

Background: PP2A regulates cardiac excitability and physiology.
Results: PP2A regulation in heart occurs through integrative transcriptional, translational, and post-translational control of three classes of subunits (17 genes) to control holoenzyme synthesis, localization, and maintenance; pathways are mechanistically altered in heart disease.
Conclusion: Multiple mechanisms are present for acute and chronic regulation of specific PP2A populations.
Significance: Results provide molecular insight into cardiac PP2A regulation.
Keywords: Cardiovascular Disease, Cell Biology, Cell Compartmentation, Physiology, Trafficking, Ankyrin, Spectrin
Abstract
Kinase/phosphatase balance governs cardiac excitability in health and disease. Although detailed mechanisms for cardiac kinase regulation are established, far less is known regarding cardiac protein phosphatase 2A (PP2A) regulation. This is largely due to the complexity of the PP2A holoenzyme structure (combinatorial assembly of three subunit enzyme from >17 subunit genes) and the inability to segregate “global” PP2A function from the activities of multiple “local” holoenzyme populations. Here we report that PP2A catalytic, regulatory, and scaffolding subunits are tightly regulated at transcriptional, translational, and post-translational levels to tune myocyte function at base line and in disease. We show that past global read-outs of cellular PP2A activity more appropriately represent the collective activity of numerous individual PP2A holoenzymes, each displaying a specific subcellular localization (dictated by select PP2A regulatory subunits) as well as local specific post-translational catalytic subunit methylation and phosphorylation events that regulate local and rapid holoenzyme assembly/disassembly (via leucine carboxymethyltransferase 1/phosphatase methylesterase 1 (LCMT-1/PME-1). We report that PP2A subunits are selectively regulated between human and animal models, across cardiac chambers, and even within specific cardiac cell types. Moreover, this regulation can be rapidly tuned in response to cellular activation. Finally, we report that global PP2A is altered in human and experimental models of heart disease, yet each pathology displays its own distinct molecular signature though specific PP2A subunit modulatory events. These new data provide an initial view into the signaling pathways that govern PP2A function in heart but also establish the first step in defining specific PP2A regulatory targets in health and disease.
Introduction
In metazoans, the delicate balance of protein phosphorylation is tightly synchronized by the competing activities of protein kinases and phosphatases. Dysregulation of protein phosphorylation has been linked to mechanical dysfunction and arrhythmias in a host of cardiovascular diseases including atrial fibrillation, sinus node disease, heart failure, and myocardial infarction (1–6). Consequently, great emphasis has been placed on defining levels and activity of protein kinases in cardiovascular disease. Furthermore, pharmacological inhibitors of kinase activity have significantly enhanced our ability to treat cardiovascular disease phenotypes (7–9). Notably, the success of β-adrenergic receptor blockers in the treatment of arrhythmia, hypertension, and heart failure are attributed to the ability to dampen several kinase pathways involved in disease. Nonetheless, kinase activity represents only one facet of the system responsible for regulating protein phosphorylation levels.
Protein phosphatase 2A (PP2A)2 is a serine/threonine phosphatase found across metazoan cell types. The PP2A holoenzyme is composed of three subunits that facilitate PP2A scaffolding (A subunit), regulation (B subunit), and catalytic activity (C subunit). Thus, potential diversity of PP2A modulation of cellular function arises from specific combinatorial holoenzyme products (2 A subunit genes, 13 B subunit genes, 2 C subunit genes) (10). PP2A function is critical for the regulation of a host of targets in both excitable and non-excitable cells ranging from ion channels and transporters, regulatory enzymes, transcription factors, and cytoskeletal proteins (11–13). Moreover, the physiological importance of PP2A is clearly illustrated by its role in human disease. For example, PP2A dysfunction is linked with numerous forms of oncogenic regulation and Alzheimer disease (14, 15) and the design of PP2A-based inhibitors is a major area of research in cancer chemotherapies. In myocytes, PP2A activity is linked with multiple targets important in membrane excitability and excitation-contraction coupling including the ryanodine receptor (RyR2), connexin43, Cav1.2, troponin, Na+/Ca2+ exchanger (NCX), and phospholamban (16–22). Although manipulation of PP2A activity or expression in animal or cell models produces defects in myocyte physiology and cardiac phenotypes (19, 20, 23–26), the role and regulation of this critical enzyme family in cardiovascular disease is still largely elusive. In fact, we lack even a basic fundamental understanding of the expression, activity, and regulation of protein phosphatases in heart and/or in cardiovascular disease.
Recently, work from our group and others has provided functional data that link dysfunction in local PP2A subunit targeting and activity with highly penetrant and potentially fatal human cardiac arrhythmia (27–35). These clinical and translational findings have raised fundamental questions regarding the scope and regulation of PP2A subunit in human heart and in cardiovascular disease. Here we identify the mechanisms underlying PP2A holoenzyme regulation in heart and in cardiovascular disease. We provide new data on the expression of all 17 PP2A genes in human heart and demonstrate a surprising diversity in the expression and distribution across multiple species and in different cardiac chambers. Moreover, we find that all components of the PP2A holoenzyme (scaffolding, catalytic, and regulatory subunits) are subject to a non-uniform and model-specific pattern of remodeling in animal models of cardiovascular disease and in human heart failure. Mechanistically, we identify the PP2A regulatory subunits as critical for conferring PP2A target specificity in vivo by targeting the holoenzyme to specific subcellular domains, suggesting an important disconnect between assays of global phosphatase activity and local control of cardiomyocyte function. Finally, we demonstrate that cardiac PP2A subunits are subject to multiple post-translational modifications that have functional consequences for assembly of the holoenzyme in human heart failure. Collectively, our findings define an unexpectedly expansive network for regulation of local PP2A enzyme activity afforded by 1) expression of large number of regulatory subunits (thirteen) with distinct and well defined subcellular localization patterns, 2) differential regulation of all holoenzyme components across heart regions, species, and disease models, and 3) multiple post-translational modifications of the catalytic subunit (controlled by expression levels of upstream regulatory enzymes PME-1 and LCMT-1) that alter assembly/disassembly of specific PP2A holoenzyme populations in disease. Together these results highlight the importance of evaluating PP2A global function in the context of complexly regulated local signaling domains.
EXPERIMENTAL PROCEDURES
Tissue Preparation
Cardiac tissue from human, canine, and murine hearts was flash-frozen with liquid N2 and ground into a fine powder using a chilled mortar and pestle. The resulting powder was then resuspended in homogenization buffer (1 mm NaHCO3, 5 mm EDTA, 1 mm EGTA, 2 mm Na3VO4, 1 mm NaF, 1 mm PMSF, and protease inhibitor mixture (Sigma)) and further homogenized using a chilled Dounce homogenizer. Samples were flash-frozen in liquid N2 and stored at −80 °C for immunoblots.
Immunoblots
After quantification tissue lysates were analyzed on Mini-PROTEAN tetra cell (Bio-Rad) on a 4–15% precast TGX gel (Bio-Rad) in Tris/glycine/SDS buffer (Bio-Rad). For each lysate, 15 μl of sample (volumes were normalized for varying protein concentrations) was loaded with 15 μl of a 20:1 mixture of Laemmle sample buffer (Bio-Rad) and β-mercaptoethanol. Samples were boiled at 100 °C for 5 min before loading. Gels were transferred to a nitrocellulose membrane using the Mini-PROTEAN tetra cell (Bio-Rad) in Tris/glycine buffer with 10% methanol (v/v, Bio-Rad). Membranes were blocked for 1 h at room temperature using a 3% BSA solution and incubated with primary antibody overnight at 4 °C. Antibodies included: PP2A-C subunit (1:500, Millipore 05-421), PPP2R5E (1:1000, Sigma HPA006034), PP2A subunit B isoform PR55α (1:500, Sigma SAB4200241), PP2A subunit B isoform B56δ (Sigma SAB4200255), PPP2R5A (1:500, Abcam ab72028) or PP2A-B56α (1:200, Santa Cruz sc-136045), PPP2R5C (1:500 Abcam ab94633), PP2Aα phosphor-Tyr-307 (1:500 Abcam, ab32104), PP2A methyl Leu-309 (1:500, Abcam ab66597), PPP2R5B (Abcam ab1366), anti-PPP2R3A (Sigma HPA035829), PP2A/A (Calbiochem 539509). Secondary antibodies included donkey anti-mouse-HRP, donkey-anti-rabbit-HRP, and donkey-anti-goat-HRP (Jackson ImmunoResearch Laboratories). Densitometry was performed using Adobe Photoshop software, and all data were normalized to GAPDH levels present in each sample.
Co-immunoprecipitation Experiments
Canine samples were flash-frozen in liquid nitrogen and ground into a fine powder. Samples were resuspended in homogenization buffer (0.32 m sucrose, 2.5 mm EGTA, 5 mm EDTA, 50 mm Tris, and 10 mm NaCl, pH 7.47) and further homogenized by mechanical agitation with a Dounce homogenizer. Triton-100 (1% final) was added to each sample, and the samples were sonicated. Lysates were centrifuged for 15 min at 3000 × g. The supernatant was incubated with 40 μl of TrueBlot anti-rabbit Ig beads (eBioscience) for 60 min at 4 °C. Samples were again centrifuged at 3000 × g for 2 min, and 200 μl of sample was loaded into each of two tubes (two tubes per sample). One tube was incubated with 4 μl of anti-PP2A B55α Ig (Calbiochem 539509) at 4 °C for 1 h followed by the addition of 40 μl of anti-rabbit Ig beads overnight at 4 °C. The second tube containing 200 μl of sample lysate was incubated with TrueBlot anti-rabbit IgG beads overnight at 4 °C. 20 and 10 μl of each sample were set aside to be used as a 10 and 5% input loading control. After overnight incubation with anti-rabbit IgG beads, the supernatant was removed from the beads, and the beads were washed 3 times with wash buffer (1× PBS, 0.1% Triton, and 150 mm NaCl) before immunoblotting with PP2A/A antibody.
Immunofluorescence
Cardiomyocytes were isolated, cultured, and processed for immunofluorescence as described previously (35–37). For neonatal cardiomyocytes, staining experiments were performed on adherent cells. For adult cells, staining experiments were performed in solution. Secondary antibodies included anti-rabbit and anti-mouse Igs conjugated to AlexaFluor488 or -568 (Invitrogen). After secondary antibody treatment, cells were extensively washed and covered with Vectashield imaging medium (Vector Laboratories), and coverslips (#1) were applied. Images were collected on a confocal microscope (510 Meta; Carl Zeiss, Inc.) with a 63× oil 1.40 NA or 40× oil 1.30 NA lens (pinhole equals 1.0 airy disc; Carl Zeiss, Inc.) using imaging software (release Version 4.0 SP1; Carl Zeiss, Inc.). Images were collected using similar confocal protocols at room temperature. Images were imported into Photoshop CS (Adobe) for cropping and linear contrast adjustment.
Statistics
p values were determined with a paired Student's t test (2-tailed) or analysis of variance when appropriate for continuous data. The Bonferroni test was used for post-hoc testing. The null hypothesis was rejected for p < 0.05. Values are expressed as the mean ± S.E.
Cardiomyocyte Preparations
Neonatal and adult mouse cardiomyocytes were prepared as previously described (35, 38). Murine hearts from wild type and ankyrin-B-deficient C57Bl/6 mice were obtained after animals were euthanized by acute CO2 asphyxiation followed by cervical dislocation in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health and Institutional Animal Care and Use Committees-approved protocols.
siRNA
siRNA recognizing PP2A/A, PP2A/C, PPP2R3A, PPP2R4, PPP2R5C, and PPP2R5E (siGENOME, Dharmacon) were individually transfected into neonatal cardiomyocytes using Effectene (39).
mRNA Analysis
PCR reactions were done in a volume of 20 μl using Platinum Taq Polymerase High Fidelity. A touchdown PCR protocol was used for each primer set to reduce any nonspecific sequence amplification. Amplification primers included: PP2A/C_5′ GTTCAGCAACGAGCTGGACCAGTG; PP2A/C_3′ CCACCACGGTCATCTGGATCTGACCA; PP2A/Aα_5′ ACCTCTCAGCTGACTGTCGGGAGAATGTGATCATGTCCC; PP2A/Aα_3′ CTCCGGACTGGCCAAGACCTTGGGGATGATTGTGGA; PP2A/Aβ_5′ CGATCGCGGTTTTAATCGACGAGCTCCGCAATGAAGACGTGC; PP2A/Aβ_3′ ATACTGACACAAGCTTCCACAGCAAGGAGGGCGCACTGAATCC; PPP2R2A_5′ GCTGGAGGAGGGAATGATATTCAGTGGTGTTTTTCTCAGGT; PPP2R2A_3′ GTAGGATCTCTATACCTTCCATCCTCCTCTTTCAAGTTAT; PPP2R2B_5′ GATCCTGCCACCATCACAACCCTGCGGGTGCCT; PPP2R2B_3′ GTCACACAGCCGGATTGTCCCTTTGCTGCGGC; PPP2R2C_5′ CDCCDCCCACTCACTCCTGTCCACCAACGAT; PPP2R2C_3′ GGACGGCTTGATGTCCACGATGTTGAAGCTCCTGTCGG; PPP2R2D_5′ TGGTGCTTCTCGCAGGTCAGGGGGGCCATCGACGA; PPP2R2D_3′ GACCCGTAGCGCCGTGATCCTAAATGGGTCTCGAAGTC; PPP2R5A_5′ GAGTATGTTTCAACTAATCGTGGTGTAATTGTTGAATCAGCG; PPP2R5A_3′ TCCCATAAATTCGGTGCAGAACAGTCTTCAGG; PPP2R5B_5′ ATGGAGACGAAGCTGCCCCCTGCAAGCACCCCCACTAGCCCCTCCTCC; PPP2R5B_3′ GACGGGCTCGATGAGGACACCCCGGGTGCTCCCCACACT; PPP2R5C_5′ CAGTGACAACGCAGCGAAGATTCTGCCCATCAT; PPP2R5C_3′ CTAGCGGCCGTCCTGGGAGGCCAGCTCATCGGCCC; PPP2R5D_5′ GGCCCGGCTTAATCCCCAGTATCCCATGTTCCGAGCCCCTCC; PPP2R5D_3′ TCAGAGAGCCTCCTGGCTGGCAGTTAGGAACTCTTCCGCCCG; PPP2R5E_5′ CCGGCTATTGTGGCGTTGGTGTACAATGTGTTGAAGGC; PPP2R5E_3′ TAAAGTTGGAATTATTCCATCACGTCTACGTCTAAGACCTCTCTTTAA;PPP2R3A_5′ GGATGTGGTGGATACCCACCCTGGTCTCACGTTCCT;PPP2R3A_3′ CTCATACATGGAGAGTACACCGTCTCCATCCACATCCAT; PPP2R3B_5′ ATGCCGCCCGGCAAAGTGCTGCAGCCGGTCCTG; PPP2R3B_3′ CGTTCGTGGGGCTGGAGGCGGCGCCAAGGG; PPP2R3C_5′ TCGTCGGCGCCTAGCGACGCCCAACACCTG; PPP2R3C_3′ ATCGCTTCCTCTCCAATCATAGGTGGTGTCTGGTGTTTGTCCAGC; PPP2R4_5′ GCTGAGGGCGAGCGGCAGCCGCCGCCA; PPP2R4_3′ GCCAGATGGGTAGGGACCACTGTGGCCACC.
H202 Treatment of Cardiomyocytes and Fibroblasts
Neonatal murine fibroblasts and cardiomyocytes were separated by differential adherence during the isolation procedure, and purified populations were confirmed by α-actinin staining. After 24 h of culture at 37 °C and 5% CO2 in either defined growth media (DMEM/F-10 supplemented with insulin (1 μg/ml), transferrin (5 μg/ml), LiCl (1 nm), NaSeO4 (1 nm), ascorbic acid (25 μg/ml), thyroxine (1 nm)) for the cardiomyocytes or complete medium (DMEM/Ham's F-10, 10% FBS, and 10% HS) for fibroblasts, 75 μm H2O2 was applied to the cells for 60 min. After H2O2 treatment, cells were lysed in Laemmle buffer.
Human Tissue Samples
Left ventricular (LV) tissue was obtained from explanted hearts of patients undergoing heart transplantation through The Cooperative Human Tissue Network, Midwestern Division at Ohio State University. Institutional approval for use of human subjects was obtained from the Institutional Review Board of Ohio State University. LV tissue from healthy donor hearts not suitable for transplantation was obtained through the Iowa Donors Network and the National Disease Research Interchange. The investigation conforms to the principles outlined in the Declaration of Helsinki. Age and sex were the only identifying information acquired from tissue providers.
Institutional Approval for Animals
All animal studies in this study were approved by the appropriate (Ohio State University/Columbia University) institutional review board (Institutional Animal Care and Use Committees).
Canine Ischemic and Non-ischemic Heart Failure Models
Myocardial infarction was produced in healthy mongrel dogs by total coronary artery occlusion, as described previously (38). A cardiectomy was performed 5 days after surgery. Thin tissue slices from visible epicardial border zone and from a remote area away from the infarct (LV base) were flash-frozen for analysis. HF via chronic tachypacing was induced as described (40, 41). Cardiac tissues were snap-frozen in liquid nitrogen and stored at −80 °C until used. Control dogs were sacrificed in parallel. All animals were used according to approved Institutional Animal Care and Use Committees protocols (Columbia University and Ohio State University).
RESULTS
Defining PP2A Family Subunits in Human Heart
The human genome encodes at least 17 different PP2A subunits (Fig. 1, A and B; 2 A subunits, 13 B subunits, 2 C subunits). However, despite intense study of PP2A function in metazoans, surprisingly little is known regarding the differential expression or activity of PP2A subunits across tissues or cell types. In fact, there are no studies that characterize the detailed expression and/or regulation of the PP2A subunit family in human heart. We identified mRNA for PP2A scaffolding subunit (A subunits α and β) in human LV (Fig. 1C). Additionally, the message for the PP2A catalytic subunit was identified (Fig. 1C). Notably, we identified mRNA expression of 12 of the 13 PP2A regulatory subunit genes present in the human genome (Fig. 1C). Catalytic subunit (PPP2AC), two scaffolding subunits (PPP2R1A, PPP2R1B), and multiple regulatory subunits (PPP2R2A, PPP2R2B, PPP2R5A, PPP2R5B, PPP2R5C, PPP2R5D, PPP2R5E, PPP2R3A, PPP2R3B, PPP2R3C, and PPP2R4) were detected in human heart. The message for PPP2R2C, not present in heart, was observed in human brain (Fig. 1C). All appropriate controls for PP2A subunit mRNAs were negative. Together these data suggest that the human heart has the potential to assemble a combinatorial library of PP2A holoenzymes with potentially unique targets/functions.
FIGURE 1.
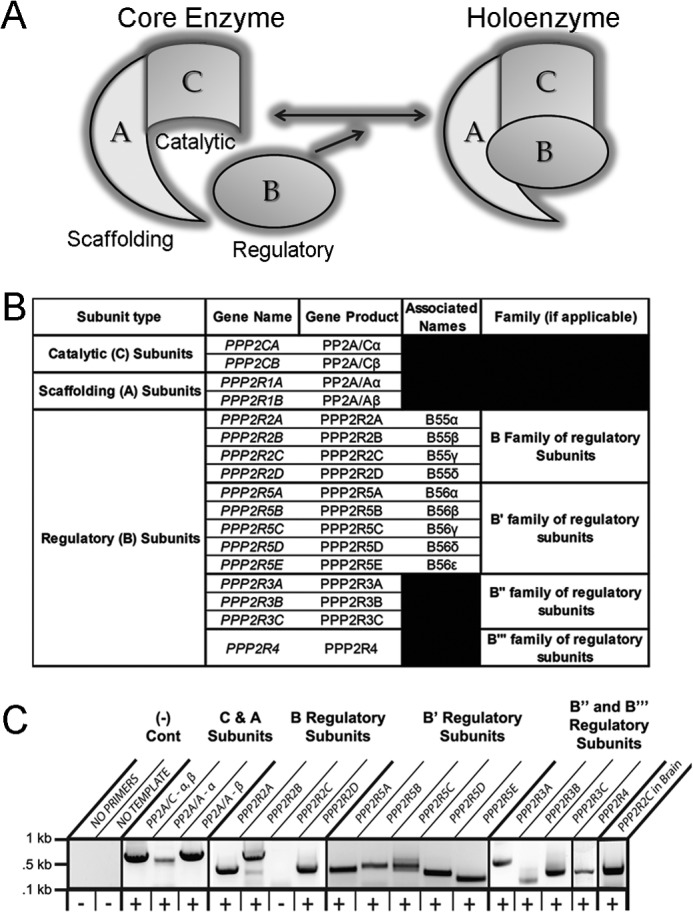
Defining PP2A family subunits in human heart. A, PP2A holoenzyme is shown. The model of PP2A holoenzyme consists of a scaffolding, catalytic, and regulatory subunit. B, human PP2A isoform descriptions include subunit type, gene name, related names, and PP2A subfamily. C, cardiac PP2A subunit transcripts are shown. Primers designed to amplify the specific subunit noted above were used to determine the presence of PP2A subunit transcripts in non-failing human heart. PCR products are shown, and a plus sign indicates the detection of a PCR product.
Expression of PP2A Subunits in Human Heart
We next tested PP2A subunit protein expression in human heart. As predicted, we observed both PP2A catalytic and scaffolding subunits at the appropriate molecular weights in human LV (Fig. 2A). Additionally, we observed nine different PP2A regulatory subunits (PPP2R2A, PPP2R2B, PPP2R3A, PPP2R4, PPP2R5A, PPP2R5B, PPP2R5C, PPP2R5D, and PPP2R5E) at their appropriate molecular weights by immunoblot (Fig. 2A). Other subunits were not detected. Notably, subsequent immunoblot analyses across human heart chambers revealed differences in PP2A subunit expression. For example, both PP2A catalytic and scaffolding subunit expression were significantly higher in right atria and ventricle when compared with left atria and ventricle (Fig. 2, A and B, n = 3, p < 0.05). We observed no statistical differences in relative protein expression of regulatory subunits between the four human chambers. Finally, we tested whether PP2A subtype expression was conserved across species. Immunoblots of human, canine, rat, and mouse LV confirmed the presence of both PP2A catalytic and scaffolding subunits (PP2A-A, PP2A-C) as well as regulatory subunits (PPP2R2A, PPP2R2B, PPP2R3A, PPP2R4, PPP2R5A, PPP2R5B, PPP2R5C, PPP2R5D, and PPP2R5E; Fig. 2B). Although we did not observe differences in overall expression patterns of the catalytic or scaffolding subunits, several regulatory subunits had variable interspecies expression levels. PPP2R5A and PPP2R5E expression levels were significantly higher in rat and mouse than in human and dog (Fig. 2D, n = 3, p < 0.05), and PPP2R2A and PPP2R5C was significantly lower in rat and mouse than in human and dog (Fig. 2D; n = 3, p < 0.05). These new findings illustrate the complexity and diversity of PP2A subunit expression in mammalian hearts. Furthermore, the data suggest that variable expression of the regulatory subunits may confer signaling specificity across regions, species, and disease states.
FIGURE 2.

Differential expression of PP2A subunits in human heart chambers and animal species. A and B, shown is PP2A subunit expression in human heart chambers. Expression of PP2A subunits across human heart chambers is shown. For panel A, * denotes p < 0.05 for each subunit relative to expression in human LV (n = 3). C and D, shown is PP2A subunit expression across species. Expression of PP2A subunits in healthy human, canine, rat, and mouse LV is shown. In D, * denotes p < 0.05 for each subunit relative to human LV (n = 3). In A–D, band densities were normalized to GAPDH, and data are shown relative to expression levels in non-failing human LV.
PP2A Regulatory Subunits Display Specific Localized Expression in Myocytes
As illustrated above, diversity in cardiac PP2A holoenzyme activity may arise from differential organ or chamber mRNA and/or protein expression. However, an additional level of target specificity may arise through differential subcellular distribution of specific PP2A holoenzymes. We tested this hypothesis by evaluating the subcellular localization of PP2A subunits in primary adult mouse myocytes. We observed broad subcellular distribution of PP2A scaffolding and catalytic subunits throughout the cytosol and nucleus of ventricular myocytes. In fact, immunostaining for both A and C subunits was present throughout the entire myocyte (Fig. 3, A and B). In contrast, we observed remarkable specificity in the localization of PP2A regulatory subunits in ventricular myocytes. For example, PPP2R5E was localized specifically to the Z-line/T-tubule region as demonstrated by co-distribution with α-actinin (Fig. 3, I–J), whereas PPP2R3A was localized to both Z- and M-lines of the cardiomyocyte (Fig. 3, C and D). PPP2R5C was primarily localized to the myocyte nucleus, with a secondary population overlying the cardiac Z-line (Fig. 3, G and H). Finally, PPP2R4 was concentrated in the cardiomyocyte nucleus and in fact localized with nuclear speckles (Fig. 3, E and F). Importantly, PP2A antibodies utilized for these studies as well as immunoblots were verified for specificity in primary neonatal cardiomyocytes ± subunit-specific siRNAs (supplemental Fig. 1). Together, these findings illustrate an additional layer of complexity of PP2A holoenzyme regulation, namely differential regulatory subunit localization, for specific subcellular targets. These findings will be important in defining the target specificity and function of each PP2A holoenzyme in vivo.
FIGURE 3.

Differential subcellular localization of PP2A regulatory subunits in heart. Isolated adult mouse cardiomyocytes were analyzed by confocal microscopy for distribution of PP2A subunits. A, PP2A/A. B, PP2A/C. C, PPP2R3A is in red; the asterisk denotes the site of the nucleus, arrows indicate Z-line, and arrowheads indicate M-line. D, PPP2R3A is in red, and α-actinin is in blue. E, PPP2R4 is in (green). F, PPP2R4 is in green, and α-actinin is in red. G, PPP2R5C is in red. H, PPP2R5C is in red, and α-actinin is in green. I, PPP2R5E is in blue. J, PPP2R5E is in blue, and α-actinin is in purple. Each experiment was done in triplicate using myocytes isolated from three different WT mice. Bar = 10 μm.
PP2A Subunit Regulation in Human Heart Failure
We next tested if expression levels of PP2A subunits were altered in human heart failure. In particular, our goal was to determine whether variability in expression of the regulatory subunits might offer a specific signature of cardiac pathology. We first investigated PP2A subunit expression in human ischemic heart failure samples. Although PP2A scaffolding subunit levels were unchanged in LV tissue, we observed a nearly 2-fold increase in PP2A catalytic subunit expression when compared with non-failing hearts (Fig. 4, A–C); n = 5 non-failing and ischemic HF, p < 0.05). We observed differential expression of regulatory subunits in ischemic heart failure with elevated levels of PPP2R5A, PPP2R5B, PPP2R5E, PPP2R3A, and PPP2R4 (Fig. 4, A and C; n = 5 non-failing and ischemic HF, p < 0.05). Other subunit levels were not statistically different between non-failing and diseased hearts (Fig. 4; n = 5 non-failing and ischemic HF, N.S.).
FIGURE 4.

Differential PP2A subunit regulation in ischemic and non-ischemic heart disease. Shown is PP2A subunit expression in non-failing (NF) human LV and in LV of human hearts in end stage ischemic heart failure (IHF) (A) or in end stage non-ischemic heart failure (nIHF) (B). C and D, densitometry analysis describes PP2A subunit expression levels in non-failing human LV and in the LV of human hearts in end stage ischemic heart failure (IHF) (C) or in end stage non-ischemic heart failure (nIHF) (D). In all experiments, GAPDH was utilized as a loading control. n = 5 for all experiments, and the asterisk denotes p < 0.05 compared with human LV.
We also examined PP2A subunit expression in LV samples from non-ischemic failing and non-failing hearts. Although we observed similar data for PP2A catalytic subunit (increased ∼2-fold), PP2A scaffolding subunit levels were also elevated in non-ischemic failing LV (Fig. 4, B and D; n = 5 non-failing and non-ischemic, p < 0.05 for A and C subunits). Regulatory subunit overexpression was observed for PPP2R5A, PPP2R5B, PPP2R5E, PPP2R3A, and PPP2R4, similar to ischemic heart failure data (Fig. 4B; n = 5 non-failing and non-ischemic HF, p < 0.05). Consistent with ischemic heart failure data, we observed no difference in the expression of PPP2R2A, PPP2R2B, PPP2R5C, or PPP2R5D gene products (n = 5 non-failing and non-ischemic HF, N.S.). These data provide the first insight into the regulation of PP2A subtype family proteins in human heart disease and demonstrate complexity in the regulation of specific PP2A holoenzymes in specific pathologies. Moreover, these data suggest that past evaluations of global PP2A function in disease may more appropriately represent a combination of differential activities of a large population of individual holoenzymes.
Differential PP2A Subunit Transcriptional Regulation in Human Heart Failure
We determined if PP2A subunits displayed differential transcriptional regulation in human heart failure. Human PP2A subunit mRNA levels were evaluated from samples from non-failing LV tissue and tissue from individuals with ischemic or non-ischemic heart failure. PP2A/A transcript (both α and β splice forms) was significantly decreased in LV ischemic heart failure samples compared with non-failing tissue (Fig. 5A; n = 3 for non-failing, n = 3 for failing, p < 0.05). Moreover, consistent with protein data, we observed a significant increase in PPP2R3A and PPP2R5B transcript levels in ischemic heart failure samples compared with non-failing samples (Fig. 5A; n = 3 for non-failing, n = 3 for HF, p < 0.05). Notably, we observed no significant difference in mRNA levels of all other PP2A subunits analyzed between non-failing and ischemic heart failure samples (Fig. 5A; n = 3 for non-failing, n = 3 for HF, p = N.S.). Consistent with ischemic heart failure data, subunit transcriptional regulation was not widely observed in non-ischemic heart failure samples. In fact, only PPP2R5D displayed a significant increase in non-ischemic heart failure samples when compared with non-failing tissue (Fig. 5B; n = 3 for non-failing, n = 3 for HF, p < 0.05). Together, these data from both ischemic and non-ischemic human heart failure suggest that PP2A subunit expression in cardiovascular disease is differentially regulated by both transcriptional and translational mechanisms, further adding to the complexity of holoenzyme regulation in human pathophysiology.
FIGURE 5.

Transcriptional regulation of PP2A subunits in human heart failure. A, shown are PP2A isoform mRNA levels in LV samples from human non-failing (black bars) and ischemic heart failure. B, PP2A isoform mRNA levels in LV samples from human non-failing (black bars) and non-ischemic heart failure are shown. n = 3 for all experiments, and an asterisk denotes p < 0.05 compared with human non-failing sample. Values were normalized to GAPDH as an internal amplification control and are expressed as levels compared with non-failing samples.
PP2A Subunit Regulation in Canine Cardiovascular Disease Models
Large and small animal experimental models have been essential for understanding the cell and molecular pathogenesis of cardiovascular disease. Therefore, we examined PP2A subunit expression in two different canine models of human heart disease. First, we examined PP2A subunit expression after myocardial infarction (5 days after total coronary artery occlusion), a leading cause of death worldwide (42). Similar to ischemic and non-ischemic human heart failure samples, we observed elevated levels of PP2A catalytic subunit in infarct border zone tissue compared with remote tissue of a well validated canine ventricular tachycardia post myocardial infarction model(43) (Fig. 6, A and C; n = 5 non-failing and post-occlusion, p < 0.05). Additionally, PP2A scaffolding subunit levels were also significantly elevated, although only moderately at 5 days post-occlusion (Fig. 6, A and C; n = 5 non-failing and post-occlusion, p < 0.05). PP2A regulatory subunits with altered expression included PPP2R5A, PPP2R5D, and PPP2R4 at 5 days post-occlusion. At this early time point, we observed no difference in the expression of regulatory subunit products of PPP2R2A, PPP2R2B, PPP2R2B, PPP2R5C, PPP2R5E, or PPP2R3A (Fig. 6, A and C; n = 5 non-failing and post-occlusion, N.S.).
FIGURE 6.

Differential PP2A subunit regulation in canine cardiovascular disease. Shown is PP2A subunit expression in canine LV. Experiments in panels A and C represent data from control versus border zone (BZ) tissue 5 days post coronary artery occlusion (n = 5, p < 0.05). Experiments in panels B and D represent data from non-failing versus heart failure. In all experiments, GAPDH was used as a loading control (n = 5, p < 0.05). MI, myocardial infarction.
We also evaluated PP2A subunit expression in a well validated long term tachy-pacing-induced non-ischemic model of canine heart failure (41). At 4 months of heart failure, we observed elevated levels of both scaffolding and catalytic subunits in experimental samples compared with non-failing tissue (Fig. 6, B and D; n = 5 non-failing and HF, p < 0.05). PPP2R2B, PPP2R5B, PPP2R5D, PPP2R5E, PPP2R3A, and PPP2R4 gene products were significantly elevated in failing canine LV compared with non-failing canine tissue (Fig. 6 B and D; n = 5 non-failing and HF, p < 0.05). In contrast, levels of PPP2R2A, PPP2R5A, and PPP2R5C were unchanged between non-failing and heart failure samples (Fig. 6, B and C; n = 5 non-failing and HF, N.S.). Collectively, data in these canine ischemic and non-ischemic disease models demonstrate significant regulation of the PP2A enzyme through regulation of both PP2A catalytic and scaffolding subunits. However, these data also reveal that full holoenzyme regulation may be tightly regulated for target specificity in each disease through regulation of regulatory subunit expression. Our data further demonstrate that each specific disease pathology (ischemic versus non-ischemic, acute versus chronic, etc.) likely has its own PP2A subunit expression signature presumably corresponding with selective regulation of specific targets.
Select PP2A Subunit Regulation in Mouse Model of Human Catecholamine-induced Arrhythmia
Dysregulation in cardiac sympathetic tone has been linked to both genetic and acquired forms of human ventricular arrhythmia (18). In fact, dysfunction in select myocyte pathways that alter the phosphoprotein axis have been identified as a primary cause of multiple forms of congenital catecholaminergic polymorphic ventricular tachycardia (44). One notable pathway is the ankyrin-B-pathway that coordinates the subcellular localization of membrane-associated ion channels, transporters, and signaling molecule that is altered in human catecholaminergic polymorphic ventricular tachycardia as well as sinus node disease, heart rate variability, and atrial fibrillation (31, 33, 35, 36). We hypothesized that PP2A subunit levels might be altered in a murine model of human ankyrin-B catecholaminergic polymorphic ventricular tachycardia. Consistent with investigated forms of human and canine heart disease, we observed significant increases in both PP2A catalytic and scaffolding subunits in adult ankyrin-B+/− LV compared with littermates (supplemental Fig. 2; n = 5 wild type and ankyrin-B+/−, p < 0.05). Additionally, we observed alterations in expression of PP2A regulatory subunits PPP2R5A, PPP2R5B, and PPP2R5E in ankyrin-B+/− hearts (supplemental Fig. 2; n = 5 wild type and ankyrin-B+/−, p < 0.05). Although there were trends for differences in other PP2A regulatory subunits, we observed no significant difference in expression of PPP2R2A, PPP2R2B, PPP2R3A, PPP2R4, PPP2R5C, or PPP2R5D between WT and ankyrin-B+/− mice (supplemental Fig. 2; n = 5 wild type and ankyrin-B+/−, p = N.S.). Thus, consistent with a role for aberrant regulation of the kinase/phosphatase signaling axis in sympathetic-mediated ventricular tachycardia, our results indicate that multiple arms of the PP2A signaling cascade are differentially regulated in an animal model of catecholaminergic polymorphic ventricular tachycardia.
Mechanisms Underlying Post-translational Regulation of Cardiac PP2A Function in Human Cardiovascular Disease
Although not studied in heart, post-translational regulation of PP2A activity is implicated in holoenzyme regulation in other organ systems and non-cardiac disease states (10, 45). We, therefore, investigated potential new mechanisms for cardiac phosphatase regulation via post-translational modifications at base line and in heart disease. Notably, the C terminus of the PP2A catalytic subunit interacts with an interface of the A and B subunits. This C-terminal region is altered by both phosphorylation and methylation that serve to “switch” the active and inactive state of the full holoenzyme through altering the recruitment and docking of the regulatory subunit with the A and C subunits (Fig. 7A) (45). We examined the phosphorylation status of the catalytic subunit of PP2A at residue Tyr-307, a site liked with inactivation of the phosphatase (10, 12, 46). Notably, we observed phosphorylated PP2A/C Tyr-307 in non-failing human heart (Fig. 7, B–D). Moreover, we observed a significant increase in the expression level of the phosphorylated (inactivated) catalytic subunit being expressed in non-ischemic heart failure samples compared with non-failing heart (Fig. 7, B and C; n = 4, p < 0.05). In contrast, we observed no difference in methylation of PP2A catalytic subunit at residue Leu-309, a site regulated by the opposing activities of LCMT-1 and phosphatase methylesterase 1 (PME-1) to control recruitment of the PPP2R2A and PPP2R2B regulatory subunits (46) (Fig. 7, A and C, n = 4, p = N.S.). Total PP2A catalytic subunit expression was increased in non-ischemic heart failure (Fig. 7, B and C, n = 4, p < 0.05). However, in parallel, levels of PP2A-phospho-Tyr-307 (inactivated) were increased. Thus, we normalized expression of phosphorylated and methylated forms of the PP2A catalytic subunit to the expression level of the total amount of the PP2A/C subunit expressed in each sample. Notably, the ratio of PP2A subunit Tyr-307 to total PP2A/C was increased in human heart failure samples compared with non-failing heart (Fig. 7D; n = 4, p < 0.05), favoring inactivation of the holoenzyme. Moreover, the ratio of methylated catalytic subunit to total catalytic subunit was significantly decreased in human heart failure compared with non-failing samples (Fig. 7D; n = 4, p < 0.05), a modification further disabling the functional holoenzyme. Notably, we observed similar findings in samples from human ischemic heart failure versus non-failing heart samples (Fig. 7, E–G; n = 4 HF, n = 5 non-failing; p < 0.05) and canine heart failure samples versus control samples (Fig. 7, H–J; n = 4 control, n = 4 HF, p < 0.05).
FIGURE 7.
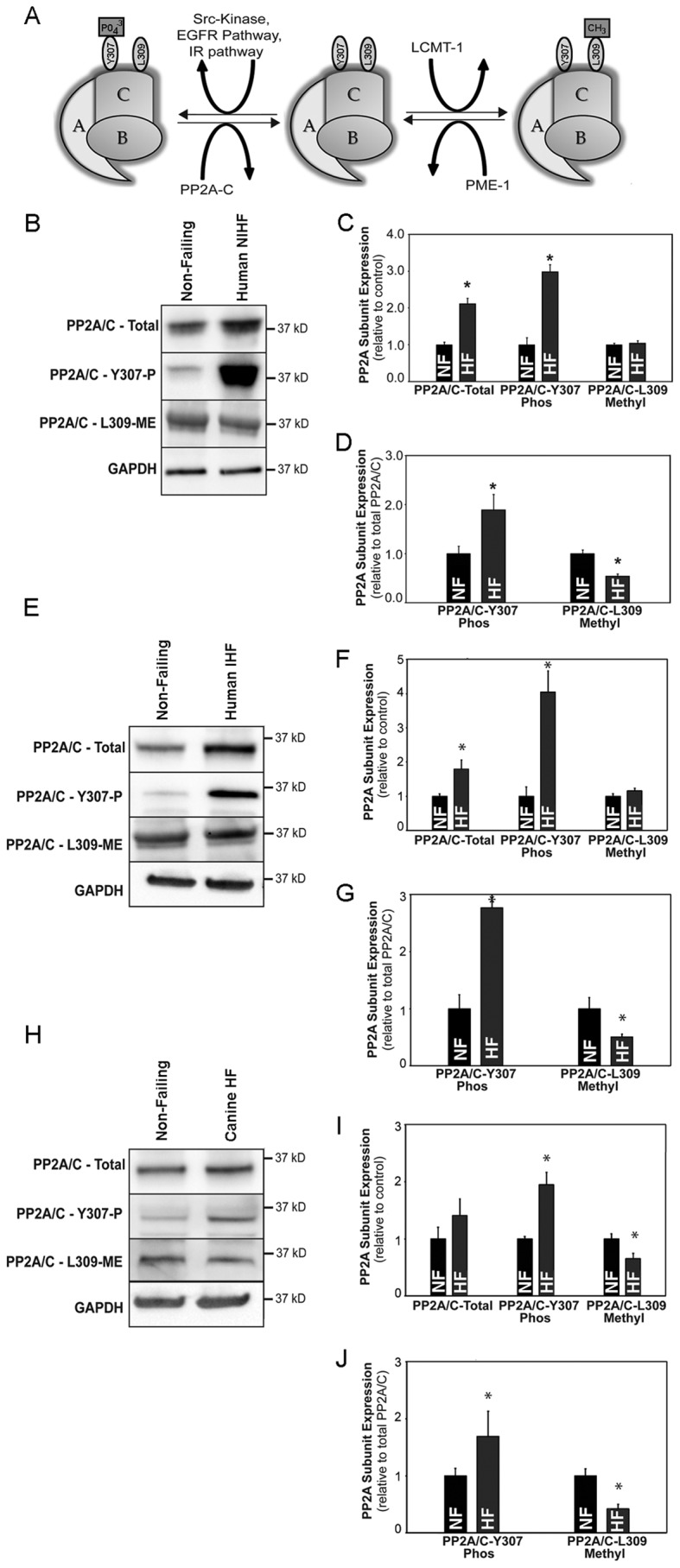
Post-translational regulation of PP2A subunits at base line and in disease. A, a model of post-translational modifications of the PP2A catalytic subunit is shown. Tyr-307 phosphorylation of PP2A/C results in generalized inhibition of phosphatase activity; Leu-309 methylation (regulated by LCMT-1 and PME-1) results in the enhanced recruitment of the PPP2R2A and PPP2R2B into the holoenzyme. Shown are expression levels of total, phosphorylated (Thr-307), and methylated (Leu-309) PP2A catalytic subunit in LV samples of non-failing (NF) and non-ischemic human heart failure (B and C; n = 5; p < 0.05), non-failing and ischemic human heart failure (E and F, n = 5 non-failing, n = 4 ischemic HF, p < 0.05), and canine control and heart failure samples (H–I, n = 4/group, p < 0.05). D, G, and J, adjusted phosphorylated and methylated catalytic subunit activities based on total PP2A catalytic subunit expression in non-failing and heart failure samples from human and canine heart are shown. GAPDH was used as an internal loading control. IR, insulin response.
In response to increased catalytic phosphorylation and decreased catalytic subunit methylation, select PP2A regulatory subunits (B55α encoded by PPP2R2A) dissociate from the core PP2A enzyme (45–47). Based on our observations of altered PP2A catalytic post-translational regulation in heart failure (increased phosphorylation, decreased methylation; Fig. 7), we hypothesized that PP2A core subunits would display reduced association with B55α in heart failure. To test this hypothesis, we performed co-immunoprecipitation experiments using control and heart failure samples from canine LV. Consistent with our hypothesis, we observed dissociation of B55α from the core PP2A scaffolding and catalytic subunits in canine heart failure samples compared with control LV samples (Fig. 8). These data demonstrate that whereas global PP2A catalytic subunit levels may increase in heart failure, levels of specific local PP2A holoenzyme populations (i.e. B55α) will be reduced due to alterations in post-translational regulation. In summary, our findings provide the initial data on PP2A subunit post-translational regulation in heart and provide data on the regulation of these critical subunits in human heart failure.
FIGURE 8.
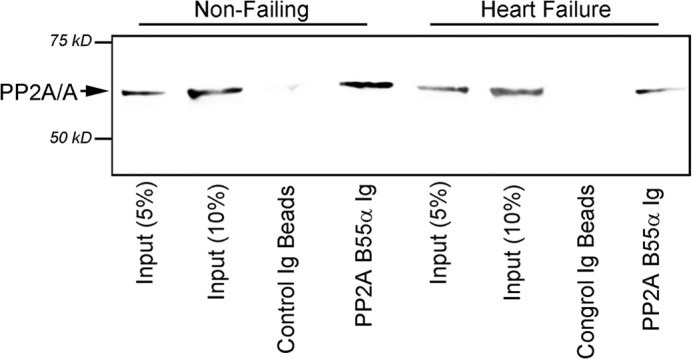
Decreased assembly of PP2A enzyme with B55 regulatory subunit in heart failure. Shown are co-immunoprecipitation experiments from non-failing and failing canine LV tissue lysate using B55α-specific Ig (lanes 1–2, and 5 and 6 represent 5 and 10% of experimental input). Note that PP2A B55α Ig immunoprecipitated reduced PP2A/A subunit in failing canine tissue compared with non-failing tissue even though PP2A/A and PP2A/C levels were elevated in the canine heart failure model. Molecular weight markers are shown in the figure. Identical data were observed in multiple biochemical experiments.
PP2A Subunit Post-translational Regulation Is Cardiac Cell Type-specific
Although excitable cardiomyocytes comprise the majority of cells in the heart, a host of other cell types including cardiac fibroblasts are present in heart and alter cardiac physiology and response in disease. Experimentally, these non-myocyte populations may affect interpretation of our analysis of global cardiac PP2A regulation. We, therefore, evaluated the presence of PP2A subunits in purified populations of primary cardiomyocytes (Fig. 9, A and B) and cardiac fibroblasts (Fig. 9, C and D). Mouse cardiac cells were utilized to generate large and homogenous cell populations. By immunoblot, both myocyte and fibroblast cell populations showed robust expression of PP2A core subunits (Fig. 9, A–D). Additionally, both cell populations displayed both post-translational PP2A C subunit methylation and phosphorylation (Fig. 9, A–D).
FIGURE 9.
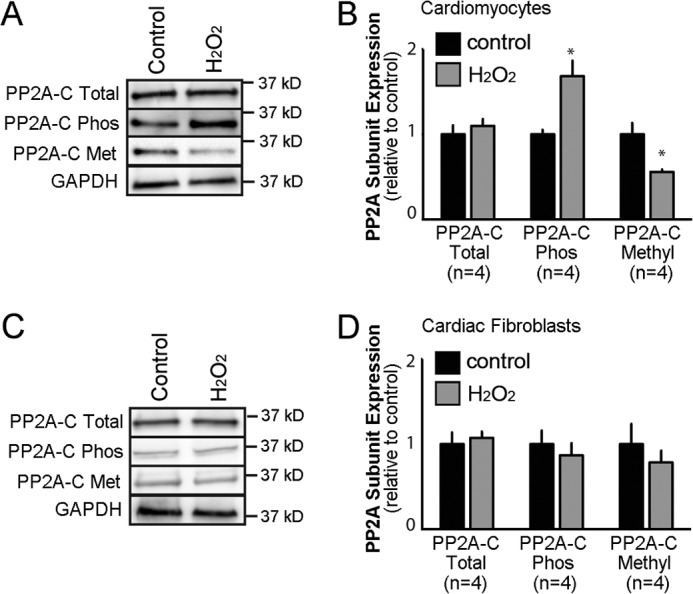
Cardiac cell type-specific regulation of PP2A post-translational regulation. A, shown is expression of the PP2A catalytic subunit in purified populations of primary ventricular cardiomyocytes ± treatment with H2O2 to induce ROS production. Immunoblots also depict relative expression of PP2A catalytic subunit phosphorylation and methylation ±H2O2. GAPDH was utilized as internal loading control. B, shown are mean expression levels of the catalytic subunit and modified forms ± H2O2 from purified primary cardiomyocytes (n = 4; * represents p < 0.05 for each antibody ± H2O2). C, shown is expression of PP2A catalytic subunit in purified populations of primary cardiac fibroblasts ± H2O2. Immunoblots also depict relative expression of PP2A catalytic subunit phosphorylation and methylation ± H2O2. GAPDH was utilized as internal loading control. D, mean expression levels are shown of the catalytic subunit and modified forms ± H2O2 from purified primary fibroblasts (n = 4; p = N.S. for each group ± H2O2).
We next examined whether both myocyte and fibroblast cell populations would show display similar responses to disease-associated agonists. Consistent with earlier heart failure data, we observed increased cardiomyocyte PP2A catalytic subunit phosphorylation and decreased PP2A catalytic subunit methylation in response to elevated reactive oxygen species (ROS), a hallmark of human heart failure (Fig. 9, A and B, n = 4 control, n = 4 ROS exposure, p < 0.05). However, unlike purified myocyte experiments, we observed no statistical difference in PP2A catalytic subunit methylation or phosphorylation in purified cardiac fibroblasts in response to elevated ROS (Fig. 9, C and D, n = 4 control, n = 4 ROS exposure, p = N.S.). Together, these data demonstrate that although PP2A subunits likely exist in most cardiac cell types, the mechanisms for PP2A subunit post-translational regulation are cell type-specific.
PP2A Holoenzyme Post-translational Regulation Is Modified Through Upstream Pathways
Methylation status of the PP2A catalytic subunit (via LCMT-1 and PME-1) alters the activity of specific PP2A subclasses. We hypothesized that down-regulation of LCMT-1 expression and/or increased expression of PME-1 may explain the observed decrease in PP2A catalytic subunit methylation in human heart failure. Consistent with this hypothesis, we observed decreased expression of LCMT-1 in both ischemic and non-ischemic heart disease (Fig. 10, A, B, E, and F; p < 0.05 versus non-failing human heart tissue). Similar results were obtained in experimental samples from chronic canine heart disease versus non-failing (Fig. 10, C, E, and F; p < 0.05 versus non-failing canine heart samples). In contrast, LCMT-1 expression was significantly increased in ankyrin-B+/− mouse hearts (mouse model of ventricular arrhythmia) compared with wild-type littermates (Fig. 10, D–F; p < 0.05 versus wild-type hearts). Conversely, we observed no statistical difference in expression of PME-1 in ischemic and non-ischemic human heart failure samples compared with non-failing heart tissue (Fig. 10, A, B, E, and F; p = N.S. heart failure versus non-failing). Similarly, canine and murine disease models also showed no change in PME-1 expression (Fig. 10, C–F, p = N.S.). Mechanistically, these data support a model where PP2A holoenzyme post-translational regulation is modulated upstream through the transcriptional control of methyltransferase activity in disease.
FIGURE 10.

Decreased catalytic subunit methylation is linked with decreased LCMT-1 expression. A–D, shown are representative immunoblots of LCMT-1 and PME-1 expression in whole heart lysates from ischemic heart failure, non-ischemic human heart failure, canine pacing-induced heart failure, and a mouse model of a human inheritable ventricular arrhythmia syndrome. NF, non-failing. E and F, shown is densitometry analysis indicating the expression level of LCMT-1 and PME-1 expression in ischemic heart failure, non-ischemic human heart failure, canine pacing-induced heart failure, and a mouse model of a human inheritable ventricular arrhythmia syndrome relative to wild type. In all experiments, GAPDH was used as a loading control. N values are noted in the figure, and p < 0.05 was considered statistically significant.
DISCUSSION
Work over the past five decades has illustrated the importance of adrenergic balance in normal cardiac physiology as well as adrenergic imbalance a host of cardiovascular pathologies (48). For example, increased activity of GSK-3β has been linked to cardiomyocyte cell death after ischemia and reperfusion (49) and increased PKA, PKC, and calmodulin kinase II activity have been linked to a host of congenital and acquired cardiac pathologies (50–52). Although our knowledge of how aberrant regulation of kinase activity contributes to human cardiovascular disease continues to expand and has seeded the development of new compounds for cardioprotection, phosphatase activity and its regulation in disease remain less well understood. Here, we provide new data that support 1) the complexity of PP2A protein family regulation in heart and in cardiovascular disease and 2) the potential diversity of individual populations of PP2A holoenzymes in myocytes. Our findings demonstrate that PP2A subunits show heterogeneity in their mRNA and protein expression patterns in heart and myocytes with distinct expression profiles across cardiac chambers, species, and disease states (Figs. 1, 2, 4–6). Steady state expression of the large family of regulatory subunits in particular is highly variable and may serve as a molecular rheostat to tune signaling specificity. Specificity is further regulated by subunit specific expression within different subcellular domains (Fig. 3), and ultimately local signaling is controlled by multiple post-translational modifications and upstream regulatory proteins and is cell type-specific. In different cardiac disease pathologies, we demonstrate remodeling of not only subunit expression but also subunit post-translational phosphorylation and methylation. Moreover, our data demonstrate that specific cardiac pathologies display specific combinations of subunit regulatory patterns associated with not only the severity of the phenotype but also with the time course of the cardiac insult. Ultimately, these data provide a new data of the complex nature of protein phosphatase regulation in health and disease, strongly supporting not only distinct but spatially and temporally regulatory phospho-targets.
An unexpected finding from this study was the diversity in PP2A regulatory (B) subunit regulation between cardiac chambers, cell types, subcellular domains, and disease phenotypes. Based on our findings, we predict that PP2A regulatory subunits provide a primary regulatory step for PP2A function in heart. Although PP2A scaffolding (A) and catalytic (C) subunits are expressed throughout all chambers and across cellular domains, we identified diversity across the regulatory subunit family. For example, although PPP2R5E was expressed at the M-line, PPP2R4 was enriched in the nucleus of the cell. These findings are unanticipated given the high sequence conservation between subunits. Our past work has demonstrated that ankyrin proteins play a key role in the subcellular targeting of the PPP2R5A regulatory subunit through a non-conserved C-terminal domain (30). Moreover, ankyrin-B levels are altered in human and experimental forms of heart failure (53), suggesting that PPP2R5A (B56α) may also altered in disease models. Consistent with this hypothesis, we observed altered PPP2R5A protein expression and distribution in response to disease conditions (Figs. 4 and 6 and supplemental Fig. 3). It will be important in the future to more clearly define how different disease pathologies result in alterations in PP2A subunit regulation. In summary, based on the relationship between ankyrin-B and PPP2R5A, we predict that the C-terminal domain of each regulatory protein may also play similar roles in holoenzyme targeting and specificity in vivo.
Whereas local PP2A activity appears to be highly linked with regulatory subunit function, post-translational regulation of PP2A catalytic subunits offers a second mechanism to tune local function. Post-translational regulation of PP2A has not been previously studied in heart or in heart disease. In human heart failure the relative decrease in the methylated catalytic subunit and increase in the phosphorylated catalytic subunit suggest that specific subsets of regulatory subunits may be selectively excluded from the PP2A enzyme (45–47). In fact, in our experiments we observed loss of the B55 family of PP2A regulatory subunit from the PP2A A/C core enzyme in disease. There are competing interpretations to these data. The first is that pathological processes associated with heart failure alter the expression or enzymatic availability of LCMT-1 and PME-1 (enzymes that regulate catalytic subunit methylation) leading to exclusion of regulatory subunits from active holoenzyme formation. A second interpretation is that shifting PP2A enzymatic activity toward targets favored by the B′ and B″ family of regulatory subunits is a physiologic response to heart failure. Our studies of LCMT-1 and PME-1 cell-type and disease-linked expressional regulation offer yet another level of PP2A regulatory complexity.
Based on our new data, post-translational regulation of the PP2A catalytic subunit appears to be altered in a relatively rapid time course by physiological and pathological stress (i.e. ROS generation within cardiomyocytes). Moreover, in light of the fact that we observed no change in catalytic subunit expression in cardiac fibroblasts following an identical time course, our data suggest that catalytic subunit methylation may be a central mechanism for acute regulation of only a select population of cardiac cells in disease. Furthermore, these results suggest that the differential regulation of PP2A identified in this study occurs in cardiomyocytes rather than another of the many cell types present in cardiac tissue. In summary, our initial experiments support a highly organized regulatory network (multiple orders of magnitude) for controlling PP2A holoenzyme function in heart. Additional studies on upstream regulatory molecules (e.g. LCMT-1/PME-1) will be necessary to further define regulation in vivo and whether these alterations are adaptive or maladaptive.
Cardiac phosphatase activity is not limited to PP2A. In fact, work over the past 15 years has also implicated critical roles for type 1 protein phosphatase (PP1) at base line and in cardiovascular disease. In fact, the diversity of PP2A regulation presented in this study has a level of complexity similar to that discovered for PP1. In mechanisms similar to PP2A, PP1 confers tissue and subcellular specificity also through multiple isoforms (catalytic diversity for PP1) and numerous regulator and endogenous inhibitors (54). Moreover an increase in PP1 is linked with a number of experimental models of heart failure (55–60), and the molecule has been studied as a potential target for therapeutic application due to its specific subcellular compartmentalization. Our new data support the findings from these PP1 data of the complex mechanisms that support phosphatase activity. Future experiments will be critical to uncover the mechanisms that underlie spatial and temporal dual regulation of PP1 and PP2A in vivo.
One limitation of this study is the lack of data for local PP2A function in specific subcellular domains. For years, the literature has utilized “broad-stroke” functional kinase and phosphatase assays to assess the phosphor-substrate of a tissue or cell. For example, the use of the phosphatase inhibitors such as okadaic acid, cantharidin, and calyculin A exposed the physiological importance of protein phosphatases and led to the discovery of key regulatory phosphoproteins (57, 61–64). However, the use of these inhibitors as inotropic therapies has been prevented due to their global and non-compartmentalized inhibitory effects. Our data suggest these global assays, although portraying the phospho-balance of an entire cell population, may have limited use in predicting the regulation of subcellular targets. Our findings suggest that monitoring the combination of the activity of specific regulatory subunits in parallel with the post-translational status of the PP2A catalytic subunit may offer a more sensitive, cell-specific and subcellular-specific assay for defining local holoenzyme activity. Importantly, although our data illustrate differences in the mRNA and protein expression profiles and localization of PP2A subunits, it is important to note that these data denote qualitative differences as each subunit was detected using a specific mRNA or antibody probe. For example, our mRNA data show increased mRNA levels of PP2A/Aβ versus PP2A/Aα. However, work in other tissues suggest differences in these ratios but also a disconnect between PP2A/A subunit mRNA and protein levels (65–67). It will be critical in the future to design targeted probes that may be able to more definitively define the relative and local concentrations of each PP2A holoenzyme in vivo.
In summary, our data describe multiple layers of complexity underlying the diversity in tissue, cellular, and subcellular target phospho-regulation in cardiac muscle and in heart disease. Our findings illustrate differential regulation of all PP2A subtype classes (scaffolding, catalytic, regulatory) in health and disease and reveal a high order of complexity in relation to the subunit-specific expression and regulation in each cardiac signature whether at base line or in disease. These data offer insight into the complexity of cardiac phospho-protein regulation but also suggest new therapeutic targets to modify specific cellular and subcellular targets for cardioprotection. Moreover, our data suggest that other excitable organs (nervous system, skeletal muscle, beta cells) may also harbor similar regulatory networks for control of cell signaling and physiology.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants HL084583 and HL083422 (to P. J. M.), HL079031, HL62494, and HL70250 (to M. E. A.), HL089836 (to C. A. C.), HL066140 (to P. A. B.), and HL096805 and HL114893 (to T. J. H.). This work was also supported in part by a grant from the Saving Tiny Hearts Society (to P. J. M.), an American Heart Association Established Investigator Award (to P. J. M.), the Gilead Sciences Research Scholars Program (to T. J. H.), and the Fondation Leducq Award to the Alliance for Calmodulin Kinase Signaling in Heart Disease (to P. J. M., M. E. A., and T. J. H.).

This article contains supplemental Figs. 1–3.
- PP2A
- protein phosphatase 2A
- PP1
- type 1 protein phosphatase
- LV
- left ventricular
- HF
- heart failure
- LCMT-1
- leucine carboxymethyltransferase 1
- PME-1
- phosphatase methylesterase 1
- ROS
- reactive oxygen species
- N.S.
- not significant.
REFERENCES
- 1. He B. J., Joiner M. L., Singh M. V., Luczak E. D., Swaminathan P. D., Koval O. M., Kutschke W., Allamargot C., Yang J., Guan X., Zimmerman K., Grumbach I. M., Weiss R. M., Spitz D. R., Sigmund C. D., Blankesteijn W. M., Heymans S., Mohler P. J., Anderson M. E. (2011) Oxidation of CaMKII determines the cardiotoxic effects of aldosterone. Nat. Med. 17, 1610–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chelu M. G., Sarma S., Sood S., Wang S., van Oort R. J., Skapura D. G., Li N., Santonastasi M., Müller F. U., Schmitz W., Schotten U., Anderson M. E., Valderrábano M., Dobrev D., Wehrens X. H. (2009) Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J. Clin. Invest. 119, 1940–1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Swaminathan P. D., Purohit A., Soni S., Voigt N., Singh M. V., Glukhov A. V., Gao Z., He B. J., Luczak E. D., Joiner M. L., Kutschke W., Yang J., Donahue J. K., Weiss R. M., Grumbach I. M., Ogawa M., Chen P. S., Efimov I., Dobrev D., Mohler P. J., Hund T. J., Anderson M. E. (2011) Oxidized CaMKII causes cardiac sinus node dysfunction in mice. J. Clin. Invest. 121, 3277–3288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bers D. M. (2011) Ca2+-calmodulin-dependent protein kinase II regulation of cardiac excitation-transcription coupling. Heart Rhythm 8, 1101–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reiken S., Gaburjakova M., Gaburjakova J., He Kl K. L., Prieto A., Becker E., Yi Gh G. H., Wang J., Burkhoff D., Marks A. R. (2001) β-Adrenergic receptor blockers restore cardiac calcium release channel (ryanodine receptor) structure and function in heart failure. Circulation 104, 2843–2848 [DOI] [PubMed] [Google Scholar]
- 6. Shan J., Kushnir A., Betzenhauser M. J., Reiken S., Li J., Lehnart S. E., Lindegger N., Mongillo M., Mohler P. J., Marks A. R. (2010) Phosphorylation of the ryanodine receptor mediates the cardiac fight or flight response in mice. J. Clin. Invest. 120, 4388–4398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Packer M. (1985) Sudden unexpected death in patients with congestive heart failure. A second frontier. Circulation 72, 681–685 [DOI] [PubMed] [Google Scholar]
- 8. Reiken S., Wehrens X. H., Vest J. A., Barbone A., Klotz S., Mancini D., Burkhoff D., Marks A. R. (2003) Beta blockers restore calcium release channel function and improve cardiac muscle performance in human heart failure. Circulation 107, 2459–2466 [DOI] [PubMed] [Google Scholar]
- 9. Shelton R. J., Clark A. L., Goode K., Rigby A. S., Houghton T., Kaye G. C., Cleland J. G. (2009) A randomized, controlled study of rate versus rhythm control in patients with chronic atrial fibrillation and heart failure, (CAFE-II Study). Heart 95, 924–930 [DOI] [PubMed] [Google Scholar]
- 10. Janssens V., Goris J. (2001) Protein phosphatase 2A. A highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 353, 417–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Virshup D. M., Shenolikar S. (2009) From promiscuity to precision. Protein phosphatases get a makeover. Mol. Cell 33, 537–545 [DOI] [PubMed] [Google Scholar]
- 12. Sents W., Ivanova E., Lambrecht C., Haesen D., Janssens V. (2012) The biogenesis of active protein phosphatase. 2A holoenzymes. A tightly regulated process creating phosphatase specificity. FEBS J., in press [DOI] [PubMed] [Google Scholar]
- 13. Dai S., Hall D. D., Hell J. W. (2009) Supramolecular assemblies and localized regulation of voltage-gated ion channels. Physiol. Rev. 89, 411–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eichhorn P. J., Creyghton M. P., Bernards R. (2009) Protein phosphatase 2A regulatory subunits and cancer. Biochim. Biophys Acta 1795, 1–15 [DOI] [PubMed] [Google Scholar]
- 15. Rudrabhatla P., Pant H. C. (2011) Role of protein phosphatase 2A in Alzheimer's disease. Curr. Alzheimer Res. 8, 623–632 [DOI] [PubMed] [Google Scholar]
- 16. Ai X., Pogwizd S. M. (2005) Connexin 43 down-regulation and dephosphorylation in nonischemic heart failure is associated with enhanced colocalized protein phosphatase type 2A. Circ. Res. 96, 54–63 [DOI] [PubMed] [Google Scholar]
- 17. Xu H., Ginsburg K. S., Hall D. D., Zimmermann M., Stein I. S., Zhang M., Tandan S., Hill J. A., Horne M. C., Bers D., Hell J. W. (2010) Targeting of protein phosphatases PP2A and PP2B to the C terminus of the L-type calcium channel Ca v1.2. Biochemistry 49, 10298–10307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marx S. O., Reiken S., Hisamatsu Y., Jayaraman T., Burkhoff D., Rosemblit N., Marks A. R. (2000) PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor). Defective regulation in failing hearts. Cell 101, 365–376 [DOI] [PubMed] [Google Scholar]
- 19. Deshmukh P. A., Blunt B. C., Hofmann P. A. (2007) Acute modulation of PP2a and troponin I phosphorylation in ventricular myocytes. Studies with a novel PP2a peptide inhibitor. Am. J. Physiol. Heart Circ. Physiol. 292, H792–H799 [DOI] [PubMed] [Google Scholar]
- 20. Wijnker P. J., Boknik P., Gergs U., Müller F. U., Neumann J., dos Remedios C., Schmitz W., Sindermann J. R., Stienen G. J., van der Velden J., Kirchhefer U. (2011) Protein phosphatase 2A affects myofilament contractility in non-failing but not in failing human myocardium. J. Muscle Res. Cell Motil. 32, 221–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schulze D. H., Muqhal M., Lederer W. J., Ruknudin A. M. (2003) Sodium/calcium exchanger (NCX1) macromolecular complex. J. Biol. Chem. 278, 28849–28855 [DOI] [PubMed] [Google Scholar]
- 22. Kohr M. J., Davis J. P., Ziolo M. T. (2009) Peroxynitrite increases protein phosphatase activity and promotes the interaction of phospholamban with protein phosphatase 2a in the myocardium. Nitric Oxide 20, 217–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gergs U., Boknik P., Buchwalow I., Fabritz L., Matus M., Justus I., Hanske G., Schmitz W., Neumann J. (2004) Overexpression of the catalytic subunit of protein phosphatase 2A impairs cardiac function. J. Biol. Chem. 279, 40827–40834 [DOI] [PubMed] [Google Scholar]
- 24. Ai X., Jiang A., Ke Y., Solaro R. J., Pogwizd S. M. (2011) Enhanced activation of p21-activated kinase 1 in heart failure contributes to dephosphorylation of connexin 43. Cardiovasc. Res. 92, 106–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marshall M., Anilkumar N., Layland J., Walker S. J., Kentish J. C., Shah A. M., Cave A. C. (2009) Protein phosphatase 2A contributes to the cardiac dysfunction induced by endotoxemia. Cardiovasc. Res. 82, 67–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brewis N., Ohst K., Fields K., Rapacciuolo A., Chou D., Bloor C., Dillmann W., Rockman H., Walter G. (2000) Dilated cardiomyopathy in transgenic mice expressing a mutant A subunit of protein phosphatase 2A. Am. J. Physiol. Heart Circ. Physiol. 279, H1307–H1318 [DOI] [PubMed] [Google Scholar]
- 27. DeGrande S., Nixon D., Koval O., Curran J. W., Wright P., Wang Q., Kashef F., Chiang D., Li N., Wehrens X. H., Anderson M. E., Hund T. J., Mohler P. J. (2012) CaMKII inhibition rescues proarrhythmic phenotypes in the model of human ankyrin-B syndrome. Heart Rhythm, 9, 2034–2041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Belevych A. E., Sansom S. E., Terentyeva R., Ho H. T., Nishijima Y., Martin M. M., Jindal H. K., Rochira J. A., Kunitomo Y., Abdellatif M., Carnes C. A., Elton T. S., Györke S., Terentyev D. (2011) MicroRNA-1 and -133 increase arrhythmogenesis in heart failure by dissociating phosphatase activity from RyR2 complex. PLoS ONE 6, e28324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Terentyev D., Belevych A. E., Terentyeva R., Martin M. M., Malana G. E., Kuhn D. E., Abdellatif M., Feldman D. S., Elton T. S., Györke S. (2009) miR-1 overexpression enhances Ca2+ release and promotes cardiac arrhythmogenesis by targeting PP2A regulatory subunit B56α and causing CaMKII-dependent hyperphosphorylation of RyR2. Circ. Res. 104, 514–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bhasin N., Cunha S. R., Mudannayake M., Gigena M. S., Rogers T. B., Mohler P. J. (2007) Molecular basis for PP2A regulatory subunit B56α targeting in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 293, H109–H119 [DOI] [PubMed] [Google Scholar]
- 31. Cunha S. R., Hund T. J., Hashemi S., Voigt N., Li N., Wright P., Koval O., Li J., Gudmundsson H., Gumina R. J., Karck M., Schott J. J., Probst V., Le Marec H., Anderson M. E., Dobrev D., Wehrens X. H., Mohler P. J. (2011) Defects in ankyrin-based membrane protein targeting pathways underlie atrial fibrillation. Circulation 124, 1212–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cunha S. R., Mohler P. J. (2008) Obscurin targets ankyrin-B and protein phosphatase 2A to the cardiac M-line. J. Biol. Chem. 283, 31968–31980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Le Scouarnec S., Bhasin N., Vieyres C., Hund T. J., Cunha S. R., Koval O., Marionneau C., Chen B., Wu Y., Demolombe S., Song L. S., Le Marec H., Probst V., Schott J. J., Anderson M. E., Mohler P. J. (2008) Dysfunction in ankyrin-B-dependent ion channel and transporter targeting causes human sinus node disease. Proc. Natl. Acad. Sci. U.S.A. 105, 15617–15622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mohler P. J., Le Scouarnec S., Denjoy I., Lowe J. S., Guicheney P., Caron L., Driskell I. M., Schott J. J., Norris K., Leenhardt A., Kim R. B., Escande D., Roden D. M. (2007) Defining the cellular phenotype of “ankyrin-B syndrome” variants. Human ANK2 variants associated with clinical phenotypes display a spectrum of activities in cardiomyocytes. Circulation 115, 432–441 [DOI] [PubMed] [Google Scholar]
- 35. Mohler P. J., Schott J. J., Gramolini A. O., Dilly K. W., Guatimosim S., duBell W. H., Song L. S., Haurogné K., Kyndt F., Ali M. E., Rogers T. B., Lederer W. J., Escande D., Le Marec H., Bennett V. (2003) Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature 421, 634–639 [DOI] [PubMed] [Google Scholar]
- 36. Cunha S. R., Bhasin N., Mohler P. J. (2007) Targeting and stability of Na+/Ca2+ exchanger 1 in cardiomyocytes requires direct interaction with the membrane adaptor ankyrin-B. J. Biol. Chem. 282, 4875–4883 [DOI] [PubMed] [Google Scholar]
- 37. Mohler P. J., Davis J. Q., Bennett V. (2005) Ankyrin-B coordinates the Na/K ATPase, Na+/Ca2+ exchanger, and InsP3 receptor in a cardiac T-tubule/SR microdomain. PLoS Biol. 3, e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hund T. J., Wright P. J., Dun W., Snyder J. S., Boyden P. A., Mohler P. J. (2009) Regulation of the ankyrin-B-based targeting pathway following myocardial infarction. Cardiovasc. Res. 81, 742–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gudmundsson H., Hund T. J., Wright P. J., Kline C. F., Snyder J. S., Qian L., Koval O. M., Cunha S. R., George M., Rainey M. A., Kashef F. E., Dun W., Boyden P. A., Anderson M. E., Band H., Mohler P. J. (2010) EH domain proteins regulate cardiac membrane protein targeting. Circ. Res. 107, 84–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gudmundsson H., Curran J., Kashef F., Snyder J. S., Smith S. A., Vargas-Pinto P., Bonilla I. M., Weiss R. M., Anderson M. E., Binkley P., Felder R. B., Carnes C. A., Band H., Hund T. J., Mohler P. J. (2012) Differential regulation of EHD3 in human and mammalian heart failure. J. Mol. Cell Cardiol. 52, 1183–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nishijima Y., Feldman D. S., Bonagura J. D., Ozkanlar Y., Jenkins P. J., Lacombe V. A., Abraham W. T., Hamlin R. L., Carnes C. A. (2005) Canine nonischemic left ventricular dysfunction. A model of chronic human cardiomyopathy. J. Card. Fail. 11, 638–644 [DOI] [PubMed] [Google Scholar]
- 42. Roger V. L., Go A. S., Lloyd-Jones D. M., Benjamin E. J., Berry J. D., Borden W. B., Bravata D. M., Dai S., Ford E. S., Fox C. S., Fullerton H. J., Gillespie C., Hailpern S. M., Heit J. A., Howard V. J., Kissela B. M., Kittner S. J., Lackland D. T., Lichtman J. H., Lisabeth L. D., Makuc D. M., Marcus G. M., Marelli A., Matchar D. B., Moy C. S., Mozaffarian D., Mussolino M. E., Nichol G., Paynter N. P., Soliman E. Z., Sorlie P. D., Sotoodehnia N., Turan T. N., Virani S. S., Wong N. D., Woo D., Turner M. B. (2012) Executive summary. Heart disease and stroke statistics-2012 update. A report from the American Heart Association. Circulation 125, 188–197 [DOI] [PubMed] [Google Scholar]
- 43. Baba S., Dun W., Cabo C., Boyden P. A. (2005) Remodeling in cells from different regions of the reentrant circuit during ventricular tachycardia. Circulation 112, 2386–2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lehnart S. E., Ackerman M. J., Benson D. W., Jr., Brugada R., Clancy C. E., Donahue J. K., George A. L., Jr., Grant A. O., Groft S. C., January C. T., Lathrop D. A., Lederer W. J., Makielski J. C., Mohler P. J., Moss A., Nerbonne J. M., Olson T. M., Przywara D. A., Towbin J. A., Wang L. H., Marks A. R. (2007) Inherited arrhythmias. A National Heart, Lung, and Blood Institute and Office of Rare Diseases workshop consensus report about the diagnosis, phenotyping, molecular mechanisms, and therapeutic approaches for primary cardiomyopathies of gene mutations affecting ion channel function. Circulation 116, 2325–2345 [DOI] [PubMed] [Google Scholar]
- 45. Longin S., Zwaenepoel K., Louis J. V., Dilworth S., Goris J., Janssens V. (2007) Selection of protein phosphatase 2A regulatory subunits is mediated by the C terminus of the catalytic subunit. J. Biol. Chem. 282, 26971–26980 [DOI] [PubMed] [Google Scholar]
- 46. Janssens V., Longin S., Goris J. (2008) PP2A holoenzyme assembly. In cauda venenum (the sting is in the tail). Trends Biochem. Sci. 33, 113–121 [DOI] [PubMed] [Google Scholar]
- 47. Chen J., Martin B. L., Brautigan D. L. (1992) Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science 257, 1261–1264 [DOI] [PubMed] [Google Scholar]
- 48. Anderson M. E., Brown J. H., Bers D. M. (2011) CaMKII in myocardial hypertrophy and heart failure. J. Mol. Cell. Cardiol. 51, 468–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Miura T., Miki T. (2009) GSK-3β, a therapeutic target for cardiomyocyte protection. Circ. J. 73, 1184–1192 [DOI] [PubMed] [Google Scholar]
- 50. Braz J. C., Gregory K., Pathak A., Zhao W., Sahin B., Klevitsky R., Kimball T. F., Lorenz J. N., Nairn A. C., Liggett S. B., Bodi I., Wang S., Schwartz A., Lakatta E. G., DePaoli-Roach A. A., Robbins J., Hewett T. E., Bibb J. A., Westfall M. V., Kranias E. G., Molkentin J. D. (2004) PKC-α regulates cardiac contractility and propensity toward heart failure. Nat. Med. 10, 248–254 [DOI] [PubMed] [Google Scholar]
- 51. Zhang R., Khoo M. S., Wu Y., Yang Y., Grueter C. E., Ni G., Price E. E., Jr., Thiel W., Guatimosim S., Song L. S., Madu E. C., Shah A. N., Vishnivetskaya T. A., Atkinson J. B., Gurevich V. V., Salama G., Lederer W. J., Colbran R. J., Anderson M. E. (2005) Calmodulin kinase II inhibition protects against structural heart disease. Nat. Med. 11, 409–417 [DOI] [PubMed] [Google Scholar]
- 52. Ling H., Zhang T., Pereira L., Means C. K., Cheng H., Gu Y., Dalton N. D., Peterson K. L., Chen J., Bers D., Brown J. H., Heller Brown J. (2009) Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J. Clin. Invest. 119, 1230–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kashef F., Li J., Wright P., Snyder J., Suliman F., Kilic A., Higgins R. S., Anderson M. E., Binkley P. F., Hund T. J., Mohler P. J. (2012) Ankyrin-B protein in heart failure, Identification of a new component of metazoan cardioprotection. J. Biol. Chem. 287, 30268–30281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nicolaou P., Kranias E. G. (2009) Role of PP1 in the regulation of Ca2+ cycling in cardiac physiology and pathophysiology. Front Biosci. 14, 3571–3585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Neumann J., Eschenhagen T., Jones L. R., Linck B., Schmitz W., Scholz H., Zimmermann N. (1997) Increased expression of cardiac phosphatases in patients with end-stage heart failure. J. Mol. Cell Cardiol. 29, 265–272 [DOI] [PubMed] [Google Scholar]
- 56. Mishra S., Gupta R. C., Tiwari N., Sharov V. G., Sabbah H. N. (2002) Molecular mechanisms of reduced sarcoplasmic reticulum Ca2+ uptake in human failing left ventricular myocardium. J. Heart Lung Transplant 21, 366–373 [DOI] [PubMed] [Google Scholar]
- 57. Huang B., Wang S., Qin D., Boutjdir M., El-Sherif N. (1999) Diminished basal phosphorylation level of phospholamban in the postinfarction remodeled rat ventricle: role of β-adrenergic pathway, Gi protein, phosphodiesterase, and phosphatases. Circ. Res. 85, 848–855 [DOI] [PubMed] [Google Scholar]
- 58. Gupta R. C., Mishra S., Rastogi S., Imai M., Habib O., Sabbah H. N. (2003) Cardiac SR-coupled PP1 activity and expression are increased and inhibitor 1 protein expression is decreased in failing hearts. Am. J. Physiol. Heart Circ. Physiol. 285, H2373–H2381 [DOI] [PubMed] [Google Scholar]
- 59. Pathak A., del Monte F., Zhao W., Schultz J. E., Lorenz J. N., Bodi I., Weiser D., Hahn H., Carr A. N., Syed F., Mavila N., Jha L., Qian J., Marreez Y., Chen G., McGraw D. W., Heist E. K., Guerrero J. L., DePaoli-Roach A. A., Hajjar R. J., Kranias E. G. (2005) Enhancement of cardiac function and suppression of heart failure progression by inhibition of protein phosphatase 1. Circ. Res. 96, 756–766 [DOI] [PubMed] [Google Scholar]
- 60. Yamada M., Ikeda Y., Yano M., Yoshimura K., Nishino S., Aoyama H., Wang L., Aoki H., Matsuzaki M. (2006) Inhibition of protein phosphatase 1 by inhibitor-2 gene delivery ameliorates heart failure progression in genetic cardiomyopathy. FASEB J. 20, 1197–1199 [DOI] [PubMed] [Google Scholar]
- 61. Linck B., Boknik P., Knapp J., Müller F. U., Neumann J., Schmitz W., Vahlensieck U. (1996) Effects of cantharidin on force of contraction and phosphatase activity in nonfailing and failing human hearts. Br. J. Pharmacol. 119, 545–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Neumann J., Bokník P., Herzig S., Schmitz W., Scholz H., Wiechen K., Zimmermann N. (1994) Biochemical and electrophysiological mechanisms of the positive inotropic effect of calyculin A, a protein phosphatase inhibitor. J. Pharmacol. Exp. Ther. 271, 535–541 [PubMed] [Google Scholar]
- 63. Neumann J., Herzig S., Boknik P., Apel M., Kaspareit G., Schmitz W., Scholz H., Tepel M., Zimmermann N. (1995) On the cardiac contractile, biochemical and electrophysiological effects of cantharidin, a phosphatase inhibitor. J. Pharmacol. Exp. Ther. 274, 530–539 [PubMed] [Google Scholar]
- 64. Neumann J., Boknik P., Herzig S., Schmitz W., Scholz H., Gupta R. C., Watanabe A. M. (1993) Evidence for physiological functions of protein phosphatases in the heart. Evaluation with okadaic acid. Am. J. Physiol. 265, H257–H266 [DOI] [PubMed] [Google Scholar]
- 65. Zhou J., Pham H. T., Ruediger R., Walter G. (2003) Characterization of the Aα and Aβ subunit isoforms of protein phosphatase 2A. Differences in expression, subunit interaction, and evolution. Biochem. J. 369, 387–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hemmings B. A., Adams-Pearson C., Maurer F., Müller P., Goris J., Merlevede W., Hofsteenge J., Stone S. R. (1990) α- and β-forms of the 65-kDa subunit of protein phosphatase 2A have a similar 39-amino acid repeating structure. Biochemistry 29, 3166–3173 [DOI] [PubMed] [Google Scholar]
- 67. Lüss H., Klein-Wiele O., Bokník P., Herzig S., Knapp J., Linck B., Müller F. U., Scheld H. H., Schmid C., Schmitz W., Neumann J. (2000) Regional expression of protein phosphatase type 1 and 2A catalytic subunit isoforms in the human heart. J. Mol. Cell Cardiol. 32, 2349–2359 10 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.