

Background: HIV-1 p17 binds heparin and heparan sulfate proteoglycans of the cell surface.
Results: Heparin/p17 interaction occurs through heparin sulfate groups and a linear basic motif of p17 N terminus, also involved in p17/CXCR1 interaction.
Conclusion: Targeting the basic motif inhibits p17-receptors interaction and consequent biological activities.
Significance: Heparin-like molecules represent template for the development of new treatments of p17-dependent/AIDS-associated pathologies.
Keywords: AIDS, Heparin, Heparin Binding Protein, HIV-1, Surface Plasmon Resonance (SPR)
Abstract
Once released by HIV+ cells, p17 binds heparan sulfate proteoglycans (HSPGs) and CXCR1 on leukocytes causing their dysfunction. By exploiting an approach integrating computational modeling, site-directed mutagenesis of p17, chemical desulfation of heparin, and surface plasmon resonance, we characterized the interaction of p17 with heparin, a HSPG structural analog, and CXCR1. p17 binds to heparin with an affinity (Kd = 190 nm) that is similar to those of other heparin-binding viral proteins. Two stretches of basic amino acids (basic motifs) are present in p17 N and C termini. Neutralization (Arg→Ala substitution) of the N-terminal, but not of the C-terminal basic motif, causes the loss of p17 heparin-binding capacity. The N-terminal heparin-binding motif of p17 partially overlaps the CXCR1-binding domain. Accordingly, its neutralization prevents also p17 binding to the chemochine receptor. Competition experiments demonstrated that free heparin and heparan sulfate (HS), but not selectively 2-O-, 6-O-, and N-O desulfated heparins, prevent p17 binding to substrate-immobilized heparin, indicating that the sulfate groups of the glycosaminoglycan mediate p17 interaction. Evaluation of the p17 antagonist activity of a panel of biotechnological heparins derived by chemical sulfation of the Escherichia coli K5 polysaccharide revealed that the highly N,O-sulfated derivative prevents the binding of p17 to both heparin and CXCR1, thus inhibiting p17-driven chemotactic migration of human monocytes with an efficiency that is higher than those of heparin and HS. Here, we characterized at a molecular level the interaction of p17 with its cellular receptors, laying the basis for the development of heparin-mimicking p17 antagonists.
Introduction
The matrix protein p17 plays important roles in HIV-1 life cycle (1) regulating both early and late stages of viral replication such as P55GAG transport to the membrane (2) and envelope glycoproteins incorporation into nascent virions (3). p17 is also released by HIV-infected cells, being detectable in plasma (4), brain (5), and lymph nodes (6) even in patients successfully treated with highly active antiretroviral therapy (6). Extracellular p17 deregulates the functions of many immune cells involved in AIDS pathogenesis (7): it reduces MIP-1α secretion in activated monocytes (8), induces activated T cell proliferation enhancing HIV replication (9), increases IFN-γ and TNF-α production decreasing the ability of IL-4 to down-regulate the secretion of such cytokines in activated peripheral blood mononuclear and natural killer cells (7, 10). Three distinct cellular receptors for p17 have been identified: the chemokine receptors CXCR1 and CXCR2, which mediate p17-driven monocyte migration (11) and endothelial cells proangiogenic activation (12), respectively, and heparan sulfate proteoglycans (HSPGs),2 tentatively associated to p17-driven cytokine up-regulation in CD4+ lymphocytes (13, 14).
HSPGs typically consist of a core protein and of glycosaminoglycan (GAG) chains made up of unbranched anionic polysaccharides resembling heparin and composed of repeating disaccharides units formed by sulfated uronic acid and hexosamine residues (15). They are expressed at densities ranging between 105 and 106/cell on the surface of almost all eukaryotic cells where they play co-receptor function for a wide array of heparin-binding cytokines. HSPGs, as well as their structural analog heparin, can be also found in a free form in the extracellular environment (16) where they act as antagonists, sequestering heparin-binding cytokines and hampering their cell surface interactions. The wide distribution and strong interactive capacity of HSPGs made them attractive adhesion molecules for viruses during their evolution. In effect, virus attachment to host cell is often mediated by their interaction with cellular HSPGs (17). Conversely, free heparin-like molecules can inhibit viral infection. Accordingly, a long list of heparin-like polyanions have been so far developed as effective antiviral compounds in vitro (18). However, the therapeutical exploitation of these compounds as antiviral drugs is limited by the fact that HSPGs act also as receptors for a wide array of cytokines involved in critical biological events such as cell growth and migration and the maintenance of normal tissue structure and function (16). These considerations call for the development of specific heparin-like compounds able to discriminate between viral and eukaryotic proteins. This, in turn, calls for the fine characterization of the interaction of each viral protein with heparin/HSPGs.
In general, the binding of heparin/HSPGs to viral proteins occurs through negatively charged sulfate groups of the GAG and clusters of positively charged amino acids of the protein also termed “basic motifs” (18). Both the sulfation degree and disposition of sulfate groups along the GAG chain are important in determining the interactive capacity (18). In heparin-binding proteins, unique or multiple basic motifs can be found composed of linear stretches of basic amino acids as in HIV-1 Tat or of conformational cluster of scattered basic amino acids that crowd together in the properly folded protein as in fibroblast growth factor 2 (FGF2) (19).
HIV-infected cells secrete three proteins (Tat, gp120, and p17) that, amazingly, share heparin-binding capacity. Tat/heparin interaction occurs with a dissociation constant (Kd) equal to 30–60 nm (20, 21) and is mainly mediated by a linear basic motif 49RKKRRQRRR57 located at the C terminus of the protein (22), with a minor contribution of three other basic amino acids (Lys12, Lys41, and Arg78) spatially enclosed in the native protein (23). At least some 2-O-, 6-O-, and N-positions along the GAG chain need to be sulfated to allow Tat binding (24), and a hexasaccharide is the minimal heparin length that retains Tat-binding capacity (25).
gp120 binds heparin with a Kd equal to 200–600 nm (26, 27). Distinct basic domain are present in the protein, among which the C-terminal “variable loop 3” (18, 28) is implicated in gp120 binding to cell surface HSPGs and consequent HIV infection (26). On the heparin chain, O-, but not N-sulfated groups are necessary for the binding to gp120 (29–31). An octa/decasaccharide is the minimal heparin size that retains gp120-binding capacity (32).
At variance with Tat and gp120, no data are available about the molecular bases of p17/heparin interaction. The structure of monomeric (33) and trimeric (34) p17 were determined, demonstrating highly similar three-dimensional structures (35). p17 folds into a compact core domain consisting of five α-helices and three stranded β-sheets. α-Helices 1–4 form a compact globular domain, whereas the C-terminal helix 5 is partially unfolded, projecting away from the protein, readily accessible for interactions such as that between p17 to P55GAG capsid domains. p17 possesses two basic motifs that may mediate its binding to heparin: in the monomer as well as in the trimer, they are both exposed on the protein surface, one in the N-terminal globular domain of the protein spanning residues 25–34, the other in its partially unfolded C-terminal tail spanning residues 109–115 (see Fig. 1, A and B). Both of the motifs are not involved in mediating oligomerization of p17, remaining exposed onto the protein surface also when it takes its trimeric form (see also Fig. 1B) (34, 35). Here, by integrating computational modeling, site directed mutagenesis of p17, chemical desulfation of heparin, and real time biomolecular interaction analysis by surface plasmon resonance (SPR), we characterized the interaction of p17 to heparin at a molecular level.
FIGURE 1.

Shown is the localization of basic motifs in monomeric (A) and trimeric (B) p17. The proteins are shown as Connolly surface. The chains are colored in green, yellow, and magenta. The N-terminal and C-terminal basic motifs are labeled in blue and cyan, respectively. Predicted binding modes between the N-terminal basic motif of p17 and heparin di- (C), tetra- (D), and hexasaccharide (E) are shown. Shown are stick oligosaccharides (green) and the side chains of p17 that interact with them (cyan). Protein is shown as a ribbon, and the hydrogen bonds are represented by magenta dotted lines. F, prediction of the binding mode of the heparin hexasaccharide at the three N-terminal sites. The modeled trimeric form of p17 is shown as Connolly surface, A chain, B chain, and C chain are colored in cyan, magenta, and yellow, respectively. The N-terminal basic motifs of each chain are colored in blue. The ligands are shown in the Corey, Pauling, and Koltun space filling-model.
MATERIALS AND METHODS
Molecular Modeling
The structures of monomeric p17 were obtained from the Protein Data Bank (PDB) code 1TAM. The structural models of the heparin di-, tetra-, and hexasaccharides were built using the PyMOL program based on their NMR solution structure (PDB code 1HPN). The results for the 1C4 conformers were here reported, but similar results were obtained with the 2SO conformers (data not shown). Docking of oligosaccharides to p17 was performed with Autodock (version 4.0) (36, 37), which allows flexibility in the heparin structure but uses a rigid body protein approximation to speed up the calculation. Two-step docking simulation was employed. First, a blind docking protocol was used on the monomeric form to predict the bound conformation with no a priori knowledge of binding site locations on the molecules (38, 39). To this aim, a grid box encompassing the entire p17 structure was generated to take all possible heparin binding sites into account. In a second set of experiments, a smaller grid, focused on the binding region identified in the blind docking was used to predict the binding mode of the heparin oligosaccharides and to optimize the ligand-protein interactions in the potential heparin binding site. In particular, affinity maps for all the atom types present, as well as an electrostatic map, were computed with a grid spacing of 0.375 Å. The search was carried out with the Lamarckian Genetic Algorithm, the number of generations, energy evaluations, and docking runs were set to 500,000, 2,500,000, and 100, respectively. Eventually, the evaluation with the lowest binding energy was used to analyze ligand pose and the interaction models of heparin oligosaccharides with the monomeric form of p17.
We also performed docking simulations of heparin with the trimeric p17 complex. Relevant to this, the only available three-dimensional structure of trimeric p17 was generated on a protein containing the N-terminal basic motif GKKQYKLKHI (Swiss Prot code P12497, PDB code 1HIW) (34), thus different from the p17 here used for experiments in vitro and for molecular modeling with the monomer containing the N-terminal basic motif GKKYKLKHI (Swiss Prot code P04585, PDB code 1TAM). Thus, a homology modeling approach was used to generate the three-dimensional structure of the trimeric form of the p17 studied experimentally. Briefly, the PDB code 1HIW was used as a template; sequence alignment of template and target (∼93%) was given as input to Modeler program (40) to build three-dimensional structures (Fig. 1F). The three-dimensional model was subjected to energy minimization using the Gromacs program (41) and evaluated using PROCHECK (42). The model obtained was submitted to docking simulations and grids focused on three N-terminal binding sites were used to predict the binding mode of heparin oligosaccharide in the relative positions.
All calculations were performed on a Linux Cluster of 32 nodes, each SuperMicro equipped with two processors: the 2.50 GHz INTEL(R) Quad-core and the 16 GB Xeon(R), for 256 total processors and 512 GB of RAM. Figures were realized with the PyMOL Molecular Graphics System (DeLano Scientific, San Carlos, CA).
Recombinant Proteins
The coding sequence of HIV-1 matrix protein p17 clade B isolate BH10 (amino acids 1–132) was amplified by PCR with specific primers, allowing the cloning of the p17 sequence into the BamH1 site of the expression vector pGEX-2T (GE Healthcare). p17 N-terminal Lys→Ala mutant, obtained by the substitution of lysines of the N-terminal basic motif with alanine, and the p17 C-terminal K→A mutant, obtained by the same mutations in the C-terminal basic motif, were cloned into the BamH1 site of the pGEX-2T vector using QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). p17Δ36, obtained by the deletion of the C-terminal sequence 97–132, was prepared as described (43). See Fig. 3A for a schematic representation of the p17 mutants. p17 proteins and the GST motif alone were purified as described (43). Human recombinant FGF2 was expressed in Escherichia coli and purified as described (44). Synthetic 86-amino acid HIV-1 Tat was from Xeptagen (Venezia, Italy). The absence of endotoxin contamination (<0.25 endotoxin units/ml) in protein preparations was assessed by Limulus amoebocyte assay (Associates of Cape Cod, Inc., East Falmouth, MA). Heat denaturation of proteins was obtained by incubation at 95 °C for 5 min.
FIGURE 3.

SPR analysis of the interaction of p17 mutants with heparin. A, schematic representation of the p17 mutants used in the present work. The N- and C-terminal basic motifs of p17 (white boxes) were deleted or neutralized (black boxes) by substituting positively charged lysine with alanine (underlined). Blank-subtracted sensorgrams showing the binding of p17 N-terminal (N-ter) Lys→Ala (500 nm) (B), p17 C-terminal (C-ter) Lys→Ala (500, 250, 125, 62.5, and 31.25 nm) (C) and p17Δ36 (950, 900, 800, 500, 250, and 100 nm) (D) to a heparin-coated sensorchip. The response (in RU) was recorded as a function of time.
Glycosaminoglycans
Type I HS was from Opocrin (Corlo, Italy). Conventional heparin (13.6 kDa) was obtained from a commercial batch preparation of unfractionated sodium heparin from beef mucosa (Laboratori Derivati Organici S.p.A., Milan, Italy) purified from contaminants (up to 95%) according to described methodologies. Selectively desulfated heparins were a gift of Dr. B. Casu (Ronzoni Institute) (45). Size-defined heparin-derived oligosaccharides were from Iduron (Manchester, UK). The capsular E. coli K5 polysaccharide has the same structure as the heparin precursor N-acetyl heparosan (46). K5 derivatives were obtained by N-deacetylation/N-sulfation and/or O-sulfation of a single batch of K5 polysaccharide as described (47). For further details on the GAGs used in this study, see Table 1.
TABLE 1.
Structural features of the GAGs utilized in the present study
| MWa | SO3−/COO− | Glc-NSO3− | Glc-6SO3− | IdoA-2SO3− | GlcA-OSO3−b | GlcA2,3SO3− | Unsulfated GlcA | |
|---|---|---|---|---|---|---|---|---|
| % | % | % | % | % | ||||
| Heparin/HS | ||||||||
| Unmodified | 13,600 | 2.14 | 78 | 77 | 59 | |||
| 2-O-Desulfated | 9,600 | 1.60 | 78 | 80 | <5 | |||
| 6-O-Desulfated | 10,300 | 1.50 | 100 | 0 | 50 | |||
| N-Desulfated/N-acetylated | 12,300 | 1.50 | 10 | 80 | 60 | |||
| Heparan sulfate | 18,000 | 1.17 | 42 | 44 | 31 | |||
| K5 polysaccharides | ||||||||
| Unsulfated | 30,000 | 0.00 | 0 | 0 | 0 | 0 | 100 | |
| K5NS | 15,000 | 1.00 | 100 | 0 | 0 | 0 | 100 | |
| K5OSL | 14,000 | 1.41 | 0 | 90 | <10 | 0 | >90 | |
| K5OSH | 11,000 | 3.77 | 0 | 100 | 0 | 100 | 0 | |
| K5NOSL | 13,000 | 1.70 | 100 | 90 | <10 | 0 | >90 | |
| K5NOSH | 15,000 | 3.84 | 100 | 100 | 30 | 70 | 0 | |
a MW, molecular weight.
b GlcA2SO3− or GlcA3SO3−.
SPR Binding Assay
SPR measurements were performed on a BIAcore X instrument (GE Healthcare). For the study of p17/heparin interaction, biotinylated heparin was immobilized onto a SA sensorchip containing preimmobilized streptavidin, allowing the immobilization of 159 resonance units (RU), equal to 11.7 fmol/mm2 of heparin. A sensorchip precoated with streptavidin alone was used to evaluate nonspecific binding and for blank subtraction (21). Increasing concentrations of p17 or of its mutants in 10 mm HEPES, pH 7.4 containing 150 mm NaCl, 3 mm EDTA, and 0.005% surfactant P20 (HBS-EP) were injected over the heparin or streptavidin surfaces for 4 min and then washed until dissociation. After each run, the sensorchip was regenerated by injection of 2.0 m NaCl in HBS-EP.
For the study of p17/CXCR1 interaction, anti-GST antibodies were immobilized onto a CM5 sensorchip using standard amine-coupling chemistry. Then, recombinant human CXCR1 with a C-terminal GST tag (10 μg/ml in 50 mm HEPES, pH 7.0, containing 0.01% cholesteryl hemisuccinate Tris salt, 0.1% CHAPS, and 0.33 mm synthetic phospholipid blend (dioleoyl) DOPC:DOPS (7:3 (w/w), Avanti polar lipids) was injected over the anti-GST surface, allowing the immobilization of 1,600 RU, equal to 25 fmol/mm2 of the receptor. A sensor chip coated with anti-GST antibodies was used as a negative control and for blank subtraction (11). Increasing concentrations of p17 or of its mutants in HBS-EP were injected over the CXCR1-coated surfaces for 15 s and then washed until dissociation.
For competition experiments, p17 (500 nm) and increasing concentrations of the different GAGs were preincubated for 10 min at 25 °C and then injected over the heparin or CXCR1 surfaces. Alternatively, p17 was injected over the sensor chips and allowed to reach an equilibrium binding. Then, increasing concentrations of the various GAGs were injected, and p17 detachment from the surfaces was evaluated. Similar results were obtained in the two experimental approaches (data not shown).
Kinetic parameters were calculated from the sensorgram overlays by using the nonlinear fitting single site model software package BIAevaluation (version 3.2). Only sensorgrams whose fitting gave χ2 values close to 10 were used (48). Also, Kd was calculated by being fitted with the proper form of Scatchard's equation for the plot of the bound RU at equilibrium versus the ligand concentration in solution. All fittings were performed by the software Solar, a least-square minimization procedure based on the Levemburg-Marquardt algorithm.
Isolation of Human Monocytes and Chemotaxis Assay
Blood was collected from healthy donors who gave informed consent according to the Helsinki Declaration. All procedures were done under sterile conditions using reagents prepared in endotoxin-free water for clinical use. Human monocytes were isolated from whole blood by Ficoll + Percoll gradient sedimentation (49) and resuspended in migration buffer (phosphate-buffered saline, pH 7.2, containing 1 mm CaCl2, 1 mm MgCl2, and 10% fetal calf serum). Monocyte migration was assessed in 3-μm pore-size Transwells (BD Biosciences). Aliquots (100 μl) of cells (2 × 106/ml) in migration buffer were added to the top well in the absence or in the presence of heparin, HS, or K5NOSH (at 1 and 10 μg/ml). Aliquots (600 μl) of medium with or without p17 (59 nm) or N-formyl-l-methionyl-l-leucyl-l-phenylalanine (fMLP, 10 nm) were added to the bottom well. Chemotaxis was performed for 90 min at 37 °C. Then, the filters were removed. After fixation with 1.5% paraformaldehyde, migrated cells were counted in five high-power fields by light microscopy at 10× magnification.
RESULTS
Molecular Docking Studies
To identify the heparin-binding site in the monomeric p17, we performed blind docking simulations in order to predict the binding mode and obtain an estimate of the total interaction energy of the complexes. For all three heparin oligosaccharides used (di-, tetra-, and hexasaccharide), the lowest energy docking was predicted to be located in the N-terminal basic motif of p17 instead of the C-terminal one, although both these motifs are exposed onto the protein surface both in its monomeric and trimeric form. Comparable results were obtained by using either the 1C4 or 2SO conformers of the three heparin oligosaccharides (data not shown). Thus, local docking simulations were carried out to predict the binding mode of the heparin oligosaccharide and to optimize the ligand-protein interactions in the potential heparin binding site identified. As shown in Fig. 1, C–F, and Table 2, p17 is predicted to interact with heparin by an extensive hydrogen-bonding network occurring mainly between the lysines of the N-terminal basic motif of p17 and the negatively charged sulfate groups of heparin. In particular, the heparin disaccharide may form five hydrogen bonds with residues Lys-27, Lys-28, and Lys-30 of p17; the heparin tetrasaccharide may form 10 hydrogen bonds with residues Lys-27, Lys-28, Lys-30, and Lys-32 localized at the beginning of the second α-helix; the heparin hexasaccharide may instead bind p17 with 12 hydrogen bonds involving the same residues of the tetrasaccharide plus residue Arg-39 located in the middle of the α-helix. Thus, heparin di- and tetrasaccharide may bind to basic amino acids that are contiguous in the basic motif of p17, whereas the heparin hexasaccharide is predicted to bind another residue (Arg-39) that in the folded structure is close to the linear basic motif but in the sequence is distant.
TABLE 2.
Docking results obtained with Autodock for heparin oligosaccharides binding to the N-terminal basic motif of monomeric p17
| Oligosaccharide probe molecules | Groups of heparin involved in hydrogen bond |
Total interaction energy of the complex | ||||
|---|---|---|---|---|---|---|
| 2-O-Sulfate | N-Sulfate | 6-O-Sulfate | Carboxyl | 3-O-Sulfate | ||
| kcal/mol | ||||||
| Disaccharide 1C4 | Lys27 | Lys28, Lys30 | Lys27 | −13.57 | ||
| No. of hydrogen bonds | 1 | 3 | 1 | |||
| Tetrasaccharide 1C4 | Lys28, Lys32 | Lys30 | Lys26 | Lys27 | Lys28 | −19.06 |
| No. of hydrogen bonds | 3 | 2 | 2 | 2 | 1 | |
| Hexasaccharide 1C4 | Lys27, Arg39 | Lys27 | Lys28, Lys30 | Lys32 | Lys32 | −22.28 |
| No. of hydrogen bonds | 4 | 1 | 4 | 2 | 1 | |
We also evaluated the binding mode of heparin oilgosaccharides with p17 in its trimeric form. As already described under “Materials and Methods,” the three-dimensional structure of the trimeric form of the p17 used experimentally (containing the N-terminal basic motif GKKYKLKHI) has not been resolved, prompting to achieve a three-dimensional homology model of its trimeric form by using as a template the available x-ray structure of a p17 trimer containing a N-terminal basic motif GKKQYKLKHI (Swiss Prot code P12497, PDB code 1HIW). As inferred by previous papers (34, 35), the putative heparin-binding N-terminal basic motif GKKYKLKHI remains exposed onto the p17 surface also in the trimeric form (Fig. 1B). Molecular docking of the hexasaccharide against modeled complex of p17 was performed on three N-terminal sites. The conformations and interactions of docked hexasaccharides in the binding sites of p17 are shown in Fig. 1F. Also, in the trimeric complex of p17, the residues involved in the hydrogen bonds network are the basic residues of the N-terminal motif, Arg-39, and sulfate groups of heparin. The structural analysis of the trimeric form complexed with the ligands suggests a hexasaccharide binding pocket of ∼26.5 Å long. The distance between the heparin binding pockets of chain A and B, between chain B and C, and between chain C and A are 26, 23.1, and 17.5 Å, respectively (Fig. 1F). These structural considerations suggest that, to interact with more than one binding site, the heparin chain should be composed of at least 18 saccharide units.
Biochemical Characterization of p17/Heparin Interaction
SPR was exploited to evaluate the capacity of p17 to bind sensorchip-immobilized heparin, a model that mimics the binding of proteins to cell surface-associated HSPGs (20). p17 interacts with immobilized heparin but not with streptavidin (Fig. 2A). When increasing concentrations of p17 were injected onto the heparin surface, the sensorgram overlay shown in Fig. 2B was obtained that allowed the calculation of association (kon) and dissociation (koff) rates and of Kd (as koff/kon ratio, Table 3). Also, equilibrium binding data were used to generate the saturation curve and Scatchard plot regression shown in Fig. 2C and used to calculate a Kd value independent from kinetic parameters (Table 3). In conclusion, p17 binds immobilized heparin with a Kd (136–190 nm) that is similar to those of other heparin-binding proteins (Table 3).
FIGURE 2.

SPR analysis of p17-heparin interaction. A, sensorgrams showing the binding of native p17 (500 nm) to a heparin- or streptavidin-coated sensorchip. B, blank-subtracted sensorgrams overlay showing the binding of increasing concentrations of native p17 (1000, 500, 250, 125, 62.5, and 31.25 nm) to a heparin-coated sensorchip. Black lines represent the experimental data. Red lines represent the fits. In A and B, the response (in RU) was recorded as a function of time. C, saturation curve obtained using the values of RU bound at equilibrium from injection of increasing concentrations of p17 onto a heparin-coated sensorchip. C, inset: Scatchard plot analysis of the equilibrium binding data shown in C. The correlation coefficient of the linear regression was equal to −0.96.
TABLE 3.
Binding parameters of the interaction of p17 and p17 mutants to heparin and CXCR1
kon and koff are reported. Kd value was either derived from the koff/kon ratio and by Scatchard plot analysis of the equilibrium binding data. The results shown are representative of other two that gave similar results. For a comparison, the binding parameters of native or heat-denatured FGF2 and Tat are reported. n.d., not determinable; n.c., not calculated.
| kon | koff | Kd(kon/koff) | Kd at equilibrium | |
|---|---|---|---|---|
| 1/ms | 1/s | m | m | |
| Control proteins-heparin | ||||
| Native FGF2 | 8.14 × 103 | 3.20 × 10−4 | 3.9 × 10−8 | n.c. |
| Heat-denatured FGF2 | n.d. | n.d. | n.d. | n.d. |
| Native Tat | 4.58 × 104 | 2.07 × 10−3 | 4.5 × 10−8 | n.c. |
| Heat-denatured Tat | 5.89 × 103 | 1.37 × 10−3 | 2.3 × 10−7 | n.c. |
| p17-heparin | ||||
| Native p17 | 3.39 × 105 | 0.0643 | 1.9 × 10−7 | 1.36 × 10−7 |
| Heat-denatured p17 | 4.32 × 105 | 0.217 | 5.03 × 10−7 | 4.23 × 10−7 |
| p17 N-terminal Lys→Ala | n.d. | n.d. | n.d. | n.d. |
| p17 C-terminal Lys→Ala | 8.14 × 105 | 0.247 | 3.05 × 10−7 | 1.02 × 10−6 |
| p17Δ36 | 1.70 × 104 | 1.72 × 10−3 | 1.01 × 10−7 | 3.13 × 10−7 |
| p17-CXCR1 | ||||
| Native p17 | 5.89 × 104 | 0.102 | 1.73 × 10−6 | n.c. |
| Heat-denatured p17 | n.d. | n.d. | n.d. | n.d. |
| p17 N-terminal Lys→Ala | n.d. | n.d. | n.d. | n.d. |
| p17 C-terminal Lys→Ala | 3.22 × 105 | 0.128 | 3.97 × 10−7 | n.c. |
To evaluate whether a proper three-dimensional configuration was necessary to p17 to bind heparin, the protein was incubated for 5 min at 95 °C and immediately analyzed in SPR for its capacity to bind to immobilized heparin. FGF2 and Tat were used as controls because their interactions with heparin were already demonstrated to depend on conformational and linear basic domain and motif, respectively (50, 51). As expected, incubation at 95 °C for 5 min causes the complete loss of FGF2 heparin-binding capacity (Table 3). At variance, it does not hamper the heparin-binding capacity of both Tat and p17, inducing only a decrease of the affinity of these interactions (Table 3). Important to note, p17 loses its capacity to bind to CXCR1 when used immediately after its heating (Table 3), indicating its effective unfolding. In conclusion, p17 interaction with heparin depends mainly on linear basic motifs rather than on conformational motifs.
Although p17 possess two distinct stretches of basic amino acids (25GKKKYKLKHI34 and 109NKSKKKA115) that may act as heparin-binding motifs, the blind docking simulations indicated that only the N-terminal one mediates p17 binding to heparin (see above). Thus, two p17 mutants were produced in which the lysines within the N- or C-terminal basic motifs were neutralized by substitution with alanine (p17 N-terminal Lys→Ala and p17 C-terminal Lys→Ala, respectively). Also, taking advantage of the unfolded nature of the C terminus of p17, we produced a deletion mutant (p17Δ36) lacking the last 36-amino acid portion containing the C-terminal basic motif (Fig. 3A). As shown in Fig. 3B, p17 N-terminal Lys→Ala completely loses its capacity to bind immobilized heparin, whereas p17 C-terminal Lys→Ala retains its heparin-binding capacity (Fig. 3C and Table 3). This latter result was paralleled by the observation that also the deletion mutant p17Δ36 retains its capacity to bind heparin (Fig. 3D and Table 3). In conclusion, the N-terminal but not the C-terminal basic motif of p17 mediates the interaction of the protein to heparin/HSPGs.
The capacity of heparin to bind to different proteins depends on its degree of sulfation, disposition of the sulfate groups and length of the saccharidic chain (18, 52). To identify the biochemical features of heparin involved in its binding to p17, we exploited a SPR competition assay already used to evaluate the HSPGs antagonist capacity of various polyanionic compounds (20). As shown in Fig. 4A, both heparin and HS displace p17 from its binding to sensorchip-immobilized heparin in a dose-dependent manner, with the former exerting a stronger inhibition in respect to the latter (ID50 = 18 and 80 nm, respectively). As HS is less sulfated than heparin (SO3−/COO− ratio = 0.93 and 2.14, respectively, see Table 1), it can be concluded that the degree of sulfation is important in determining the p17-binding capacity of heparin/HS.
FIGURE 4.

Effect of chemically modified heparins or size-defined oligosaccharides on the interaction of p17 to immobilized heparin. Selectively desulfated heparins (A) or size-defined heparin oligosaccharides (B) were evaluated for their capacity to inhibit the interaction of p17 with sensorchip-immobilized heparin. In A, the responses were plotted as percentage of the binding of p17 measured in the absence of free GAGs. Each point is the mean ± S.E. of three separate determinations. In B, the responses were plotted as ID50 of inhibition. The experiment shown is representative of another one that gave similar results. UMFH, unmodified heparin. B, inset, the logarithm of the potency (ID50) of the various size-defined heparin oligosaccharides in inhibiting the binding of p17 to immobilized heparin were plotted against their length (monomer units). The correlation coefficient of the linear regression was equal to −0.93.
Next, a series of selectively desulfated heparin (Table 1) was evaluated for their capacity to bind p17. Selective desulfation of 2-O, 6-O, and N positions abolishes the capacity of heparin to interact with p17 (Fig. 4A), indicating that all of the sulfate groups of heparin are important for its p17-binding capacity.
Finally, a series of size-defined heparin oligosaccharides were evaluated for their capacity to compete with sensorchip-immobilized full-length heparin for the binding to p17. As shown in Fig. 4C, at the doses tested, all of the oligosaccharides except the disaccharide compete with immobilized heparin for the binding to p17 with potencies that increase logarithmically with their size. Taken together, these results indicate that the p17-binding capacity of heparin is greatly influenced by the length of its saccharidic chain.
Biochemical Characterization of p17-CXCR1 Interaction
In addition to HSPGs, also CXCR1 acts as p17 receptor on leukocytes (11). Interestingly, the extracellular N-terminal domain of CXCR1 is highly negatively charged (53), and the N-terminal basic motif 25GKKKYKLKHI34 of p17 partially overlaps with the p17 receptor binding domain 9SGGELDRWEKIRLRPGGKKK28 (54). These observations prompted us to investigate whether neutralization of the basic amino acids of the N-terminal basic motif of p17 affected the interaction of the protein to the chemokine receptor. The interaction of wild type p17 with CXCR1 has been already characterized (11). Here, we observed that p17 N-terminal Lys→Ala, but not p17 C-terminal Lys→Ala, completely loses its capacity to bind to CXCR1 (Fig. 5 and Table 3), indicating that, in addition to heparin/HSPGs, the N-terminal basic motif of p17 is also required for its binding to CXCR1.
FIGURE 5.

SPR analysis of the interaction of p17 mutants with CXCR1. Blank-subtracted sensorgrams showing the binding of wild type p17 (continuous line), p17 C-terminal (C-ter) Lys→Ala (dashed line), and p17 N-terminal (N-ter) Lys→Ala (dotted line) (all at 2 μm) to a CXCR1-coated sensorchip. The response in RU was recorded as a function of time.
Biotechnological Heparins as p17 Antagonists
K5 derivatives produced by chemical sulfation of N- or O-positions are endowed with defined sulfation patterns and with the capacity to hamper different biological activities of both Tat and gp120 (31, 55). Here, we assessed the p17-binding and antagonist capacity of a panel of K5 derivatives including the following: N-sulfated K5 (K5NS), O-sulfated K5 with low and high degrees of sulfation (K5OSL and K5OSH) and N,O-sulfated K5 with low and high degrees of sulfation (K5NOSL and K5NOSH) (Tab. 1). As shown in Fig. 6A, K5NOSH inhibits the binding of p17 to immobilized heparin with a potency that is higher than that of unmodified heparin (ID50 = 2.5 and 18 nm, respectively). K5NS, K5OSL, K5NOSL and K5OSH are less effective (ID50 = 30.0, 60.0, 56.0 and 70.0 nm respectively). If compared with the other sulfated derivatives, unsulfated K5 exerts a very limited inhibition (less than 40%) when assayed at 100 nm, suggesting that the contribution of the backbone sugar in p17 interaction is limited in respect to the contribution of sulfate groups. Except for K5NS and K5OSH (discussed below), a clear correlation exists between the SO3−/COO− ratio and the p17-heparin inhibitory capacity of the GAGs tested (Fig. 6A, inset), further sustaining the importance of the sulfation degree in the p17-binding capacity of heparin/HSPGs.
FIGURE 6.

Effect of biotechnological heparins on the interaction of p17 to its receptors. K5 derivatives were evaluated for their capacity to inhibit the interaction of p17 with heparin (A) or CXCR1 (B) immobilized to a sensorchip. The responses were plotted as percentage of the binding of p17 measured in the absence of any free GAG. Each point is the mean ± S.E. of three separate determinations. A, inset, the potency (ID50) of the various GAGs in inhibiting the binding of p17 to immobilized heparin were plotted against their SO3−/COO− ratio. The correlation coefficient of the linear regression was equal to −0.96.
We then evaluated the ability of the K5 derivatives to inhibit the interaction of p17 to surface-immobilized CXCR1. Preliminary assays demonstrated that the GAGs tested do not bind directly to sensorchip-immobilized CXCR1 (data not shown). As shown in Fig. 6B, K5NOSH inhibits p17-CXCR1 interaction with the highest efficiency (ID50 = 2.6 nm) followed by K5NS, K5OSL, K5NOSL, and K5OSH (ID50 = 48.0, 57.0, and 90.0 nm, respectively). Instead, K5OSL, and unsulfated K5 were ineffective. Heparin and HS are weak inhibitors of the p17-CXCR1 interaction (ID50 = 70.0 and 250.0 nm, respectively). There is no statistical correlation between the SO3−/COO− ratio and the p17-CXCR1 inhibitory capacity of the various GAGs tested (data not shown).
Both HSPGs and CXCR1 have been implicated in p17-induced activation of leukocytes (11, 14). Because K5NOSH inhibits the interaction of p17 with both heparin and CXCR1, we evaluated its capacity to prevent p17-dependent migration of monocytes. Heparin and HS, which efficiently inhibit p17 binding to heparin but not to CXCR1, were used as controls. As shown in Fig. 7, K5NOSH efficiently inhibits monocyte migration to p17 both at 1 and 10 μg/ml, whereas heparin shows a significant reduced inhibitory potency, as it is active only at 10 μg/ml. HS was ineffective, however, at both the concentrations tested.
FIGURE 7.
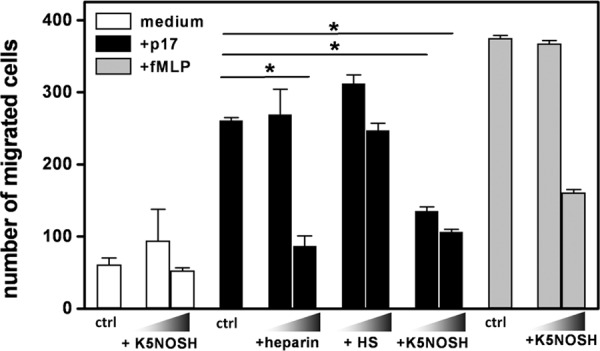
Effect of heparin, HS and K5NOSH on monocyte migration induced by p17. Monocytes in the absence (control, ctrl) or in the presence of heparin, HS and K5NOSH (all at 1 and 10 μg/ml) were added to the top well, whereas medium alone or containing p17 (59 nm) or fMLP (10 nm) was added to the bottom well. Bars represent the mean ± S.D. of three independent experiments performed in duplicate. Statistical analysis was performed using GraphPad Prism software (version 5, GraphPad Software, Inc.) and one-way analysis of varaince and Bonferroni's post test was used to compare data. *, p < 0.01 statistically different compared with p17-stimulated cells. NT, not treated cells.
To exclude that K5NOSH aspecifically inhibit cell migration, the assay was repeated with fMLP instead of p17. As shown in Fig. 7, K5NOSH does not affect fMLP-driven migration of monocytes when tested at 1 μg/ml, exerting a 50% inhibition only at doses ten times higher. In addition, K5NOSH does not affect the basal monocytes migration measured in the presence of medium alone.
DISCUSSION
HIV-infected cells release Tat, gp120, and p17 proteins that, acting in a cytokine-like manner, target uninfected leukocytes (56, 57), neurons (58–60), and endothelial cells (20, 60, 61) inducing various biological effects that, in turn, favor viral spread and the onset of AIDS-associated diseases. Some of these effects depend on the interaction of the viral proteins with HSPGs expressed on the surface of target cells (18, 52, 62), making this class of receptors a promising nodal targets for the development of “multitarget” anti-HIV drugs. This aim requires the fine characterization of the molecular bases of the interaction of the viral proteins with heparin/HSPGs, a study that has been already performed for Tat and gp120, leading to the development of heparin-like compounds endowed with gp120 and/or Tat antagonist capacity (18, 52, 62). Differently, the characterization of the interaction of p17 with heparin/HSPGs has not been performed yet. Here, we report about the biochemical features of p17 and of heparin that govern their interaction.
p17 unfolding does not hamper its binding to heparin, causing only a 3-fold reduction of the affinity of the interaction. Interestingly, a similar behavior has been observed also for Tat, whose interaction with heparin depends mainly on a 49RKKRRQRRR57 linear basic motif with additional but limited contribution given by three other spatially enclosed basic amino acids (Lys12, Lys41, and Arg78) (22, 23). It is thus tentative to hypothesize that also for p17, the main contribution to heparin interaction derives from linear basic motifs, with a minor role played by tridimensional structures. At variance, p17 unfolding hampers its binding to CXCR1 indicating that, for this interaction, the N-terminal basic domain (along with additional structures) need to be presented to the chemokine receptor in a proper tridimensional conformation.
In effect, p17 possess two linear basic motifs: the first in its N-terminal globular part, the second in its partially unfolded C-terminal tail, a region readily accessible to interactions both in the monomeric and trimeric forms of p17. Nevertheless, neutralization or deletion of the C-terminal basic motif does not abolish the heparin-binding capacity of p17, although neutralization of the N-terminal basic motif causes a complete loss of p17 heparin-binding capacity, indicating that this motif is the main responsible for the interaction of p17 with the GAG. Interestingly, independent studies have revealed that the N-terminal but not the C-terminal basic motif of p17 is required to other p17 biological activities, such as HIV particle production and infectivity (3, 63).
As already mentioned, however, heat denaturation induces a 3-fold decrease of the p17 affinity for heparin, suggesting that, in addition to the linear N-terminal basic motif, additional points of interaction with the GAG may originate after p17 folding. In effect, molecular docking studies predicted that the binding of heparin to p17 is characterized by an extensive hydrogen-bonding network of interactions occurring mainly between the negatively charged groups of heparin and the contiguous lysines of the N-terminal basic motif. However, an additional contribution to the interaction seems to be given by an arginine localized at position 39 in the second α-helix that is distant in the sequence but close to the stretch of lysines in the folded structure. Thus, heat denaturation could unfold the α helix, distancing Arg39 from the N-terminal basic motif, reducing the width of the zone of interaction and thus the affinity of the binding. Further site-directed mutagenesis experiments and SPR analyses are required to formally prove this possibility.
Different biochemical features of heparin impact its capacity to bind p17. (i) Sulfate groups, rather than the carbonic backbone of heparin, mediate its interaction with p17. Indeed, unsulfated K5 polysaccharide shows a very limited p17 binding capacity. Also, among the GAGs tested, a correlation exists between their sulfation degree and their capacity to inhibit p17/heparin interaction. (ii) Other than the sulfation degree, also the specific disposition of the sulfate groups along the heparin chain impacts its p17-binding capacity: in effect, the p17 antagonist capacity of K5NS is higher than that of K5OSL despite its lower SO3−/COOH (1.0 and 1.41, respectively, Table 1). Also, K5OSH and K5OSL exert a similar p17 antagonist activity despite the fact that the former is endowed with a SO3−COOH ratio that is three times higher that the latter (3.77 and 1.41, respectively (Table 1 and Fig. 4B). Thus, N-sulfate groups seem to be particularly important for the interaction of K5 derivatives with p17, whereas an increase in O-sulfation does not contribute further to their p17-binding capacity. Accordingly, by plotting the percent of sulfation of the different positions in the GAGs (see Table 1) versus their capacity to inhibit the binding of p17 to immobilized heparin, a moderate correlation (r = −0.70) can be calculated only for the Glc-NSO3− position (data not shown). (iii) Epimerization of the GAGs decreases their p17-binding capacity. Indeed, non-epimerized K5NS is a stronger p17 binder than epimerized 6-O- and 2-O-desulfated heparins, despite these two latter GAGs retain N-sulfation and a higher sulfation degree (SO3−/COOH = 1.0 for K5NS and to 1.5 for the two desulfated heparins, see Table 1). (iv) The p17-binding capacity of heparin depends on the length of its saccharidic chain. Indeed, size-defined heparin oligosaccharides, all endowed with a similar degree and disposition of sulfate groups, compete with immobilized heparin for the binding to p17 with potencies that increase logarithmically with the length of their saccharidic chain. Actually, in the competition binding assay, the hexa- and tetra-, but not the disaccharide prevents p17 binding to immobilized heparin. However, in a direct binding assay with p17 immobilized to the sensorchip, we were able to demonstrate that also the disaccharide binds the protein (data not shown), indicating that its lack of effect in the competition assay is due to its low inhibitory potency and the dilution of the sample available. In conclusion, the results from the binding assays support the data from computational modeling that predict that also very short heparin sequences can bind to p17.
According to the results of the molecular docking models, a heparin hexasaccharide accommodates a single p17 basic motif (Fig. 1). However, docking simulations of the hexasaccharide against the trimeric p17 predict a length of ∼26.5 Å of the N-terminal binding site and a distance between two binding sites of ∼23 Å, suggesting that a heparin chain should be composed of at least 18 monosaccharides to interact simultaneously with more than one heparin binding pockets favoring cooperative p17 bindings and its oligomerization, as already demonstrated for HIV-1 Tat and other chemokines (25, 64). In effect, we observed that the p17 antagonist capacity of heparin oligosaccharides increases logarithmically with their length (Fig. 4B) and that full-length heparin effectively induces p17 oligomerization.3 Further computational studies with longer heparin chains should be needed to appropriately characterize the interaction of heparin with the trimeric p17 complex.
p17 binds heparin with an affinity (Kd = 190 nm) that is similar to that of gp120 (Kd = 200–600 nm) and only three times lower than that of Tat (Kd = 30–60 nm), two HIV proteins that exploit HSPGs to mediate important biological effects (see Introduction). On the other hand, p17 binds to HSPGs on leukocytes, and although no formal demonstration has been provided, this interaction has been tentatively associated to the modulation of inflammatory cytokines expression (14). Even if the possibility that p17-HSPGs interaction directly induces some biological activity in the cell cannot be ruled out, it is likely that HSPGs may instead act as typical co-receptors, which, by binding p17, cause its conformational modifications/oligomerization that, in turn, ameliorate its binding to signaling receptors such as CXCR1. Relevant to this point, SPR analysis revealed that p17 binds both CXCR1 and heparin with Kd values that are significantly higher that the concentrations required to induce leukocyte migration (11), suggesting that, in vivo, the “productive” interaction of p17 with cells consists of the formation of a p17-HSPG-CXCR1 ternary complex, in which the affinity of the interactions is increased, as already demonstrated for other heparin-binding cytokines (65). This possibility is also sustained by the observation that in the trimeric form of p17 the N-terminal basic motif remains exposed onto the protein surface (34), readily accessible to receptors for multiple interactions. Further experiments are required to clarify the complex relationship existing between p17, HSPGs and chemochine receptors at the surface of target cells.
The involvement of p17 N-terminal basic motif in the binding to both heparin and CXCR1 is not surprising. Indeed, the basic motif of Tat is involved in the binding to HSPGs, integrins and VEGFR2 (61). Interestingly, selected K5 derivatives prevent Tat interaction with all these receptors, inhibiting the consequent biological activities (55). Here, we have shown that K5NOSH prevents the binding of p17 to both HSPGs and CXCR1, inhibiting p17-driven monocyte chemotaxis. Thus, the basic motif of p17 (as well as those of other viral proteins) can be considered as nodal motifs implicated in multiple interactions, thus emerging as preferential aim for the development of multitarget drugs interfering with different receptors (18). Finally, it is also worth noting that K5 derivatives are able to interfere with other HIV proteins (Tat and gp120, see above), suggesting the possibility to use K5 derivatives as template for the development of drugs endowed with an interesting anti-HIV multitarget action, directed against both different receptors of a given HIV protein and against different HIV proteins (18).
Acknowledgments
We thank P. Bergese and L. Ravelli for Solar (the software for Langmuir analysis), M. Moscatelli for the setting up of computational studies, and G. Zoppetti for helpful discussion.
This work was supported by the Istituto Superiore di Sanita (AIDS Grants 40H.51 (to M. R.) and 40G.16 (to A. C.)), the Italian Ministry of University and Scientific Research (to M. R. and A. C.), the Bonino-Pulejo Foundation (Messina, Italy; to A. C.), the Accordo Quadro RL-CNR “Nanoscienze per materiali e applicazioni biomediche,” the Interomics Flagship Project, and Italian Ministry of University and Scientific Research Fondo per gli Investimenti della Ricerca di Base-Protocollo HIRMA Grant RBAP11YS7K (to L. M.).
A. Bugatti, C. Giagulli, P. D'ursi, A. Caruso, and M. Rusnati, manuscript in preparation.
- HSPG
- heparan sulfate proteoglycan
- FGF2
- fibroblast growth factor 2
- HS
- heparan sulfate
- RU
- resonance units
- SPR
- surface plasmon resonance
- GAG
- glycosaminoglycan
- PDB
- Protein Data Bank
- fMLP
- N-formyl-l-methionyl-l-leucyl-l-phenylalanine
- K5NS
- N-sulfated K5.
REFERENCES
- 1. Fiorentini S., Riboldi E., Facchetti F., Avolio M., Fabbri M., Tosti G., Becker P. D., Guzman C. A., Sozzani S., Caruso A. (2008) HIV-1 matrix protein p17 induces human plasmacytoid dendritic cells to acquire a migratory immature cell phenotype. Proc. Natl. Acad. Sci. U.S.A. 105, 3867–3872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bryant M., Ratner L. (1990) Myristoylation-dependent replication and assembly of human immunodeficiency virus 1. Proc. Natl. Acad. Sci. U.S.A. 87, 523–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cannon P. M., Matthews S., Clark N., Byles E. D., Iourin O., Hockley D. J., Kingsman S. M., Kingsman A. J. (1997) Structure-function studies of the human immunodeficiency virus type 1 matrix protein, p17. J. Virol. 71, 3474–3483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Budka H. (1990) Human immunodeficiency virus (HIV) envelope and core proteins in CNS tissues of patients with the acquired immune deficiency syndrome (AIDS). Acta Neuropathol. 79, 611–619 [DOI] [PubMed] [Google Scholar]
- 5. Popovic M., Tenner-Racz K., Pelser C., Stellbrink H. J., van Lunzen J., Lewis G., Kalyanaraman V. S., Gallo R. C., Racz P. (2005) Persistence of HIV-1 structural proteins and glycoproteins in lymph nodes of patients under highly active antiretroviral therapy. Proc. Natl. Acad. Sci. U.S.A. 102, 14807–14812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fiorentini S., Giagulli C., Caccuri F., Magiera A. K., Caruso A. (2010) HIV-1 matrix protein p17: a candidate antigen for therapeutic vaccines against AIDS. Pharmacol. Ther. 128, 433–444 [DOI] [PubMed] [Google Scholar]
- 7. De Francesco M. A., Baronio M., Fiorentini S., Signorini C., Bonfanti C., Poiesi C., Popovic M., Grassi M., Garrafa E., Bozzo L., Lewis G. K., Licenziati S., Gallo R. C., Caruso A. (2002) HIV-1 matrix protein p17 increases the production of proinflammatory cytokines and counteracts IL-4 activity by binding to a cellular receptor. Proc. Natl. Acad. Sci. U.S.A. 99, 9972–9977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. De Francesco M. A., Poiesi C., Ricotta D., Manca N. (2006) HIV p17 reverses the anti-inflammatory activity of IL-4 on IL-15 stimulated monocytes and modulates their ability to secrete MIP-1 α. Virus Res. 118, 170–177 [DOI] [PubMed] [Google Scholar]
- 9. De Francesco M. A., Caruso A., Fallacara F., Canaris A. D., Dima F., Poiesi C., Licenziati S., Corulli M., Martinelli F., Fiorentini S., Turano A. (1998) HIV p17 enhances lymphocyte proliferation and HIV-1 replication after binding to a human serum factor. Aids 12, 245–252 [DOI] [PubMed] [Google Scholar]
- 10. Vitale M., Caruso A., De Francesco M. A., Rodella L., Bozzo L., Garrafa E., Grassi M., Gobbi G., Cacchioli A., Fiorentini S. (2003) HIV-1 matrix protein p17 enhances the proliferative activity of natural killer cells and increases their ability to secrete proinflammatory cytokines. Br. J. Haematol. 120, 337–343 [DOI] [PubMed] [Google Scholar]
- 11. Giagulli C., Magiera A. K., Bugatti A., Caccuri F., Marsico S., Rusnati M., Vermi W., Fiorentini S., Caruso A. (2012) HIV-1 matrix protein p17 binds to the IL-8 receptor CXCR1 and shows IL-8-like chemokine activity on monocytes through Rho/ROCK activation. Blood 119, 2274–2283 [DOI] [PubMed] [Google Scholar]
- 12. Caccuri F., Giagulli C., Bugatti A., Benetti A., Alessandri G., Ribatti D., Marsico S., Apostoli P., Slevin M. A., Rusnati M., Guzman C. A., Fiorentini S., Caruso A. (2012) HIV-1 matrix protein p17 promotes angiogenesis via chemokine receptors CXCR1 and CXCR2. Proc. Natl. Acad. Sci. U.S.A. 109, 14580–14585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Poiesi C., De Francesco M. A., Baronio M., Manca N. (2008) HIV-1 p17 binds heparan sulfate proteoglycans to activated CD4(+) T cells. Virus Res. 132, 25–32 [DOI] [PubMed] [Google Scholar]
- 14. De Francesco M. A., Baronio M., Poiesi C. (2011) HIV-1 p17 matrix protein interacts with heparan sulfate side chain of CD44v3, syndecan-2, and syndecan-4 proteoglycans expressed on human activated CD4+ T cells affecting tumor necrosis factor α and interleukin 2 production. J. Biol. Chem. 286, 19541–19548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lindahl U., Lidholt K., Spillmann D., Kjellén L. (1994) More to “heparin” than anticoagulation. Thromb. Res. 75, 1–32 [DOI] [PubMed] [Google Scholar]
- 16. Turnbull J., Powell A., Guimond S. (2001) Heparan sulfate: decoding a dynamic multifunctional cell regulator. Trends Cell Biol. 11, 75–82 [DOI] [PubMed] [Google Scholar]
- 17. Spillmann D. (2001) Heparan sulfate: anchor for viral intruders? Biochimie 83, 811–817 [DOI] [PubMed] [Google Scholar]
- 18. Rusnati M., Vicenzi E., Donalisio M., Oreste P., Landolfo S., Lembo D. (2009) Sulfated K5 Escherichia coli polysaccharide derivatives: A novel class of candidate antiviral microbicides. Pharmacol. Ther. 123, 310–322 [DOI] [PubMed] [Google Scholar]
- 19. Coltrini D., Rusnati M., Zoppetti G., Oreste P., Grazioli G., Naggi A., Presta M. (1994) Different effects of mucosal, bovine lung and chemically modified heparin on selected biological properties of basic fibroblast growth factor. Biochem. J. 303, 583–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bugatti A., Urbinati C., Ravelli C., De Clercq E., Liekens S., Rusnati M. (2007) Heparin-mimicking sulfonic acid polymers as multitarget inhibitors of human immunodeficiency virus type 1 Tat and gp120 proteins. Antimicrob. Agents Chemother. 51, 2337–2345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rusnati M., Urbinati C., Caputo A., Possati L., Lortat-Jacob H., Giacca M., Ribatti D., Presta M. (2001) Pentosan polysulfate as an inhibitor of extracellular HIV-1 Tat. J. Biol. Chem. 276, 22420–22425 [DOI] [PubMed] [Google Scholar]
- 22. Goldstein G. (1996) HIV-1 Tat protein as a potential AIDS vaccine. Nat. Med. 2, 960–964 [DOI] [PubMed] [Google Scholar]
- 23. Ai J., Xin X., Zheng M., Wang S., Peng S., Li J., Wang L., Jiang H., Geng M. (2008) A triad of lys12, lys41, arg78 spatial domain, a novel identified heparin binding site on tat protein, facilitates tat-driven cell adhesion. PLoS One 3, e2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rusnati M., Coltrini D., Oreste P., Zoppetti G., Albini A., Noonan D., d'Adda di Fagagna F., Giacca M., Presta M. (1997) Interaction of HIV-1 Tat protein with heparin. Role of the backbone structure, sulfation, and size. J. Biol. Chem. 272, 11313–11320 [DOI] [PubMed] [Google Scholar]
- 25. Rusnati M., Tulipano G., Spillmann D., Tanghetti E., Oreste P., Zoppetti G., Giacca M., Presta M. (1999) Multiple interactions of HIV-I Tat protein with size-defined heparin oligosaccharides. J. Biol. Chem. 274, 28198–28205 [DOI] [PubMed] [Google Scholar]
- 26. Moulard M., Lortat-Jacob H., Mondor I., Roca G., Wyatt R., Sodroski J., Zhao L., Olson W., Kwong P. D., Sattentau Q. J. (2000) Selective interactions of polyanions with basic surfaces on human immunodeficiency virus type 1 gp120. J. Virol. 74, 1948–1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Crublet E., Andrieu J. P., Vivès R. R., Lortat-Jacob H. (2008) The HIV-1 envelope glycoprotein gp120 features four heparan sulfate binding domains, including the co-receptor binding site. J. Biol. Chem. 283, 15193–15200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ping L. H., Nelson J. A., Hoffman I. F., Schock J., Lamers S. L., Goodman M., Vernazza P., Kazembe P., Maida M., Zimba D., Goodenow M. M., Eron J. J., Jr., Fiscus S. A., Cohen M. S., Swanstrom R. (1999) Characterization of V3 sequence heterogeneity in subtype C human immunodeficiency virus type 1 isolates from Malawi: underrepresentation of X4 variants. J. Virol. 73, 6271–6281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lopalco L., Ciccomascolo F., Lanza P., Zoppetti G., Caramazza I., Leoni F., Beretta A., Siccardi A. G. (1994) Anti-HIV type 1 properties of chemically modified heparins with diminished anticoagulant activity. AIDS Res. Hum. Retroviruses 10, 787–793 [DOI] [PubMed] [Google Scholar]
- 30. Rider C. C., Coombe D. R., Harrop H. A., Hounsell E. F., Bauer C., Feeney J., Mulloy B., Mahmood N., Hay A., Parish C. R. (1994) Anti-HIV-1 activity of chemically modified heparins: correlation between binding to the V3 loop of gp120 and inhibition of cellular HIV-1 infection in vitro. Biochemistry 33, 6974–6980 [DOI] [PubMed] [Google Scholar]
- 31. Vicenzi E., Gatti A., Ghezzi S., Oreste P., Zoppetti G., Poli G. (2003) Broad spectrum inhibition of HIV-1 infection by sulfated K5 Escherichia coli polysaccharide derivatives. Aids 17, 177–181 [DOI] [PubMed] [Google Scholar]
- 32. Vivès R. R., Imberty A., Sattentau Q. J., Lortat-Jacob H. (2005) Heparan sulfate targets the HIV-1 envelope glycoprotein gp120 coreceptor binding site. J. Biol. Chem. 280, 21353–21357 [DOI] [PubMed] [Google Scholar]
- 33. Matthews S., Barlow P., Clark N., Kingsman S., Kingsman A., Campbell I. (1995) Refined solution structure of p17, the HIV matrix protein. Biochem. Soc. Trans. 23, 725–729 [DOI] [PubMed] [Google Scholar]
- 34. Hill C. P., Worthylake D., Bancroft D. P., Christensen A. M., Sundquist W. I. (1996) Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: implications for membrane association and assembly. Proc. Natl. Acad. Sci. U.S.A. 93, 3099–3104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Massiah M. A., Worthylake D., Christensen A. M., Sundquist W. I., Hill C. P., Summers M. F. (1996) Comparison of the NMR and X-ray structures of the HIV-1 matrix protein: evidence for conformational changes during viral assembly. Protein Sci. 5, 2391–2398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Morris G. M., Goodsell D. S., Huey R., Olson A. J. (1996) Distributed automated docking of flexible ligands to proteins: parallel applications of AutoDock 2.4. J. Comput. Aided Mol. Des. 10, 293–304 [DOI] [PubMed] [Google Scholar]
- 37. Huey R., Morris G. M., Olson A. J., Goodsell D. S. (2007) A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 28, 1145–1152 [DOI] [PubMed] [Google Scholar]
- 38. Hetényi C., van der Spoel D. (2002) Efficient docking of peptides to proteins without prior knowledge of the binding site. Protein Sci. 11, 1729–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hetényi C., van der Spoel D. (2006) Blind docking of drug-sized compounds to proteins with up to a thousand residues. FEBS Lett. 580, 1447–1450 [DOI] [PubMed] [Google Scholar]
- 40. Sali A., Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 41. Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A. E., Berendsen H. J. (2005) GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 [DOI] [PubMed] [Google Scholar]
- 42. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., Thornton J. M. (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 43. Giagulli C., Marsico S., Magiera A. K., Bruno R., Caccuri F., Barone I., Fiorentini S., Andò S., Caruso A. (2011) Opposite effects of HIV-1 p17 variants on PTEN activation and cell growth in B cells. PLoS One 6, e17831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Isacchi A., Statuto M., Chiesa R., Bergonzoni L., Rusnati M., Sarmientos P., Ragnotti G., Presta M. (1991) A six-amino acid deletion in basic fibroblast growth factor dissociates its mitogenic activity from its plasminogen activator-inducing capacity. Proc. Natl. Acad. Sci. U.S.A. 88, 2628–2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Inoue Y., Nagasawa K. (1976) Selective N-desulfation of heparin with dimethyl sulfoxide containing water or methanol. Carbohydr. Res. 46, 87–95 [DOI] [PubMed] [Google Scholar]
- 46. Casu B., Grazioli G., Razi N., Guerrini M., Naggi A., Torri G., Oreste P., Tursi F., Zoppetti G., Lindahl U. (1994) Heparin-like compounds prepared by chemical modification of capsular polysaccharide from E. coli K5. Carbohydr. Res. 263, 271–284 [DOI] [PubMed] [Google Scholar]
- 47. Leali D., Belleri M., Urbinati C., Coltrini D., Oreste P., Zoppetti G., Ribatti D., Rusnati M., Presta M. (2001) Fibroblast growth factor-2 antagonist activity and angiostatic capacity of sulfated Escherichia coli K5 polysaccharide derivatives. J. Biol. Chem. 276, 37900–37908 [DOI] [PubMed] [Google Scholar]
- 48. Khalifa M. B., Choulier L., Lortat-Jacob H., Altschuh D., Vernet T. (2001) BIACORE data processing: an evaluation of the global fitting procedure. Anal. Biochem. 293, 194–203 [DOI] [PubMed] [Google Scholar]
- 49. Fontana L., Giagulli C., Minuz P., Lechi A., Laudanna C. (2001) 8-Iso-PGF2 α induces β 2-integrin-mediated rapid adhesion of human polymorphonuclear neutrophils: a link between oxidative stress and ischemia/reperfusion injury. Arterioscler. Thromb. Vasc. Biol. 21, 55–60 [DOI] [PubMed] [Google Scholar]
- 50. Coltrini D., Rusnati M., Zoppetti G., Oreste P., Isacchi A., Caccia P., Bergonzoni L., Presta M. (1993) Biochemical bases of the interaction of human basic fibroblast growth factor with glycosaminoglycans. New insights from trypsin digestion studies. Eur. J. Biochem. 214, 51–58 [DOI] [PubMed] [Google Scholar]
- 51. Rusnati M., Tulipano G., Urbinati C., Tanghetti E., Giuliani R., Giacca M., Ciomei M., Corallini A., Presta M. (1998) The basic domain in HIV-1 Tat protein as a target for polysulfonated heparin-mimicking extracellular Tat antagonists. J. Biol. Chem. 273, 16027–16037 [DOI] [PubMed] [Google Scholar]
- 52. Rusnati M., Urbinati C. (2009) Polysulfated/sulfonated compounds for the development of drugs at the crossroad of viral infection and oncogenesis. Curr. Pharm. Des. 15, 2946–2957 [DOI] [PubMed] [Google Scholar]
- 53. Fernando H., Nagle G. T., Rajarathnam K. (2007) Thermodynamic characterization of interleukin-8 monomer binding to CXCR1 receptor N-terminal domain. FEBS J. 274, 241–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fiorentini S., Marini E., Bozzo L., Trainini L., Saadoune L., Avolio M., Pontillo A., Bonfanti C., Sarmientos P., Caruso A. (2004) Preclinical studies on immunogenicity of the HIV-1 p17-based synthetic peptide AT20-KLH. Biopolymers 76, 334–343 [DOI] [PubMed] [Google Scholar]
- 55. Urbinati C., Bugatti A., Oreste P., Zoppetti G., Waltenberger J., Mitola S., Ribatti D., Presta M., Rusnati M. (2004) Chemically sulfated Escherichia coli K5 polysaccharide derivatives as extracellular HIV-1 Tat protein antagonists. FEBS Lett. 568, 171–177 [DOI] [PubMed] [Google Scholar]
- 56. Fiorentini S., Marini E., Caracciolo S., Caruso A. (2006) Functions of the HIV-1 matrix protein p17. New Microbiol. 29, 1–10 [PubMed] [Google Scholar]
- 57. Herbein G., Gras G., Khan K. A., Abbas W. (2010) Macrophage signaling in HIV-1 infection. Retrovirology 7, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Corasaniti M. T., Maccarrone M., Nistico R., Malorni W., Rotiroti D., Bagetta G. (2001) Exploitation of the HIV-1 coat glycoprotein, gp120, in neurodegenerative studies in vivo. J. Neurochem. 79, 1–8 [DOI] [PubMed] [Google Scholar]
- 59. Chauhan A., Turchan J., Pocernich C., Bruce-Keller A., Roth S., Butterfield D. A., Major E. O., Nath A. (2003) Intracellular human immunodeficiency virus Tat expression in astrocytes promotes astrocyte survival but induces potent neurotoxicity at distant sites via axonal transport. J. Biol. Chem. 278, 13512–13519 [DOI] [PubMed] [Google Scholar]
- 60. Singh I. N., Goody R. J., Dean C., Ahmad N. M., Lutz S. E., Knapp P. E., Nath A., Hauser K. F. (2004) Apoptotic death of striatal neurons induced by human immunodeficiency virus-1 Tat and gp120: Differential involvement of caspase-3 and endonuclease G. J. Neurovirol. 10, 141–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rusnati M., Presta M. (2002) HIV-1 Tat protein and endothelium: from protein/cell interaction to AIDS-associated pathologies. angiogenesis 5, 141–151 [DOI] [PubMed] [Google Scholar]
- 62. Lembo D., Donalisio M., Rusnati M., Bugatti A., Cornaglia M., Cappello P., Giovarelli M., Oreste P., Landolfo S. (2008) Sulfated K5 Escherichia coli polysaccharide derivatives as wide-range inhibitors of genital types of human papillomavirus. Antimicrob. Agents Chemother. 52, 1374–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hearps A. C., Jans D. A. (2007) Regulating the functions of the HIV-1 matrix protein. AIDS Res. Hum. Retroviruses 23, 341–346 [DOI] [PubMed] [Google Scholar]
- 64. Hoogewerf A. J., Kuschert G. S., Proudfoot A. E., Borlat F., Clark-Lewis I., Power C. A., Wells T. N. (1997) Glycosaminoglycans mediate cell surface oligomerization of chemokines. Biochemistry 36, 13570–13578 [DOI] [PubMed] [Google Scholar]
- 65. Rusnati M., Presta M. (1996) Interaction of angiogenic basic fibroblast growth factor with endothelial cell heparan sulfate proteoglycans. Biological implications in neovascularization. Int. J. Clin. Lab. Res. 26, 15–23 [DOI] [PubMed] [Google Scholar]