

Background: Although reactive oxygen species (ROS) are integral for TGF-β signaling, the source of ROS is not clear.
Results: Inhibition of TGF-β-induced mitochondrial ROS generation attenuates profibrotic gene expression.
Conclusion: ROS generated by complex III of the electron transport chain are required for TGF-β-mediated transcription in normal human lung fibroblasts.
Significance: Mitochondrial ROS might be a novel target to prevent TGF-β-mediated induced fibrosis.
Keywords: Fibroblast, Fibrosis, Lung, NADPH Oxidase, Superoxide Ion, Transforming Growth Factorβ (TGF-β)
Abstract
TGF-β signaling is required for normal tissue repair; however, excessive TGF-β signaling can lead to robust profibrotic gene expression in fibroblasts, resulting in tissue fibrosis. TGF-β binds to cell-surface receptors, resulting in the phosphorylation of the Smad family of transcription factors to initiate gene expression. TGF-β also initiates Smad-independent pathways, which augment gene expression. Here, we report that mitochondrial reactive oxygen species (ROS) generated at complex III are required for TGF-β-induced gene expression in primary normal human lung fibroblasts. TGF-β-induced ROS could be detected in both the mitochondrial matrix and cytosol. Mitochondrially targeted antioxidants markedly attenuated TGF-β-induced gene expression without affecting Smad phosphorylation or nuclear translocation. Genetically disrupting mitochondrial complex III-generated ROS production attenuated TGF-β-induced profibrotic gene expression. Furthermore, inhibiting mitochondrial ROS generation attenuated NOX4 (NADPH oxidase 4) expression, which is required for TGF-β induced myofibroblast differentiation. Lung fibroblasts from patients with pulmonary fibrosis generated more mitochondrial ROS than normal human lung fibroblasts, and mitochondrially targeted antioxidants attenuated profibrotic gene expression in both normal and fibrotic lung fibroblasts. Collectively, our results indicate that mitochondrial ROS are essential for normal TGF-β-mediated gene expression and that targeting mitochondrial ROS might be beneficial in diseases associated with excessive fibrosis.
Introduction
TGF-β is a multifunctional cytokine that regulates cellular proliferation differentiation and extracellular matrix production (1, 2). Dysregulation of TGF-β expression or signaling has been implicated in the pathogenesis of a variety of diseases, including cancer and fibrosis. TGF-β binds high-affinity cell-surface receptors known as types I, II, and III. Type III receptors bind TGF-β and transfer it to type I and II receptors. Type I and II receptors are serine/threonine protein kinases that initiate intracellular signaling by phosphorylating transcription factors such as Smad (small mothers against decapentaplegic). Once phosphorylated, Smad2 and Smad3 bind Smad4, and the resulting Smad complex then translocates into the nucleus. Within the nucleus, the Smad heterocomplex interacts with transcriptional coactivators such as p300 (3), a nuclear scaffolding protein, and histone acetyltransferases, which facilitate binding of the Smad complex to canonical Smad-binding elements of target genes to activate transcription.
In addition to Smad-mediated signaling, TGF-β acts through other pathways that are required for normal profibrotic gene expression in many systems. MAPK pathways, including p38, ERK, and JNK, can be activated by TGF-β, and in some contexts, inhibition of these pathways attenuates TGF-β-mediated transcription (4). In addition, tyrosine kinases (c-Abl (c-Abelson) and Src kinase family members) are required for extracellular matrix expression in fibroblasts stimulated with TGF-β (5, 6). The mechanisms by which these and other pathways are activated and modulate TGF-β gene transcription are incompletely understood. Many of the Smad-independent signaling pathways can be activated by intracellular reactive oxygen species (ROS)2 (7). For example, kinase signaling can be prolonged by the ROS-mediated inactivation of phosphatases (8). Several groups of investigators have reported that NOX4 (NADPH oxidase 4) is induced by TGF-β to generate intracellular ROS, which are required for the conversion of lung, kidney, and cardiac fibroblasts into a myofibroblast phenotype (9–11). The induction of NOX4 expression following TGF-β stimulation requires Smad3-mediated gene transcription, which suggests that NOX4-generated ROS act to amplify rather than initiate TGF-β-mediated gene transcription (9, 12). We have recently shown that oncogenic and hypoxic signaling requires mitochondrial generated ROS at complex III (13, 14). Unlike most oxidant-generating enzymes in the electron transport chain, complex III can release superoxide anions into the mitochondrial intermembrane space. These superoxide anions can be converted to H2O2 and released into the cytosol to activate signaling pathways. Here, we report that TGF-β induces mitochondrial complex III ROS generation, which is required for the TGF-β-mediated expression of profibrotic genes.
EXPERIMENTAL PROCEDURES
ROS Measurements
Intracellular ROS were measured using the redox-sensitive GFP (roGFP) described previously (15). Normal human lung fibroblasts (NHLFs) grown in FGMTM-2 BulletKit medium (both from Lonza) were infected with an adenovirus (10 pfu) encoding roGFP targeted to the cytosol (cyto-roGFP) or mitochondrial matrix (mito-roGFP). This roGFP protein contains two surface-exposed cysteines at positions 147 and 204 (S147C and Q204C). In the presence of an oxidant, a disulfide bond forms between the two surface-exposed cysteines, increasing the excitation at 400 nm at the expense of the peak near 490 nm. The ratio of fluorescence between 400 and 490 nm is proportional to the oxidant-induced disulfide bond formed (16). The mito-roGFP was targeted to the mitochondria by attaching the cytochrome c oxidase subunit IV mitochondrial localization sequence to the N terminus (16). Cells were harvested for analysis with a CyAn ADP flow cytometry analyzer (Dako). The mean fluorescent channel for the ratio of violet excitable to blue excitable was determined with Summit v4.2 software (Dako). The percent oxidized probe was determined as the ratio of the sample mean to the mean from the probe oxidized by 1 mm H2O2 and reduced by 1 mm DTT (17). Intracellular ROS were also measured using Amplex Red (Invitrogen) according to the manufacturer's protocol. In brief, NHLFs were lysed in 100 μm Amplex Red solution supplemented with 2 units/ml horseradish peroxidase and 200 milliunits/ml superoxide dismutase (OXIS International) and incubated in the dark for 30 min. Fluorescence was measured in a SpectraMax Gemini plate reader (Molecular Devices) with excitation of 540 nm and emission of 590 nm.
Western Immunoblotting
NHLFs were grown to 70% confluence, serum-starved for 24 h, and then incubated with recombinant human TGF-β (5 ng/ml; VWR International) and appropriate controls. Cell lysates were collected and analyzed by Western analysis. Nuclear extracts were obtained using a nuclear extract kit (Active Motif catalog no. 40010) according to the manufacturer's instructions. Immunoblotting was performed using antibodies to phosphorylated Smad2/3 (BD Biosciences), the Rieske iron-sulfur protein (RISP; MitoSciences), and the ubiquinone-binding protein QPC (Proteintech). Antibodies to Smad3 and RNA polymerase II (Santa Cruz Biotechnology) and actin (Sigma-Aldrich) were used as loading controls. Cell death was measured by propidium iodine staining (100 ng/ml) and flow cytometry.
Quantitative PCR
Quantitative mRNA expression was determined by real-time RT-PCR using SYBR Green chemistry. The following primer sequences were used: α-smooth muscle actin (α-SMA), GGCGGTGCTGTCTCTCTAT and CCAGATCCAGACGCATGATG; RPL19 (control), AGTATGCTCAGGCTTCAGAAGA and CATTGGTCTCATTGGGGTCTAAC; connective tissue growth factor (CTGF), GGCTTACCGACTGGAAGAC and AGGAGGCGTTGTCATTGG; and NOX4, CACCTCTGCCTGTTCATCTG and GGCTCTGCTTAGACACAATCC. NHLFs were pretreated with MitoQ, MitoVit E, Mito-CP, or triphenylphosphonium (TPP) for 30 min, after which TGF-β (5 ng/ml) was added, followed by incubation for 24 h. Total RNA from cells was then isolated using an Ambion RNAqueous-4PCR kit. cDNA was synthesized from 1 μg of total RNA using an Ambion RETROscript cDNA synthesis kit with random decamer primers. Quantitative real-time RT-PCR was carried out using SYBR Green chemistry. cDNAs was amplified using the Bio-Rad iCycler iQ system. Cycle threshold values were normalized for amplification of mitochondrial RPL19.
Measurements of Smad-binding Element (SBE)-Luciferase Activity
Transfection to assess Smad-mediated expression was performed using the Mirus TransIT transfection reagent (Mirus Bio LLC, Madison, WI) according to the manufacturer's protocol. SBE-luciferase is a pGL2 vector containing Smad-binding response elements upstream of firefly luciferase 2. Thymidine kinase-Renilla luciferase was cotransfected to control for transfection efficiency. Luciferase activity was measured by luminometry according to standard protocols for the Promega Dual-Luciferase reporter assay system.
shRNA and Generation of RISP and QPC Stable Cell Lines
The pLKO.1 validated lentiviral vectors were obtained from Sigma to express shRNAs targeting RISP and QPC (RISP, CCGGCCTATTTGGTAACTGGAGTAACTCGAGTTACTCCAGTTACCAAATAGGTTTTTG; and QPC, CGGGTGATCAGCTACAGCTTGTCACTCGAGTGACAAGCTGTAGCTGATCACTTTTTG). Control cell lines were generated using the identical backbone encoding a shRNA against Drosophila hypoxia-inducible factor (RISP) or the lentiviral pLKO backbone with no transgene (QPC). Stable cell lines were generated by lentiviral infection using the 293FT packaging cell line and puromycin selection. Forty-eight hours post-transfection, medium containing virus was supplemented with 8 μg/ml Polybrene (Sigma) for cell line infection and applied to cells. Both shRNA- and control transfected cells were maintained in the appropriate selection media.
Human Specimens
The collection of clinical data and specimens was approved by the Northwestern University Institutional Review Board. Samples of lung fibroblasts from fibrotic lungs were cultured and stored as described previously (18). Profibrotic gene expression was measured using the methodology and primers described above.
Statistics
The data were analyzed using Prism 4 (GraphPad Software, Inc., La Jolla, CA). All data are displayed as means ± S.E. Statistical significance was determined by paired or unpaired t tests as appropriate. p < 0.05 was considered statistically significant.
RESULTS
TGF-β Induces Mitochondrial and Cytosolic ROS
In NHLFs, TGF-β (5 ng/ml) increased intracellular H2O2 levels after 30 min (Amplex Red). The increase in H2O2 was attenuated by treatment with the ALK-5 inhibitor SB431542 (Fig. 1A). The increase in H2O2 was also attenuated by treatment with MitoQ, a mitochondrially targeted antioxidant (Fig. 1B). MitoQ is ubiquinone conjugated to a lipophilic TPP cation, which accumulates within mitochondria, where it is reduced to the antioxidant ubiquinol. To confirm that TGF-β increased mitochondrial ROS generation, we utilized a redox-sensitive GFP probe targeted to the mitochondrial matrix (mito-roGFP). TGF-β increased the oxidation of mito-roGFP in fibroblasts at 30 min, as did antimycin A, which increased mitochondrial complex III ROS generation (Fig. 1C). Most mitochondrial ROS-generating enzymes release ROS in the mitochondrial matrix; however, ROS generated at complex III of the electron transport chain can be released into the mitochondrial intermembrane space, from which they can escape into the cytosol. Consistent with mitochondrial generation of ROS at complex III, TGF-β increased oxidation of cyto-roGFP, as did antimycin A (positive control) (Fig. 1D). Another mitochondrially targeted antioxidant, MitoVit E (MVE), caused a reduction in the TGF-β-mediated oxidation of mito-roGFP and cyto-roGFP, which was similar to MitoQ (Fig. 1, E and F, respectively).
FIGURE 1.
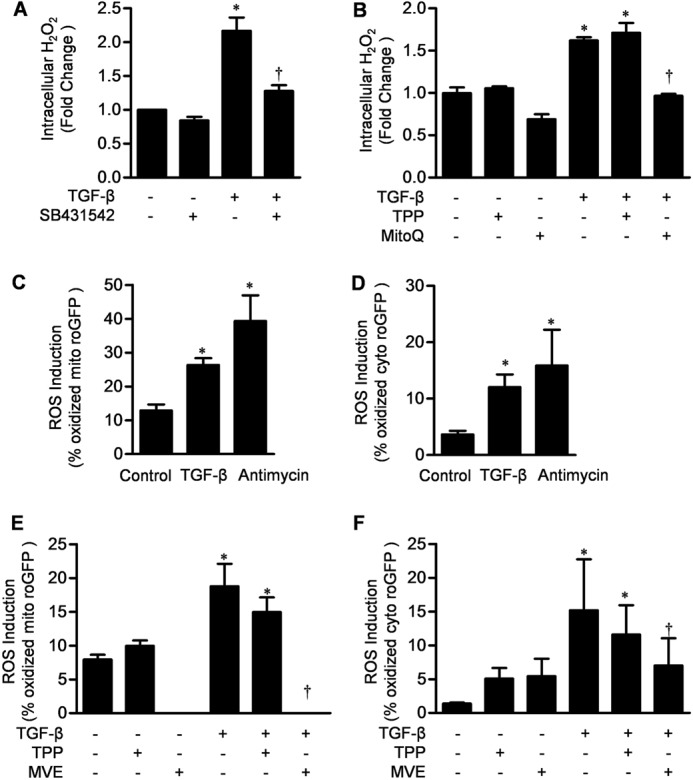
TGF-β induces mitochondrial ROS which is inhibited by a mitochondrially targeted antioxidant. A, primary cultures of NHLFs were treated with TGF-β (5 ng/ml) in the presence or absence of SB431542. B, primary cultures of NHLFs were treated with TGF-β (5 ng/ml) alone and in the presence of TPP (1 μm) or MitoQ (1 μm), and intracellular H2O2 levels were assessed using Amplex Red. C, primary NHLFs were infected with 100 pfu of adenovirus encoding mito-roGFP. Twenty-four hours after infection, the cells were treated with TGF-β1 (5 ng/ml) or antimycin A (1 μg/ml) for 1 h, and oxidation of the probe was measured by flow cytometry. D, primary NHLFs were infected with 100 pfu of adenovirus encoding cyto-roGFP. Twenty-four hours after infection, the cells were treated with TGF-β1 (5 ng/ml) or antimycin A (1 μg/ml) for 1 h, and oxidation of the probe was measured by flow cytometry. E, primary NHLFs were infected with 100 pfu of adenovirus encoding mito-roGFP. Twenty-four hours after infection, the cells were treated with TGF-β (5 ng/ml) in the presence of TPP (1 μm) or MVE (1 μm, 1-h pretreatment), and oxidation of the probe was measured by flow cytometry. F, primary NHLFs were infected with 100 pfu of adenovirus encoding cyto-roGFP. Twenty-four hours after infection, the cells were treated with TGF-β1 (5 ng/ml) in the presence of TPP (1 μm) and MVE (1 μm, 1-h pretreatment), and oxidation of the probe was measured by flow cytometry. Error bars represent mean ± S.E. (n = 3 for A–D and n = 6 for E and F). *, p < 0.05 for comparison between TGF-β and the control; †, p < 0.05 for comparison between MVE and TPP.
Mitochondrially Targeted Antioxidants Do Not Inhibit Smad3 Phosphorylation or Cell Viability but Inhibit TGF-β-induced Transcription
To determine whether the increase in mitochondrial ROS is required for Smad3 phosphorylation or nuclear translocation, NHLFs were treated with TGF-β (5 ng/ml) in the presence or absence of MVE or its control cation, TPP (both at 1 μm). The levels of phosphorylated Smad3 were similar in the untreated cells and in the cells treated with TPP or MVE (Fig. 2A). Similar results were obtained using another mitochondrially targeted antioxidant, Mito-CP (data not shown). To exclude any nonspecific toxic effects of mitochondrial antioxidants, we determined the viability of normal fibroblasts in the presence of MVE or Mito-CP with and without TGF-β. There was no significant difference in the rates of death of cells treated with MVE or TPP in the presence or absence of TGF-β (Fig. 2B; data for mito-CP not shown). To determine whether mitochondrial ROS are required for Smad-dependent transcriptional activity, NHLFs were transfected with the Smad2/3-responsive SBE-luciferase and treated with TGF-β (5 ng/ml) in the presence or absence of MitoQ or MVE (both at 1 μm). Both MitoQ and MVE diminished SBE-luciferase activity compared with the TPP control cation (Fig. 2, C and D). Furthermore, MitoQ and MVE inhibited the TGF-β-induced increase in the levels of mRNAs encoding α-SMA and CTGF, two well characterized TGF-β target genes (Fig. 2, E and F). Interestingly, MitoQ and MVE also attenuated expression of NOX4 (Fig. 2, E and F), indicating that mitochondrial ROS are required for the TGF-β-mediated transcription of NOX4. Similar results were observed using Mito-CP (data not shown). Collectively, these results indicate that mitochondrial ROS are required for the transcription of TGF-β target genes, including NOX4, downstream of the activation and nuclear translocation of Smad2/3.
FIGURE 2.

Mitochondrially targeted antioxidants inhibit TGF-β-mediated gene transcription downstream of the nuclear translocation of phosphorylated Smad3. A, primary NHLFs were treated with TGF-β (5 ng/ml) in the presence or absence of MVE or its control cation (TPP) (both 1 μm), and cytosolic and nuclear fractions were isolated and immunoblotted using antibodies to phosphorylated (p-Smad3) and total Smad3. Antibodies to actin and RNA polymerase II (RNA pol II) were used as loading controls and to ensure exclusion of cytosolic proteins. B, primary NHLFs were treated with TGF-β (5 ng/ml) with or without MVE or TPP (both 1 μm), and 24 h later, cell death was measured by propidium iodine staining. C and D, NHLFs were transfected with a plasmid containing SBE-luciferase and treated 24 h later with TGF-β (5 ng/ml) with or without MitoQ (C), MVE (D), or TPP (all 1 μm), and SBE-luciferase activity was measured 24 h later. E and F, primary NHLFs were grown to 70% confluence and incubated with TGF-β (5 ng/ml) with or without MitoQ (E), MVE (F), or TPP, and 24 h later, the levels of mRNAs encoding α-SMA, CTGF, and NOX4 were measured using quantitative RT-PCR in cell lysates. Error bars represent mean ± S.E. (n = 3 for all measures). *, p < 0.05 for comparison between TGF-β1 and the control; †, p < 0.05 for comparison between MitoQ or MVE and TPP.
Mitochondrial Complex III ROS Are Required TGF-β-dependent Gene Expression
We have previously reported that mitochondrial complex III generates ROS, which act as signaling molecules that are required for the stabilization of HIF1 during hypoxia, the proliferation of Kras-driven tumor cell lines, and the differentiation of mesenchymal stem cells (13, 19, 20). RISP and QPC are complex III proteins required for electron transport. Loss of RISP prevents complex III ROS generation, whereas loss of QPC prevents electron transport without affecting (or slightly increasing) complex III ROS generation (21). We used lentivirally delivered shRNAs to knock down expression of RISP or QPC in NHLFs (Fig. 3, A and B). The RISP knockdown cells, but not the QPC knockdown or the control transfected cells, had reduced levels of TGF-β-induced ROS production (Fig. 3, C and D). Similarly, only knockdown of RISP reduced the TGF-β-mediated transcription of α-SMA, CTGF, and NOX4 (Fig. 3, E and F).
FIGURE 3.

Mitochondrially generated ROS are necessary for TGF-β-mediated transcription. A and B, primary NHLFs were stably transfected with a shRNA against RISP, QPC, or control lentiviruses (Drosophila hypoxia-inducible factor and pLKO), and the levels of RISP and QPC were measured by immunoblotting. C and D, these cells were treated with TGF-β, and intracellular H2O2 levels were measured using Amplex Red. E and F, control and RISP and QPC knockdown fibroblasts were treated with TGF-β (5 ng/ml), and 24 h later, mRNAs encoding the TGF-β target genes α-SMA, CTGF, and NOX4 were measured by quantitative RT-PCR. Error bars represent mean ± S.E. (n = 3 for all measures). *, p < 0.05 for comparison between TGF-β and the control; †, p < 0.05 for comparison between RISP knockdown and the control.
We then sought to determine whether mitochondrial antioxidants could inhibit TGF-β-mediated transcription when electron transport was inhibited. Incubating fibroblasts expressing a stable knockdown of QPC with MVE inhibited TGF-β-induced expression of α-SMA, CTGF, and NOX4 (Fig. 4A). Analogous to the QPC knockdown, antimycin A blocks electron transport through the cytochrome b complex. When cells were treated with TGF-β in the presence of antimycin A, the transcription of α-SMA and CTGF was increased (Fig. 4B). The addition of MVE with antimycin A attenuated the TGF-β-induced transcription of α-SMA, CTGF, and NOX4 (Fig. 4C). Similar results were obtained using another mitochondrially targeted antioxidant, Mito-CP (data not shown). Collectively, these results indicate that complex III ROS regulate TGF-β-induced transcription independently of oxidative phosphorylation.
FIGURE 4.

Mitochondrial ROS are sufficient to augment TGF-β-mediated transcription in primary NHLFs. A, primary NHLFs were stably transfected with a shRNA encoding QPC or a control lentivirus and treated with TGF-β (5 ng/ml) in the presence or absence of MVE, and 24 h later, the levels of mRNAs encoding the TGF-β transcriptional targets α-SMA, CTGF, and NOX4 were measured by quantitative RT-PCR. kd, knockdown. B and C, primary NHLFs were treated with antimycin A (1 μm), which inhibits electron transport through cytochrome b (analogous to the loss of QPC), with or without TGF-β (5 ng/ml) (B) and with or without MVE (C), and 24 h later, the levels of mRNAs encoding the same TGF-β transcriptional targets were measured. Error bars represent mean ± S.E. (n = 3 for all measures). *, p < 0.05 for comparison between TGF-β and the control; †, p < 0.05 for comparison between antimycin A and the control; ‡, p < 0.05 for comparison between MVE and the control.
Fibroblasts from Patients with Lung Fibrosis Produce Mitochondrial ROS That Are Required for Profibrotic Gene Expression
Next, we examined mitochondrial ROS production and the effects of mitochondrial antioxidants on profibrotic gene expression in fibroblasts from patients with lung fibrosis. Cell lines from two patients with scleroderma-associated pulmonary fibrosis and from two patients with idiopathic pulmonary fibrosis produced significantly more mitochondrial ROS in response to TGF-β than fibroblasts from normal donors (Fig. 5A). The mitochondrially targeted antioxidant Mito-CP inhibited the TGF-β-mediated induction of α-SMA and NOX4 in all four cell lines from patients with lung fibrosis (Fig. 5, B–E). Similar effects were observed with CTGF (data not shown). Collectively, these data suggest that mitochondrial ROS are increased in fibroblasts from patients with fibrotic lung diseases and contribute to TGF-β-induced gene expression.
FIGURE 5.

Mitochondrial ROS are required for TGF-β-induced gene expression in lung fibroblasts obtained from patients with lung fibrosis and scleroderma. A, primary cultures of lung fibroblasts from each of four patients with lung fibrosis were treated with TGF-β (5 ng/ml), and 30 min later, intracellular H2O2 levels were measured using Amplex Red. Values are presented as the -fold change compared with cultures of NHLFs. B and C, primary lung fibroblasts cultured from two patients with scleroderma-associated pulmonary fibrosis were treated with TGF-β (5 ng/ml) in the presence or absence of Mito-CP or its control cation (TPP), and 24 h later, the levels of mRNAs encoding α-SMA and NOX4 were measured by quantitative RT-PCR. D and E, the experiments were repeated with lung fibroblasts from two patients with idiopathic pulmonary fibrosis. Error bars represent mean ± S.E. (n = 3 for all measures). *, p < 0.05 for comparison between fibroblasts from patients with lung fibrosis and NHLFs; †, p < 0.05 for comparison between TGF-β and the control; ‡, p < 0.05 for comparison between Mito-CP and TPP. SSc, systemic sclerosis (scleroderma); IPF, idiopathic pulmonary fibrosis.
DISCUSSION
This study supports four novel conclusions with respect to TGF-β-mediated transcription in lung fibroblasts. First, TGF-β induces the generation of mitochondrial ROS from complex III of the electron transport chain. Second, mitochondrial ROS generation is required for TGF-β-mediated gene transcription downstream of the phosphorylation and nuclear translocation of Smad3. Third, the TGF-β-induced transcription of NOX4 requires the mitochondrial generation of ROS, suggesting the presence of a feed-forward loop that increases intracellular ROS signaling after TGF-β activation. Fourth, fibroblasts from patients with lung fibrosis stimulated with TGF-β produce more mitochondrial ROS and a higher level of profibrotic gene expression compared with lung fibroblasts from normal donors. Both TGF-β-induced mitochondrial ROS generation and gene expression can be inhibited by mitochondrially targeted antioxidants in these cells.
Other groups of investigators have reported that TGF-β increases intracellular levels of H2O2 in multiple cell types, including fibroblasts (7). Consistent with these reports, we detected an increase in ROS generation 30 min after TGF-β stimulation in NHLFs (22–24). Using oxidant-sensitive fluorescent proteins localized to the mitochondria or cytosol, we were able to localize the source of these ROS to the mitochondria. Blocking mitochondrial ROS generation with a targeted antioxidant markedly reduced TGF-β-mediated gene transcription but had no effect on the phosphorylation or nuclear translocation of Smad3. We confirmed these results genetically by demonstrating that TGF-β-mediated transcription was also inhibited in cells with a stable knockdown of RISP, which is required for the generation of ROS from complex III of the mitochondrial electron transport chain. Because loss of RISP both impairs complex III ROS production and inhibits electron transport, we measured TGF-β-mediated transcription in cells with a stable knockdown QPC and in cells treated with antimycin A. Both strategies impair electron transport but either do not affect or increase complex III ROS generation. We found that TGF-β-mediated transcription was normal in these cells, strongly suggesting that the loss of RISP inhibited TGF-β-mediated transcription by preventing mitochondrial ROS generation rather than by preventing electron transport. These results were confirmed by our finding that the administration of mitochondrially targeted antioxidants effectively inhibited TGF-β-mediated transcription in QPC knockdown cells and in cells treated with antimycin A.
Thannickal and Fanburg (7) first reported the ability of TGF-β to induce H2O2 in fibroblasts. They observed an increase in ROS generation, which began 8 h after TGF-β treatment, required de novo protein synthesis, and was dependent on activity of the NADPH oxidases. Thannickal and co-workers went on to show that the administration of a siRNA against NOX4 prevented fibrosis in two murine models of lung fibrosis, bleomycin and FITC, likely by inhibiting myofibroblast activation (12). These data highlight the important role of NADPH oxidases in the pathogenesis of TGF-β-mediated lung fibrosis. We found that the TGF-β-induced generation of mitochondrial ROS is required for the transcription of NOX4. This suggests that activation of the TGF-β receptor induced both Smad activation and the mitochondrial generation of ROS, both of which are required for the transcription of NOX4. Activation of NOX4 amplifies and prolongs the ROS signal, perhaps maintaining TGF-β-mediated transcription over time.
At high levels, ROS can damage proteins and lipids, resulting in cell dysfunction or death. We have shown that at low levels, mitochondrially derived H2O2 serves as a key signaling molecule required for the cellular adaptation to environmental stress. For example, we reported that tumor cells require mitochondrial complex III oxidant generation to maintain a cancer phenotype in vivo and in vitro and that mesenchymal stem cells are reliant on these oxidants to drive differentiation. In both of these examples, the oxidants acted to increase the activity of pathways that drive growth and proliferation. We speculate that the TGF-β-dependent transformation of fibroblasts to myofibroblasts and their subsequent migration, proliferation, and matrix production require oxidant signaling to drive pathways that promote biosynthesis. Although our study does not identify these ROS targets, likely candidates include the MAPK pathways ERK1/2 and p38 or the Akt pathway. It may be that the initial activation of these pathways by mitochondrial ROS is sustained by the induction of NOX4-derived ROS.
Our findings suggest that mitochondrially derived ROS might be important targets for the treatment of fibrotic lung diseases. Bocchino et al. (25) reported that lung fibroblasts from patients with idiopathic pulmonary fibrosis produce more ROS in response to TGF-β than cells from normal donors. In lung fibroblasts from patients with idiopathic and scleroderma-induced lung fibrosis, we found that more mitochondrially derived ROS were produced in response to TGF-β compared with lung fibroblasts from normal donors and that these ROS were required for the transcription of profibrotic genes, including NOX4. Despite these higher levels of ROS, mitochondrially targeted antioxidants administered at concentrations that can be achieved in the serum were sufficient to inhibit TGF-β-mediated transcription (25). In animal models of lung fibrosis, investigators have shown that pretreatment with untargeted antioxidants, including N-acetylcysteine, can partially prevent the development of fibrosis. Our data suggest that mitochondrially targeted antioxidants might selectively inhibit TGF-β signaling. By preventing NOX4 transcription, the targeted antioxidants might also inhibit the sustained ROS signal induced by the NAD(P)H oxidase system. MitoQ is a clinically available mitochondrially targeted antioxidant, has substantial safety data in humans, and is an example of a drug that could be tested in models of fibrosis.
In summary, we have shown that the administration of TGF-β to NHLFs results in the generation of mitochondrial ROS. Mitochondrial ROS derived from complex III of the mitochondrial electron transport chain are required for TGF-β-mediated transcription of profibrotic genes in both NHLFs and lung fibroblasts from patients with idiopathic and scleroderma-induced lung fibrosis. Because the induction of NOX4 also requires mitochondrially generated ROS, we speculate that TGF-β-induced ROS generation originates from the mitochondria and is sustained and amplified by cytosolic NAD(P)H oxidases.
Acknowledgments
We thank Drs. Joy Joseph and Balaraman Kalyanaraman (Medical College of Wisconsin) for kindly providing the mitochondrially targeted antioxidants.
Footnotes
- ROS
- reactive oxygen species
- roGFP
- redox-sensitive GFP
- NHLF
- normal human lung fibroblast
- cyto-roGFP
- cytosol-targeted roGFP
- mito-roGFP
- mitochondrial matrix-targeted roGFP
- RISP
- Rieske iron-sulfur protein
- α-SMA
- α-smooth muscle actin
- CTGF
- connective tissue growth factor
- TPP
- triphenylphosphonium
- SBE
- Smad-binding element
- MVE
- MitoVit E.
REFERENCES
- 1. Jennings M. T., Pietenpol J. A. (1998) The role of transforming growth factor β in glioma progression. J. Neurooncol. 36, 123–140 [DOI] [PubMed] [Google Scholar]
- 2. Verrecchia F., Mauviel A. (2002) Transforming growth factor-β signaling through the Smad pathway: role in extracellular matrix gene expression and regulation. J. Invest. Dermatol. 118, 211–215 [DOI] [PubMed] [Google Scholar]
- 3. Bhattacharyya S., Ghosh A. K., Pannu J., Mori Y., Takagawa S., Chen G., Trojanowska M., Gilliam A. C., Varga J. (2005) Fibroblast expression of the coactivator p300 governs the intensity of profibrotic response to transforming growth factor β. Arthritis Rheum. 52, 1248–1258 [DOI] [PubMed] [Google Scholar]
- 4. Hashimoto S., Gon Y., Takeshita I., Matsumoto K., Maruoka S., Horie T. (2001) Transforming growth factor-β1 induces phenotypic modulation of human lung fibroblasts to myofibroblast through a c-Jun NH2-terminal kinase-dependent pathway. Am. J. Respir Crit. Care Med. 163, 152–157 [DOI] [PubMed] [Google Scholar]
- 5. Daniels C. E., Wilkes M. C., Edens M., Kottom T. J., Murphy S. J., Limper A. H., Leof E. B. (2004) Imatinib mesylate inhibits the profibrogenic activity of TGF-β and prevents bleomycin-mediated lung fibrosis. J. Clin. Invest. 114, 1308–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mishra R., Zhu L., Eckert R. L., Simonson M. S. (2007) TGF-β-regulated collagen type I accumulation: role of Src-based signals. Am. J. Physiol. Cell Physiol. 292, C1361–C1369 [DOI] [PubMed] [Google Scholar]
- 7. Thannickal V. J., Fanburg B. L. (1995) Activation of an H2O2-generating NADH oxidase in human lung fibroblasts by transforming growth factor β1. J. Biol. Chem. 270, 30334–30338 [DOI] [PubMed] [Google Scholar]
- 8. Heneberg P., Dráber P. (2005) Regulation of Cys-based protein tyrosine phosphatases via reactive oxygen and nitrogen species in mast cells and basophils. Current medicinal chemistry 12, 1859–1871 [DOI] [PubMed] [Google Scholar]
- 9. Sturrock A., Cahill B., Norman K., Huecksteadt T. P., Hill K., Sanders K., Karwande S. V., Stringham J. C., Bull D. A., Gleich M., Kennedy T. P., Hoidal J. R. (2006) Transforming growth factor-β1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 290, L661–L673 [DOI] [PubMed] [Google Scholar]
- 10. Cucoranu I., Clempus R., Dikalova A., Phelan P. J., Ariyan S., Dikalov S., Sorescu D. (2005) NAD(P)H oxidase 4 mediates transforming growth factor-β1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 97, 900–907 [DOI] [PubMed] [Google Scholar]
- 11. Barnes J. L., Gorin Y. (2011) Myofibroblast differentiation during fibrosis: role of NAD(P)H oxidases. Kidney Int. 79, 944–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hecker L., Vittal R., Jones T., Jagirdar R., Luckhardt T. R., Horowitz J. C., Pennathur S., Martinez F. J., Thannickal V. J. (2009) NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 15, 1077–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Weinberg F., Hamanaka R., Wheaton W. W., Weinberg S., Joseph J., Lopez M., Kalyanaraman B., Mutlu G. M., Budinger G. R., Chandel N. S. (2010) Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. U.S.A. 107, 8788–8793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Emerling B. M., Weinberg F., Snyder C., Burgess Z., Mutlu G. M., Viollet B., Budinger G. R., Chandel N. S. (2009) Hypoxic activation of AMPK is dependent on mitochondrial ROS but independent of an increase in AMP/ATP ratio. Free Radic. Biol. Med. 46, 1386–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dooley C. T., Dore T. M., Hanson G. T., Jackson W. C., Remington S. J., Tsien R. Y. (2004) Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J. Biol. Chem. 279, 22284–22293 [DOI] [PubMed] [Google Scholar]
- 16. Hanson G. T., Aggeler R., Oglesbee D., Cannon M., Capaldi R. A., Tsien R. Y., Remington S. J. (2004) Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J. Biol. Chem. 279, 13044–13053 [DOI] [PubMed] [Google Scholar]
- 17. Soberanes S., Urich D., Baker C. M., Burgess Z., Chiarella S. E., Bell E. L., Ghio A. J., De Vizcaya-Ruiz A., Liu J., Ridge K. M., Kamp D. W., Chandel N. S., Schumacker P. T., Mutlu G. M., Budinger G. R. (2009) Mitochondrial complex III-generated oxidants activate ASK1 and JNK to induce alveolar epithelial cell death following exposure to particulate matter air pollution. J. Biol. Chem. 284, 2176–2186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ramos C., Montaño M., García-Alvarez J., Ruiz V., Uhal B. D., Selman M., Pardo A. (2001) Fibroblasts from idiopathic pulmonary fibrosis and normal lungs differ in growth rate, apoptosis, and tissue inhibitor of metalloproteinase expression. Am. J. Respir. Cell Mol. Biol. 24, 591–598 [DOI] [PubMed] [Google Scholar]
- 19. Emerling B. M., Platanias L. C., Black E., Nebreda A. R., Davis R. J., Chandel N. S. (2005) Mitochondrial reactive oxygen species activation of p38 mitogen-activated protein kinase is required for hypoxia signaling. Mol. Cell. Biol. 25, 4853–4862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tormos K. V., Anso E., Hamanaka R. B., Eisenbart J., Joseph J., Kalyanaraman B., Chandel N. S. (2011) Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metab. 14, 537–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Collins Y., Chouchani E. T., James A. M., Menger K. E., Cochemé H. M., Murphy M. P. (2012) Mitochondrial redox signalling at a glance. J. Cell Sci. 125, 801–806 [DOI] [PubMed] [Google Scholar]
- 22. Amara N., Goven D., Prost F., Muloway R., Crestani B., Boczkowski J. (2010) NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFβ1-induced fibroblast differentiation into myofibroblasts. Thorax 65, 733–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Black D., Lyman S., Qian T., Lemasters J. J., Rippe R. A., Nitta T., Kim J. S., Behrns K. E. (2007) Transforming growth factor β mediates hepatocyte apoptosis through Smad3 generation of reactive oxygen species. Biochimie 89, 1464–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Michaeloudes C., Sukkar M. B., Khorasani N. M., Bhavsar P. K., Chung K. F. (2011) TGF-β regulates Nox4, MnSOD and catalase expression, and IL-6 release in airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 300, L295–L304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bocchino M., Agnese S., Fagone E., Svegliati S., Grieco D., Vancheri C., Gabrielli A., Sanduzzi A., Avvedimento E. V. (2010) Reactive oxygen species are required for maintenance and differentiation of primary lung fibroblasts in idiopathic pulmonary fibrosis. PLoS ONE 5, e14003. [DOI] [PMC free article] [PubMed] [Google Scholar]