyanont N, et al. Alzheimer’s prevention initiative: A plan to accelerate the evaluation of presymptomatic treatments. Journal of Alzheimer’s Disease. 2011;3(Suppl 26):321–329. doi: 10.3233/JAD-2011-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Chen K, Liu X, Bandy D, Yu M, Lee W, Ayutyanont N, Keppler J, Reeder SA, Langbaum JBS, Alexander GE, Klunk WE, Mathis CA, Price JC, Aizenstein HJ, DeKosky ST, Caselli RJ. Fibrillar amyloid-β burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proc Natl Acad Sci USA. 2009;106(16):6820–6825. doi: 10.1073/pnas.0900345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Caselli RJ, Yun LS, Chen K, Bandy D, Minoshima S, Thibodeau SN, Osborne D. Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. New England Journal of Medicine. 1996;96:752–758. doi: 10.1056/NEJM199603213341202. [DOI] [PubMed] [Google Scholar]
- Remes AM, Finnila S, Mononen H, Tuominen H, Takalo R, Herva R, Majamaa K. Hereditary dementia with intracerebral hemorrhages and cerebral amyloid angiopathy. Neurology. 2004;63:234–240. doi: 10.1212/01.wnl.0000129988.68657.fa. [DOI] [PubMed] [Google Scholar]
- Remes AM, Laru L, Tuominen H, Aalto S, Kemppainen N, Mononen H, Någen K, Parkkola R, Rinne JO. 11C-PIB-PET amyloid imaging in patients with APP locus duplication. Archives of Neurology. 2007 doi: 10.1001/archneur.65.4.540. (in press) [DOI] [PubMed] [Google Scholar]
- Rentz DM, Locascio JJ, Becker JA, Moran EK, Eng E, Buckner RL, Sperling RA, Johnson KA. Cognition, reserve, and amyloid deposition in normal aging. Annals of Neurology. 2010;67:353–364. doi: 10.1002/ana.21904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnick SM, Sojkova J, Zhou Y, An Y, Ye W, Holt DP, Dannals RF, Mathis CA, Klunk WE, Ferrucci L, Kraut MA, Wong DF. Longitudinal cognitive decline is associated with fibrillar amyloid-beta measured by [11C]PiB. Neurology. 2010;74:807–815. doi: 10.1212/WNL.0b013e3181d3e3e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinne JO, Brooks DJ, Rossor MN, Fox NC, Bullock R, Klunk WE, et al. 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer’s disease treated with bapineuzumab: A phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurology. 2010;9:363–372. doi: 10.1016/S1474-4422(10)70043-0. [DOI] [PubMed] [Google Scholar]
- Roe CM, Mintun MA, D’Angelo G, Xiong C, Grant EA, Morris JC. Alzheimer disease and cognitive reserve: Variation of education effect with carbon 11-labeled Pittsburgh Compound B uptake. Archives of Neurology. 2008;65:1467–1471. doi: 10.1001/archneur.65.11.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosand J, Muzikansky A, Kumar A, Wisco JJ, Smith EE, Betensky RA, Greenberg SM. Spatial clustering of hemorrhages in probable cerebral amyloid angiopathy. Annals of Neurology. 2005;58:459–462. doi: 10.1002/ana.20596. [DOI] [PubMed] [Google Scholar]
- Rosen RF, Ciliax BJ, Wingo TS, Gearing M, Dooyema J, Lah JJ, et al. Deficient high-affinity binding of Pittsburgh compound B in a case of Alzheimer’s disease. Acta Neuropathologica. 2010;119:221–233. doi: 10.1007/s00401-009-0583-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom MH, Alkalay A, Agarwal N, Baker SL, O’Neil JP, Janabi M, et al. Distinct clinical and metabolic deficits in PCA and AD are not related to amyloid distribution. Neurology. 2011;76:1789–1796. doi: 10.1212/WNL.0b013e31821cccad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovelet-Lecrux A, Frebourg T, Tuominen H, Majamaa K, Campion D, Remes AM. APP locus duplication in a Finnish family with dementia and intracerebral haemorrhage. Journal of Neurology Neurosurgery and Psychiatry. 2007;78:1158–1159. doi: 10.1136/jnnp.2006.113514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe CC, Ellis KA, Rimajova M, Bourgeat P, Pike KE, Jones G, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiology of Aging. 2010;31:1275–1283. doi: 10.1016/j.neurobiolaging.2010.04.007. [DOI] [PubMed] [Google Scholar]
- Rowe CC, Ackerman U, Browne W, Mulligan R, Pike KL, O’Keefe G, et al. Imaging of amyloid β in Alzheimer’s disease with 18F-BAY94-9172, a novel PET tracer: Proof of mechanism. Lancet Neurology. 2008;7:129–135. doi: 10.1016/S1474-4422(08)70001-2. [DOI] [PubMed] [Google Scholar]
- Rowe CC, Ng S, Ackermann U, Gong SJ, Pike K, Savage G, Cowie TF, Dickinson KL, Maruff P, Darby D, Smith C, Woodward M, Merory J, Tochon-Danguy H, O’Keefe G, Klunk WE, Mathis CA, Price JC, Masters CL, Villemagne VL. Imaging beta-amyloid burden in aging and dementia. Neurology. 2007;68:1718–1725. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
- Sabbagh MN, Fleisher A, Chen K, Rogers J, Berk C, Reiman E, Pontecorvo M, Mintun M, Skovronsky D, Jacobson SA, Sue LI, Liebsack C, Charney AS, Cole L, Belden C, Beach TG. Positron emission tomography and neuropathologic estimates of fibrillar amyloid-β in a patient with Down syndrome and Alzheimer disease. Archives of Neurology. 2011;68(11):1461–1466. doi: 10.1001/archneurol.2011.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheinin NM, Aalto S, Koikkalainen J, Lötjönen J, Karrasch M, Kemppainen N, Viitanen M, Någren K, Helin S, Scheinin M, Rinne JO. Follow-up of [11C]PIB uptake and brain volume in patients with Alzheimer disease and controls. Neurology. 2009;73(15):1186–1192. doi: 10.1212/WNL.0b013e3181bacf1b. [DOI] [PubMed] [Google Scholar]
- Shimada H, Ataka S, Tomiyama T, Takechi H, Mori H, Miki T. Clinical course of patients with familial early-onset Alzheimer’s disease potentially lacking senile plaques bearing the E693γ mutation in amyloid precursor protein. Dementia and Geriatric Cognitive Disorders. 2011;32(1):45–54. doi: 10.1159/000330017. [DOI] [PubMed] [Google Scholar]
- Shin J, Kepe V, Barrio JR, Small GW. The merits of FDDNP-PET imaging in Alzheimer’s disease. Journal of Alzheimer’s Disease. 2011;3(Suppl 26):135–145. doi: 10.3233/JAD-2011-0008. [DOI] [PubMed] [Google Scholar]
- Shin J, Lee SY, Kim SH, Kim YB, Cho SJ. Multitracer PET imaging of amyloid plaques and neurofibrillary tangles in Alzheimer’s disease. Neuroimage. 2008;43(2):236–244. doi: 10.1016/j.neuroimage.2008.07.022. [DOI] [PubMed] [Google Scholar]
- Shoghi-Jadid K, Small GW, Agdeppa ED, Kepe V, Ercoli LM, Siddarth P, Read S, Satyamurthy N, Petric A, Huang SC, Barrio JR. Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease. The American Journal of Geriatric Psychiatry. 2002;10:24–35. [PubMed] [Google Scholar]
- Small GW, Siddarth P, Burggren AC, Kepe V, Ercoli LM, Miller KJ, Lavretsky H, Thompson PM, Cole GM, Huang SC, Phelps ME, Bookheimer SY, Barrio JR. Influence of cognitive status, age, and APOE-4 genetic risk on brain FDDNP positron-emission tomography imaging in persons without dementia. Arch Gen Psychiatry. 2009;66(1):81–87. doi: 10.1001/archgenpsychiatry.2008.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small GW, Kepe V, Ercoli LM, Siddarth P, Bookheimer SY, Miller KJ, Lavretsky H, Burggren AC, Cole GM, Vinters HV, Thompson PM, Huang SC, Satyamurthy N, Phelps ME, Barrio JR. PET of brain amyloid and tau in mild cognitive impairment. The New England Journal of Medicine. 2006;355:2652–2663. doi: 10.1056/NEJMoa054625. [DOI] [PubMed] [Google Scholar]
- Snider BJ, Fagan AM, Roe C, Shah AR, Grant EA, Xiong C, Morris JC, Holtzman DM. Cerebrospinal fluid biomarkers and rate of cognitive decline in very mild dementia of the Alzheimer type. Archives of Neurology. 2009;66(5):638–645. doi: 10.1001/archneurol.2009.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snowden JS, Stopford CL, Julien CL, Thompson JC, Davidson Y, Gibbons L, et al. Cognitive phenotypes in Alzheimer’s disease and genetic risk. Cortex. 2007;43:835–845. doi: 10.1016/s0010-9452(08)70683-x. [DOI] [PubMed] [Google Scholar]
- Sojkova J, Driscoll I, Iacono D, Zhou Y, Codispoti KE, Kraut MA, Ferrucci L, Pletnikova O, Mathis CA, Klunk WE, O’Brien RJ, Wong DF, Troncoso JC, Resnick SM. In vivo fibrillar beta-amyloid detected using [11C]PiB positron emission tomography and neuropathologic assessment in older adults. Archives of Neurology. 2011;68(2):232–240. doi: 10.1001/archneurol.2010.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR, Jr, Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison-Bogorad M, Wagster MV, Phelps CH. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association work-groups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & Dementia. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Laviolette PS, O’Keefe K, O’Brien J, Rentz DM, Pihlajamaki M, Marshall G, Hyman BT, Selkoe DJ, Hedden T, Buckner RL, Becker JA, Johnson KA. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron. 2009;63:178–188. doi: 10.1016/j.neuron.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storandt M, Head D, Fagan AM, Holtzman DM, Morris JC. Toward a multifactorial model of Alzheimer disease. Neurobiol Aging. 2012 doi: 10.1016/j.neurobiolaging.2011.11.029. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storandt M, Mintun MA, Head D, Morris JC. Cognitive decline and brain volume loss as signatures of cerebral amyloid-beta peptide deposition identified with Pittsburgh compound B: Cognitive decline associated with Abeta deposition. Archives of Neurology. 2009;66(12):1476–1481. doi: 10.1001/archneurol.2009.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strozyk D, Blennow K, White LR, Launer J. CSF A beta 42 levels correlate with amyloid neuropathology in a population-based autopsy study. Neurology. 2003;60:652–656. doi: 10.1212/01.wnl.0000046581.81650.d0. [DOI] [PubMed] [Google Scholar]
- Suenaga T, Hirano A, Llena JF, Yen SH, Dickson DW. Modified Biel-schowsky stain and immunohistochemical studies on striatal plaques in Alzheimer’s disease. Acta Neuropathologica. 1990;80:280–286. doi: 10.1007/BF00294646. [DOI] [PubMed] [Google Scholar]
- Suotunen T, Hirvonen J, Immonen-Raiha P, Aalto S, Lisinen I, Arponen E, Teras M, Koski K, Sulkava R, Seppanen M, Rinne JO. Visual assessment of [(11) C]PIB PET in patients with cognitive impairment. European Journal of Nuclear Medicine and Molecular Imaging. 2010;37(6):1141–1147. doi: 10.1007/s00259-010-1382-8. [DOI] [PubMed] [Google Scholar]
- Tang-Wai DF, Graff-Radford NR, Boeve BF, Dickson DW, Parisi JE, Crook R, et al. Clinical, genetic, and neuropathologic characteristics of posterior cortical atrophy. Neurology. 2004;63:1168–1174. doi: 10.1212/01.wnl.0000140289.18472.15. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Kovacs DM, Kim TW, Moir RD, Guenette SY, Wasco W. The gene defects responsible for familial Alzheimer’s disease. Neurobiology of Disease. 1996;3:159–168. doi: 10.1006/nbdi.1996.0016. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Annals of Neurology. 1991;30(4):572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Thal DR, Rub U, Orantes M, Braak H. Phases of Ab-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- Theuns J, Marjaux E, Vandenbulcke M, Van Laere K, Kumar-Singh S, Bormans G, Brouwers N, Van den Broeck M, Vennekens K, Corsmit E, Cruts M, De Strooper B, Van Broeckhoven C, Vandenberghe R. Alzheimer dementia caused by a novel mutation located in the APP C-terminal intracytosolic fragment. Human Mutation. 2006;27:888–896. doi: 10.1002/humu.20402. [DOI] [PubMed] [Google Scholar]
- Thompson PW, Ye L, Morgenstern JL, Sue L, Beach TG, Judd DJ, Shipley NJ, Libri V, Lockhart A. Interaction of the amyloid imaging tracer FDDNP with hallmark Alzheimer’s disease pathologies. Journal of Neurochemistry. 2009;109(2):623–630. doi: 10.1111/j.1471-4159.2009.05996.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolboom N, van der Flier WM, Yaqub M, Boellaard R, Verwey NA, Blankenstein MA, Windhorst AD, Scheltens P, Lammertsma AA, van Berckel BN. Relationship of cerebrospinal fluid markers to 11C-PiB and 18F-FDDNP binding. Journal of Nuclear Medicine. 2009;50(9):1464–1470. doi: 10.2967/jnumed.109.064360. [DOI] [PubMed] [Google Scholar]
- Tolboom N, Yaqub M, van der Flier WM, Boellaard R, Luurtsema G, Windhorst AD, Barkhof F, Scheltens P, Lammertsma AA, van Berckel BNM. Detection of Alzheimer pathology in vivo using both 11C-PIB and 18F-FDDNP PET. Journal of Nuclear Medicine. 2009;50(2):191–197. doi: 10.2967/jnumed.108.056499. [DOI] [PubMed] [Google Scholar]
- Tolboom N, Yaqub M, van der Flier W, Boellaard R, Luurtsema G, Windhorst B, Barkhof F, Scheltens P, Lammertsma A, van Berckel B. Imaging beta amyloid deposition in vivo: Quantitative comparison of [18F]FDDNP and [11C]PIB. Journal of Nuclear Medicine. 2007a;48:57. [Google Scholar]
- Tomiyama T, Nagata T, Shimada H, Teraoka R, Fukushima A, Kanemitsu H, Takuma H, Kuwano R, Imagawa M, Ataka S, Wada Y, Yoshioka E, Nishizaki T, Watanabe Y, Mori H. A new amyloid beta variant favoring oligomerization in Alzheimer’s-type dementia. Annals of Neurology. 2008;63(3):377–387. doi: 10.1002/ana.21321. [DOI] [PubMed] [Google Scholar]
- van de Pol LA, van der Flier WM, Korf ES, Fox NC, Barkhof F, Scheltens P. Baseline predictors of rates of hippocampal atrophy in mild cognitive impairment. Neurology. 2007;69(15):1491–1497. doi: 10.1212/01.wnl.0000277458.26846.96. [DOI] [PubMed] [Google Scholar]
- Vandenberghe R, Van Laere K, Ivanoiu A, Salmon E, Bastin C, Triau E, Hasselbalch S, Law I, Andersen A, Korner A, Minthon L, Garraux G, Nelissen N, Bormans G, Buckley C, Owenius R, Thurfjell L, Farrar G, Brooks DJ. 18F-flutemetamol amyloid imaging in Alzheimer disease and mild cognitive impairment: a phase 2 trial. Annals of Neurology. 2010;68(3):319–329. doi: 10.1002/ana.22068. [DOI] [PubMed] [Google Scholar]
- Venneti S, Lopresti BJ, Wang G, Hamilton RL, Mathis CA, Klunk WE, Apte UM, Wiley CA. PK11195 labels activated microglia in Alzheimer’s disease and in vivo in a mouse model using PET. Neurobiology of Aging. 2009;30(8):1217–1226. doi: 10.1016/j.neurobiolaging.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villemagne VL, Okamura N, Pejoska S, Drago J, Mulligan RS, Chetelat G, O’Keefe GG, Jones G, Kung HF, Pontecorvo M, Masters CL, Skovronsky DM, Rowe CC. Differential diagnosis in Alzheimer’s disease and dementia with Lewy bodies via VMAT2 and amyloid imaging. Neuro-Degenerative Diseases. 2012 Jan 17; doi: 10.1159/000334535. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Villemagne VL, Ong K, Mulligan RS, Holl G, Pejoska S, Jones G, O’Keefe G, Ackerman U, Tochon-Danguy H, Chan JG, Reininger CB, Fels L, Putz B, Rohde B, Masters CL, Rowe CC. Amyloid imaging with (18) F-florbetaben in Alzheimer disease and other dementias. Journal of Nuclear Medicine. 2011b;52:1210–1217. doi: 10.2967/jnumed.111.089730. [DOI] [PubMed] [Google Scholar]
- Villemagne VL, Pike KE, Chételat G, Ellis KA, Mulligan RS, Bourgeat P, Ackermann U, Jones G, Szoeke C, Salvado O, Martins R, O’Keefe G, Mathis CA, Klunk WE, Ames D, Masters CL, Rowe CC. Longitudinal assessment of Aβ and cognition in aging and Alzheimer disease. Annals of Neurology. 2011a;69(1):181–192. doi: 10.1002/ana.22248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villemagne VL, McLean CA, Reardon K, Boyd A, Lewis V, Klug G, Jones G, Baxendale D, Masters CL, Rowe CC, Collins SJ. 11C-PiB PET studies in typical sporadic Creutzfeldt-Jakob disease. Journal of Neurology Neurosurgery and Phychiatry. 2009;80(9):998–1001. doi: 10.1136/jnnp.2008.171496. [DOI] [PubMed] [Google Scholar]
- Villemagne VL, Pike KE, Darby D, Maruff P, Savage G, Ng S, Ackermann U, Cowie TF, Currie J, Chan SG, Jones G, Tochon-Danguy H, O’Keefe G, Masters CL, Rowe CC. Abeta deposits in older non-demented individuals with cognitive decline are indicative of preclinical Alzheimer’s disease. Neuropsychologia. 2008;46:1688–1697. doi: 10.1016/j.neuropsychologia.2008.02.008. [DOI] [PubMed] [Google Scholar]
- Vlassenko AG, Mintun MA, Xiong C, Sheline YI, Goate AM, Benzinger TL, Morris JC. Amyloid-beta plaque growth in cognitively normal adults: longitudinal [11C]Pittsburgh compound B data. Annals of Neurology. 2011;70(5):857–861. doi: 10.1002/ana.22608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Golob E, Bert A, Nie K, Chu Y, Dick MB, Mandelkern M, Su MY. Alterations in regional brain volume and individual MRI-guided perfusion in normal control, stable mild cognitive impairment, and MCI-AD converter. Journal of Geriatric Psychiatry and Neurology. 2009;22(1):35–45. doi: 10.1177/0891988708328212. [DOI] [PubMed] [Google Scholar]
- Whitaker C, Eckman C, Almeida C, Feinstein D, Atwood C, Eckman E, Crutcher K, Hersh L, Leissring M, LaVoie M, Ertekin-Taner N, Shapiro P, Takahashi R, Yamin R, Mansourian S, Estus S, Lesne S, Turner T, Farris W, Stroebel G. Live discussion: Amyloid-beta degradation: The forgotten half of Alzheimer’s disease. 12 September 2002. Journal of Alzheimer’s Disease. 2003;5:491–497. [PubMed] [Google Scholar]
- Wilcox KC, Lacor PN, Pitt J, Klein WL. Aβ oligomer-induced synapse degeneration in Alzheimer’s disease. Cellular and Molecular Neurobiology. 2011;31(6):939–948. doi: 10.1007/s10571-011-9691-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf DS, Gearing M, Snowdon DA, Mori H, Markesbery WR, Mirra SS. Progression of regional neuropathology in Alzheimer disease and normal elderly: Findings from the Nun study. Alzheimer Disease & Associated Disorders. 1999;13:226–231. doi: 10.1097/00002093-199910000-00009. [DOI] [PubMed] [Google Scholar]
- Wolk DA, Grachev ID, Buckley C, Kazi H, Grady MS, Trojanowski JQ, Hamilton RH, Sherwin P, McLain R, Arnold SE. Association between in vivo fluorine 18-labeled flutemetamol amyloid positron emission tomography imaging and in vivo cerebral cortical histopathology. Archives of Neurology. 2011;68(11):1398–1403. doi: 10.1001/archneurol.2011.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolk DA, Price JC, Saxton JA, Snitz BE, James JA, Lopez OL, Aizenstein HJ, Cohen AD, Weissfeld LA, Mathis CA, Klunk WE, De-Kosky ST. Amyloid imaging in mild cognitive impairment subtypes. Archives of Neurology. 2009;65(5):557–568. doi: 10.1002/ana.21598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong DF, Rosenberg PB, Zhou Y, Kumar A, Raymont V, Ravert HT, Dannals RF, Nandi A, Brasić JR, Ye W, Hilton J, Lyketsos C, Kung HF, Joshi AD, Skovronsky DM, Pontecorvo MJ. In vivo imaging of amyloid deposition in Alzheimer disease using the radioligand 18F-AV-45 (florbetapir [corrected] F 18) Journal of Nuclear Medicine. 2010;51(6):913–920. doi: 10.2967/jnumed.109.069088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia W, Ostaszewski BL, Kimberly WT, Rahmati T, Moore CL, Wolfe MS, Selkoe DJ. FAD mutations in presenilin-1 or amyloid precursor protein decrease the efficacy of a gamma-secretase inhibitor: Evidence for direct involvement of PS1 in the gamma-secretase cleavage complex. Neurobiology of Disease. 2000;7:673–681. doi: 10.1006/nbdi.2000.0322. [DOI] [PubMed] [Google Scholar]
- Younkin SG. The APP and PS1/2 mutations linked to early onset familial Alzheimer’s disease increase the extracellular concentration and A beta 1–42(43) Rinshō Shinkeigaku = Clinical Neurology. 1997;37:1099. [PubMed] [Google Scholar]
- Yuan Y, Gu ZX, Wei WS. Fluorodeoxyglucose-positron-emission tomography, single-photon emission tomography, and structural MR imaging for prediction of rapid conversion to Alzheimer disease in patients with mild cognitive impairment: A meta-analysis. AJNR American Journal of Neuroradiology. 2009;30(2):404–410. doi: 10.3174/ajnr.A1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziolko SK, Weissfeld LA, Klunk WE, Mathis CA, Hoge JA, Lopresti BJ, Dekosky ST, Price JC. Evaluation of voxel-based methods for the statistical analysis of PIB PET amyloid imaging studies in Alzheimer’s disease. Neuroimage. 2006;33:94–102. doi: 10.1016/j.neuroimage.2006.05.063. [DOI] [PubMed] [Google Scholar]