

Background: Inhibition of PAI-1 may yield beneficial effects in e.g. cardiovascular diseases and cancer.
Results: The small molecule PAI-1 inhibitor, AZ3976, binds latent but not active PAI-1. The structure of the AZ3976·latent PAI-1 complex is presented.
Conclusion: AZ3976 inhibits PAI-1 by accelerating latency transition, presumably by binding a prelatent form of PAI-1.
Significance: This study provides new drug design opportunities for PAI-1 inhibitors.
Keywords: Drug Discovery, High Throughput Screening (HTS), Isothermal Titration Calorimetry, Plasminogen Regulation, Surface Plasmon Resonance (SPR), X-ray Crystallography, PAI-1 Inhibitor, Plasma Clot Lysis, Prelatent PAI-1
Abstract
A novel class of small molecule inhibitors for plasminogen activator inhibitor type 1 (PAI-1), represented by AZ3976, was identified in a high throughput screening campaign. AZ3976 displayed an IC50 value of 26 μm in an enzymatic chromogenic assay. In a plasma clot lysis assay, the compound was active with an IC50 of 16 μm. Surprisingly, AZ3976 did not bind to active PAI-1 but bound to latent PAI-1 with a KD of 0.29 μm at 35 °C and a binding stoichiometry of 0.94, as measured by isothermal calorimetry. Reversible binding was confirmed by surface plasmon resonance direct binding experiments. The x-ray structure of AZ3976 in complex with latent PAI-1 was determined at 2.4 Å resolution. The inhibitor was bound in the flexible joint region with the entrance to the cavity located between α-helix D and β-strand 2A. A set of surface plasmon resonance experiments revealed that AZ3976 inhibited PAI-1 by enhancing the latency transition of active PAI-1. Because AZ3976 only had measurable affinity for latent PAI-1, we propose that its mechanism of inhibition is based on binding to a small fraction in equilibrium with active PAI-1, a latent-like prelatent form, from which latent PAI-1 is then generated more rapidly. This mode of action, with induced accelerated latency transition of active PAI-1 may, together with supporting x-ray data, provide improved opportunities for small molecule drug design in the hunt for therapeutically useful PAI-1 inhibitors.
Introduction
Mammalian blood contains the fibrinolytic system that is capable of dissolving blood clots (1). On the surface of the clot, the inactive proenzyme plasminogen is converted to the active enzyme plasmin, which degrades fibrin into soluble fibrin degradation products. The serine proteases that activate plasminogen into plasmin are tissue-type plasminogen activator (tPA)2 and urokinase-type plasminogen activator (uPA) (2). Inhibition of the fibrinolytic system in vivo may occur either at the level of plasmin by α2-antiplasmin or α2-macroglobulin. Inhibition can also be at the level of tPA/uPA (3, 4), mainly by plasminogen activator inhibitor type-1 (PAI-1), which forms a suicide complex with tPA or uPA (5).
PAI-1 is a member of the serine protease inhibitor (serpin) superfamily. The tertiary structure of PAI-1 is composed of nine α-helices (hA–hI) and three β-sheets (A, B, and C). The key feature is the reactive center loop (RCL), a solvent-exposed unstructured loop of ∼20 amino acids containing the target sequence recognized by proteases such as tPA and uPA (5–8). In vitro, the conformation of active PAI-1 spontaneously converts from the active state to an energetically more favorable inactive latent state by moving the RCL into the central β-sheet (9–13). The conversion of PAI-1 from the active to the latent conformation appears to be unique among serpins in that it occurs spontaneously at a relatively rapid rate (14–17). For that reason, most crystal structures of PAI-1 are of latent PAI-1 or of stabilized mutants (9, 18–20). However, recently, the crystal structure of W175F PAI-1 has been determined, which may be the structure most closely representing active PAI-1 (6).
In vivo, most circulating PAI-1 in plasma is in a complex with vitronectin (VN) (21), which stabilizes PAI-1 in the active conformation (17, 21–23), competent to inhibit tPA and uPA. Still, PAI-1 ex vivo in blood has a half-life of only ∼1 h at 37 °C (24). It has been found that PAI-1 binds to the N-terminal ∼50-amino acid somatomedin B domain of VN (25–27). X-ray evidence (23) suggests that binding to this domain induces conformational changes in PAI-1 which, in addition to steric effects, may contribute to its improved stability. However, the main part of PAI-1 that is stored in the α-vesicles in platelets is not in complex with VN (28–30) and is active (31). Upon activation of platelets, this stored PAI-1 is released to protect the primary clot from fibrinolysis (28, 31) by inhibiting tPA secreted from nearby endothelium (32). Although an inhibitor for PAI-1 that neutralizes the activity of both PAI-1 in complex with VN and of free PAI-1 may yield the most effective inhibition, it is still possible that selective inhibition of free PAI-1 could provide therapeutic benefit.
Several studies with inhibiting antibodies and small molecules against PAI-1 have shown effects in different animal models of fibrinolysis (33–42). We are not aware of a PAI-1 inhibitor that has been tested in patients, but the low molecular weight PAI-1 inhibitor PAI-749 (diaplasinin) has been tested ex vivo in human blood using the Badimon chamber and was found not to have an effect on in vitro or ex vivo thrombus formation or fibrinolysis in the presence or absence of tPA (43, 44). It is fair to say that development of oral PAI inhibitors has been progressing slowly, probably due to the plasticity of this unique target with no obvious druggable sites.
Here, we describe a novel small molecule ligand for PAI-1 exhibiting profibrinolytic activities in a plasma clot lysis assay in vitro. A wide range of biophysical techniques support that the ligand accelerates PAI-1 latency transition and a crystal structure of latent PAI-1 in complex with the ligand is presented.
MATERIALS AND METHODS
Proteins
Latent glycosylated recombinant human plasminogen activator type 1, PAI-1, purified from the conditioned Chinese hamster ovary (CHO) cell medium, was originally obtained from Symbicom AB (Umeå, Sweden). Activated human PAI-1 was prepared by incubation of latent PAI-1 in 6 m guanidine-HCl, followed by gel filtration on Superdex 75 at pH 5.5 (12). In most experiments, active glycosylated human PAI-1 from a single batch with a specific activity of 75% was used. Active non-glycosylated human PAI-1 expressed in Escherichia coli was purchased from Molecular Innovations, Inc. (Novi, MI) and was shown to have a specific activity of 78%. Latent non-glycosylated human PAI-1 was expressed in E. coli and prepared in house (see below). Hereafter, if not indicated otherwise, experiments performed using PAI-1 refers to the use of the active glycosylated human PAI-1 batch displaying ∼75% specific activity (thus containing ∼25% inactive/latent PAI-1 protein), and experiments performed using latent PAI-1 refers to the use of latent non-glycosylated human PAI-1 batch with 0% specific activity. The two protein batches with glycosylated active and glycosylated latent PAI-1 have been characterized using circular dichroism spectroscopy (data not shown).
Rat PAI-1 and rat tPA were purchased from Molecular Innovations. Two-chain tissue human plasminogen activator, tPA, from melanoma cells, immunoaffinity-purified and stored in 1 m NH4HCO3 (Biopool, Umeå, Sweden), had a specific activity of 812,000 international units/mg. Stock solutions of PAI-1 and tPA, which had been kept stored at −80 °C for many years, were without apparent change in activity. Monomeric VN was purchased from Innovative Research, Inc. (Novi, MI). The solution with the rabbit anti-mouse monoclonal antibody (RamFc) was from the mouse antibody capture kit (BR-1008-38 from Biacore, GE Healthcare). The mouse anti-human tPA monoclonal IgG1 antibody 2:2 B10 (lot no. 4) was from IMCO Corp., Ltd. (Stockholm, Sweden). The mouse monoclonal antibody H4B3, specific for non-glycosylated latent PAI-1 was obtained from Molecular Innovations, Inc. As a neutral reference antibody mouse anti-FX from Innovative Research was used, showing no interaction with either PAI-1, tPA, or VN.
Additional Chemicals and Materials
From Biacore (GE Healthcare) were obtained N-ethyl-N′-(3-dimethyl aminopropyl)-carbodiimide HCl (EDC), N-hydroxy-succinimide (NHS), and 0.1 m ethanolamine hydrochloride-NaOH, pH 8.5, from the amine coupling kit BR-1000-50 and the borate buffer, pH 8.5 (BR-1003-53). EDC and NHS were dissolved in water to 0.4 and 0.1 m, respectively, and stored in 500-μl aliquots at −20 °C. For pegylation, O-(2-aminoethyl)-O-methyl polyethylene glycol 5000 (PEG) from Fluka (06679) was used. 99.99% DMSO was from Aldrich 27,685-5. Pefachrome tPA (CH3SO2-d-HHT-Gly-Arg-pNA) was obtained from Pentapharm (Basel, Switzerland). All solutions were made with deionized water that was further purified by reversed osmosis on an Elgastadt UHP (Elga, Ltd., High Wycombe Bucks, England). The buffers were filtered through Filtropur S plus 0.2 mm (Sarstedt, Nümbrecht, Germany). All other chemicals were of reagent grade.
Preparation of the Plasma Pool
Blood samples were drawn, after informed consent, from 40 healthy volunteers, with minimal trauma by free flow from a 17-gauge VenflonTM needle (Becton Dickinson Infusion Therapy AB, Helsingborg, Sweden). Nine parts blood (45 ml) were collected into 50-ml polypropylene tubes (Sarstedt, Nümbrecht, Germany) containing one part calcium chelator (5 ml of 0.105 m citrate) at room temperature. Directly after collection, the tubes were turned gently to mix anticoagulant and blood and centrifuged at 3600 × g for 20 min at room temperature, and the platelet poor plasma supernatant was pooled and pipetted into polypropylene cryotubes (Sarstedt, Nümbrecht, Germany) and stored at −80 °C until use.
Surface Plasmon Resonance, Instrument, and General Conditions
The surface plasmon resonance (SPR) experiments were performed on a Biacore 3000 and Biacore T200 optical biosensor (GE Healthcare). Sensor chip CM5 (research grade, different lots) was obtained from Biacore. Prior to use, all sensor chips were left at room temperature for 30 min to prevent condensation of water vapor on the detector side of the chip surface. The running buffer in the SPR competition experiments was made from HBS-EP buffer, 10× (BR-1006-69 from Biacore) diluted to 10 mm HEPES, 0.15 m NaCl, 3 mm EDTA, 0.05% Tween 20, and 0.5% DMSO. The running buffer in the direct binding experiments, here referred to as PBSTD buffer, was made by dilution of 10× (BR-1006-72 from Biacore) to 50 mm sodium phosphate, 100 mm NaCl (pH 7.4), and supplemented with 0.005% Tween 20 and 1% DMSO. The anionic detergent Tween 20 has previously been found not to inhibit PAI-1 (14) and was therefore used in all assays to prevent adhesion. Before use in Biacore, the running buffer was degassed for 5 min in an ultrasound bath (Branson 2510). The SPR competition experiments were run at 20 °C using a flow rate of 10 μl/min, and the SPR direct binding experiments were run at 25 °C using a flow rate of 30 μl/min. The obtained sensorgrams were processed and analyzed using Biacore 3000 or T200 evaluation software.
Protein Expression and Purification of Latent Non-glycosylated PAI-1
Synthetic genes encoding full-length human PAI-1 with an N-terminal His tag were ordered from Geneart (Invitrogen) and inserted into a modified PET vector (details available upon request). Overexpression was done in E. coli BL21 DE3 cells using shake flasks. Gene synthesis was induced using isopropyl-β-thio-galactopyranoside, and expression was done overnight at 22 °C. Cell cultures were harvested by centrifugation. Pellets were resuspended in 140 ml of 50 mm Tris-HCl buffer at pH 7.5 containing 0.5 m NaCl, 10 mm imidazole, and Complete EDTA-free (F. Hoffmann, La Roche, Ltd.) and sonicated at 70% with output level 9 for 25 s on ice using the large tip on the Sonifier 450W (Branson Ultrasonics Co. Danbury, CT). The lysate was cleared by centrifugation and purified on nickel-nitrilotriacetic acid (Superflow, Qiagen) and desalted into 20 mm Tris-HCl, pH 7.5, 150 mm NaCl. Latent PAI-1 was induced by incubating the protein for 23 h at 37 °C followed by size exclusion chromatography using Superdex 200 (GE Healthcare). The His tag was removed using tobacco etch virus protease (in-house) and purified using ion exchange (CaptoQ column, GE Healthcare) and a second size exclusion chromatography step, HiLoad 26/60 Superdex 200 (10 mm sodium acetate, pH 5.6, 150 mm NaCl, 1 mm EDTA). The homogeneity of the latent PAI-1 protein was verified using ITC, see below.
Protein Concentration Determination
The protein concentration after buffer exchange and dilution of stock solutions was determined from the absorbance spectrum between 250 and 350 nm with a NanoDrop ND-1000 (NanoDrop Technologies, Inc., Wilmington, DE) spectrophotometer according to E280 at pH 7.4 equal to 34.7, 35.8, 116, and 98 mm−1 cm−1 for active PAI-1, latent PAI-1, tPA, and VN, respectively (22, 45).
Specific Activity Determination of PAI-1
First, 5 nm active tPA, calculated from its specific activity, was preincubated with 0, 2, 4, 6, and 8 nm PAI-1 in 100 μl of running buffer for 16.5 min at 25 °C, and then the remaining tPA activity was monitored in a Spectramax® microplate reader (Molecular Devices Corp.) from the change in optical absorbance at 405 nm, A405, after addition of 100 μl of Pefachrome tPA to a final assay concentration of 0.5 mm. The specific activity of PAI-1 expressed in percent, was calculated from the plot of the rate, dA405/dt versus the ratio of total PAI-1 and tPA by linear extrapolation (S.D. 5%, n = 3).
High Throughput Screen Chromogenic Assay, Primary Screen
Small molecular weight PAI-1 inhibitors were identified using a 384-well chromogenic assay performed at room temperature, measuring residual tPA activity remaining after co-incubation of compounds from the AstraZeneca collection with PAI-1 and tPA. Compounds (0.6-μl 2 mm; final concentration, 30 μm) in 100% DMSO were predispensed to assay plates. First, 10 μl of 48 nm PAI-1 (final concentration 12 nm) in assay buffer, 50 mm Tris-HCl, pH 7.4, 100 mm NaCl, 0.01% Tween 20, 0.1% BSA at 4 °C, was added to the compound plates and incubated for 15 min at room temperature. BSA was an essential additive to maintain stability of PAI-1 in the large reagent volumes required for the screen. After incubation, 20 μl of 16 nm tPA (final concentration 8 nm) in assay buffer was added. The plate was incubated for an additional 20 min at room temperature before addition of 10 μl of the chromogenic substrate Pefachrome tPA (final assay concentration, 250 μm). The remaining tPA activity in each well was determined from A405 read at two time points, 0 and 25 min, in a Tecan Ultra microplate reader. Background A405 was taken immediately after addition of the substrate. The background A405 was subtracted from the final A405 taken after 25 min. Quality control plates were interleaved with the screening plates containing multiple wells of a maximum effect control, diaplasinin, a known PAI-1 inhibitor (46), and a minimum effect control, DMSO. The extent of PAI-1 inhibition (Unknown) was determined by relating the activity in the compound well to the medians of the diaplasinin and vehicle control wells using the following equation, % Effect = 100 × (Unknown − min)/(max − min). Positive hits were identified as any compound that inhibited PAI-1 activity by >30% and with more than three S.D. above plate medians.
The subsequent three-point concentration response screening (30, 10, and 3 μm final compound concentrations) in triplicate was used to confirm hits and give an indication of compounds with IC50 values <30 μm. Interesting clusters were subsequently selected for seven-point concentration response screening using 3:1 dilution steps and 100 μm highest final concentration. The IC50 value was calculated by non-linear regression from the plot of % inhibition versus concentration using the standard four parameter sigmoidal fit model, model 205 in the IDBS XLfit application. Concentration response screening was also carried out in presence of VN (final concentration, 14 nm).
Chromogenic Assay to Measure Inhibition of Rat PAI-1
First, 0.5-μl compounds in 100% DMSO were predispensed to 384-well Greiner assay plates enabling seven-point concentration screening with 3-fold dilution steps and a final highest compound concentration of 99 μm. Rat PAI-1, rat tPA, and Pefachrome tPA were diluted in assay buffer. First, 10-μl 20 nm rat PAI-1 was added to the assay plate and incubated for 10 min at room temperature, and then 20 μl of 10 nm rat tPA was added, and the plate was incubated for an additional 15 min before addition of 20 μl of Pefachrome tPA (final concentration, 150 μm). The remaining tPA activity in each well was determined from the A405 with a Pherastar microplate reader. Background absorbance measurements were taken immediately after addition of the substrate. The background was subtracted from the final absorbance reading taken after 1 h. Percent effect was calculated relative to the maximum effect with an excess of diaplasinin, and minimum effect controls (DMSO) were included in the assay plates. The IC50 value was calculated by non-linear regression from the plot of % inhibition versus concentration using the standard four-parameter sigmoidal fit model.
Inhibition of Human PAI-1 in Human Plasma
Plasma was thawed and incubated at 37 °C for ∼1 h. The other assay components were kept at room temperature except tPA and PAI-1, which were kept on ice. 5 μl of compound in DMSO was dispensed to 96-well Nunc F assay plates enabling 10-point concentration screening with 2-fold dilution steps and a final highest compound concentration of 82 μm. 45 μl of assay buffer containing 10 mm Hepes, pH 7.4, 0.15 m NaCl, and 33.3 mm CaCl2 was added to the assay plates. PAI-1 was diluted in 0.15 m NaCl with 0.01% Tween 20, and 25 μl was added to the assay plates. The assay plates were warmed briefly to 37 °C before adding 125 μl of plasma mixed 4:1 with human tPA diluted in 0.15 m NaCl with 0.01% Tween 20. The final concentrations for the assay components were 50% plasma, 0.65 nm PAI-1, and 1.2 nm tPA. The change in turbidity was followed from the change in A405 in a Spectramax microplate reader at 2-min intervals at 37 °C for up to 10 h. Clot longevity was calculated in seconds from t2–t1, where t1 was the time to half the amplitude on the positive slope and t2 was the time to halve the amplitude on the negative slope. The data were converted to % inhibition relative to on plate controls. DMSO in the presence of PAI-1 and tPA was used to calculate 0% inhibition, and DMSO in the presence of tPA only was used to calculate 100% inhibition. The IC50 value was calculated by non-linear regression from the plot of % inhibition versus concentration using the standard four-parameter sigmoidal fit model.
Isothermal Calorimetry
The ITC titration experiments were carried out on a MicroCal ITC-200 system (GE Healthcare) using protein that has been passed through a PD-10 column (GE Healthcare) equilibrated with 50 mm sodium phosphate (pH 7.4), 100 mm NaCl, and 2% DMSO. The concentration of AZ3976 was measured by NMR. Complete titration of 45 μm glycosylated active or 50 μm glycosylated latent PAI-1 was achieved by injecting 20 × 0.5-μl aliquots of 1 mm AZ3976 or 20 × 1-μl aliquots of 1 mm AZ3976, respectively, at 35 °C with 90 s of waiting time between subsequent injections. The thermodynamic parameters of the binding of AZ3976 were extracted by analysis of the binding isotherms applying a single site binding model using the MicroCal Origin software package (version 7.0), yielding binding enthalpy (ΔH), stoichiometry (n), entropy (ΔS), and association constant (Ka), from which the dissociation constant was calculated KD = 1/Ka.
Crystallization and Structure Determination
For crystallization, latent human non-glycosylated PAI-1 was concentrated to ∼4.0 g/liter in a buffer containing 10 mm sodium acetate, pH 5.6, 150 mm NaCl, and 1 mm EDTA. AZ3976 was dissolved in deuterated DMSO and added to the protein sample to a final concentration of 0.3 mm. Crystals were obtained by the hanging drop vapor diffusion technique: 2.5 μl of protein sample was mixed with 1 μl of a well solution containing 2.0 m NaCl, 100 mm bis-Tris, pH 6.8. The drop was allowed to equilibrate over a reservoir containing well solution. Prior to data collection, crystals were transferred to a drop of well solution supplemented with 20% glycerol as cryoprotectant. The crystals were then quickly removed from the drop and flash-frozen in liquid nitrogen. Data were collected at beam line ID23 (ESRF Grenoble) to 2.4 Å resolution.
The data were processed using MOSFLM (47), scaled, and further reduced, using the CCP4 suite of programs (Collaborative Computational Project) (48, 49). Initial phasing was done by molecular replacement using a high resolution ligand-free structure of latent PAI-1 (PDB code 1LJ5, as a starting model (18). Model rebuilding was performed within Coot (48), and refinement was performed (49). For data collection and refinement statistics, see Table 1.
TABLE 1.
Crystallographic data collection and refinement statistics
| Data collection | |
|---|---|
| Space group | P31 |
| Unit cell parameters | a = b = 119.40, c = 99.615 |
| Resolution range (Å) | 50–2.4 (2.46–2.4)a |
| No. of reflections (total/unique) | 149,812/62,179 |
| Redundancy | 5.9 (5.9) |
| Data completeness (%) | 100 (100) |
| Average I/σI | 16 (2.4) |
| Rmerge (%) | 9.9 (50) |
| Statistics for the final model | |
| Assymmetric unit content | |
| Nonhydrogen atoms | 9428 |
| Protein chains | 3 |
| AZ3976 copies | 3 |
| Water molecules | 244 |
| R factor (%) | 18.1 |
| Free R factor (%) | 15.7 |
| Wilson B factor (Å2) | 53.0 |
| Average B factors (Å2) | |
| All atoms | 45.9 |
| Protein (chains A/B/C) | 37.8/46.5/53.9 |
| Inhibitor (A/B/C) | 29.9/33.3/41.9 |
| r.m.s.d. bond length (Å)b | 0.014 |
| r.m.s.d. bond angles (°) | 1.485 |
| PDB code | 4AQH |
a Number within parenthesis refer to the highest resolution shell.
b r.m.s.d., root mean square deviation.
Immobilization of Proteins for SPR
For the SPR competition experiments, the dextran surfaces of all of the flow channels on the CM5 chip were first pegylated in borate buffer, pH 8.5. A fresh solution was made by dissolving 5.25 mg of PEG in 150 μl of borate buffer. The surface was activated for 7 min with 0.05 m NHS and 0.2 m EDC, followed by injection of dissolved PEG for 7 min, and this cycle was repeated once. Then, the pegylated CM5 sensor chip surface was activated again with the normal standardized amine coupling procedure with EDC/NHS, followed by coupling of the protein and thereafter deactivation of residual esters with 1 m ethanolamine hydrochloride, pH 8.5, and finally, HCl to remove non-covalently bound material. Two protein immobilization buffers were used: 10 mm sodium formate, pH 4.0, for coupling of VN and 10 mm sodium acetate, pH 4.0, for coupling of RamFc. Activation time, ligand concentration, and contact time were adjusted according to the desired response, the number of resonance units (RU). A flow rate of 5 μl/min, a deactivation time of 4 min, and regeneration time of 2 min with 50 mm HCl was used during immobilization. Subsequently, the surface was equilibrated with the running buffer to be used in the following experiments. On the pegylated chip with RamFc, the antibody was coupled to flow channel 1, flow channel 2 to 9000 RU, and to flow channel 4 to 4.0 kRU, but flow channel 3 was only pegylated. On the VN chip, VN was coupled to 900 RU only on flow channel 2, and the reference channel flow channel 1 was only pegylated. On the day of the binding experiment, the running buffer was degassed, and the chip was primed.
Immobilization procedures for glycosylated latent and active PAI-1 were performed using PBSTD buffer. The sensor surface was prepared using amine-coupling chemistry at 25 °C and at a flow rate of 10 μl/min. The surface was activated for 7 min with 0.05 m NHS and 0.2 m EDC, followed by injection of either latent or active PAI-1 (20 μg/ml in 10 mm sodium acetate, pH 5.0) for 2 min. Finally, 1 m ethanolamine hydrochloride, pH 8.5, was injected for 6 min to block residual activated groups. The immobilization strategy yielded similar immobilization levels of both forms of the protein to levels of 3.6–4.0 kRU. Reference surfaces were prepared accordingly, omitting the injection of protein over the activated reference surface.
Effect of AZ3976 on Binding of PAI-1 to VN and Effect of tPA
Before each new cycle, the VN surface was first regenerated with a 2-min pulse of 0.1 m HCl. First, AZ3976 in buffer was injected over the VN surface followed by active PAI-1 in buffer with compound. The effect of AZ3976 on PAI-1 binding to VN was studied in experiments where different concentrations of AZ3976 were preincubated with 10 nm PAI-1 for different time periods in running buffer at 20 °C before injection over immobilized VN. The initial binding rate was analyzed as a function of the inhibitor concentration and/or the incubation time of PAI-1 with compound. Subsequently, PAI-1 was stripped from the VN surface by injection of 5 nm tPA in the presence of the same concentration of compound as used in the PAI-1 sample using the KINJECT command.
Binding of PAI-1 to Captured tPA in Presence of AZ3976
First, on the reference channel, flow channel 1, a neutral monoclonal antibody was captured, and then the anti-human tPA antibody 2:2 B10 was captured on flow channel 2. Subsequently, tPA was injected and captured on B10. When PAI-1 then is injected over both flow channel 1 and flow channel 2, the quantification of the formation of the irreversible 1:1 complex with tPA on flow channel 2 is recorded from the difference in RU between flow channel 2 and flow channel 1. Because the Biacore response (ΔRU) is proportional to the change in mass on the chip, the PAI-1 binding rate is proportional to the change in RU, dRU/dt, and to the initial rate ΔRU/Δt for the first 10 s after start of injection. This sandwich method provides the possibility for complete regeneration of RamFc with 10 mm glycine, pH 1.7, for multiple experiments. For evaluation of the inhibition of PAI-1 in solution, AZ3976 in buffer was first injected over captured tPA, immediately followed by 10 nm PAI-1 preincubated with the compound, followed by tPA in buffer with compound. In some experiments in addition to the complex of B10 with tPA on flow channel 2, also the monoclonal antibody for latent non-glycosylated PAI-1, H4B3 was captured on flow channel 4. Subsequently, either active non-glycosylated or latent non-glycosylated human PAI-1 incubated for 1 h at 20 °C with compound was injected over all flow cells at the same time.
Binding of AZ3976 to Immobilized Latent and Active PAI-1
The compound AZ3976 was dissolved in 100% DMSO to a stock concentration of 100 mm. Dilutions into running buffer were performed immediately prior to analysis. To match the DMSO concentrations of the analyte and the running buffer, a secondary stock in running buffer was prepared by diluting the compound in DMSO to 1 mm through addition of PBSTD buffer and was further diluted in running buffer to yield a starting concentration of 20 μm for analysis. For the concentration response, this was further diluted 3-fold in running buffer with the lowest analyte concentration of 27.4 nm to yield a seven-point concentration response curve.
For the analysis, five running buffer blanks were injected to equilibrate the instrument. AZ3976 was injected for 90 s, and the dissociation was monitored for 4 min. As the complex is not tightly bound and rapidly dissociated under the experimental conditions, no regeneration step was required. The data collection rate was 10 Hz. All experiments have been repeated four times to allow for error estimations.
RESULTS
High Throughput Screen
In the primary chromogenic PAI-1 assay, 925,529 compounds from the AstraZeneca compound collection were screened. 10,331 compounds displayed >30% activity, and the subsequent three-point concentration response screening confirmed 3064 hits with IC50 values below 30 μm. Selection criteria at this and subsequent stages in the screening cascade considered parameters such as potency, ligand efficiency, lipophilic ligand efficiency, logD, and chemical structure differences to known PAI-1 inhibitors (particularly avoiding hydrophobic acids, which had failed earlier in-house optimization attempts). A set of ∼4000 hits and near neighbors to interesting clusters were selected for seven-point concentration response. Next, representatives of attractive series were selected for screening in a secondary plasma clot lysis assay. Six chemical series were then chosen for further lead identification work. One series displayed particularly favorable properties, with AZ3976 as a representative (Fig. 1). AZ3976 displayed an IC50 in the chromogenic assay of 26 μm for inhibition of PAI-1 and showed no effect on tPA alone at 100 μm. No inhibition by 100 μm AZ3976 was found in the chromogenic assay with PAI-1 in the presence of equimolar concentration of VN. AZ3976 was active in the human plasma clot lysis assay with added PAI-1, with an IC50 value of 16 μm (Fig. 2). The functional data in combination with the favorable physicochemical properties prompted additional studies to investigate the mode of action of AZ3976.
FIGURE 1.
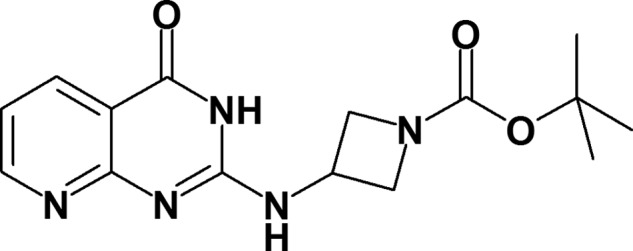
Chemical structure of AZ3976.
FIGURE 2.
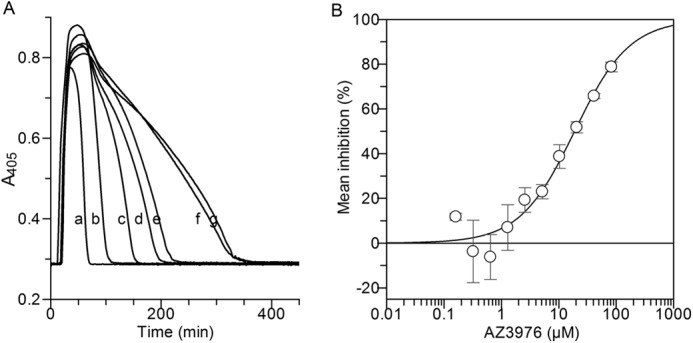
Effect of AZ3976 on clot lysis in human plasma with added PAI-1. Before addition of plasma, 1.7 nm PAI-1 in buffer was preincubated briefly at 37 °C with 0 to 0.22 mm AZ3976. The reaction was started by dilution with plasma containing tPA to final concentration of 50% plasma, 1.2 nm tPA and 0.65 nm PAI-1. Fibrin formation was monitored from the change in A405 in a microplate reader at 2-min intervals at 37 °C for up to 10 h. A, example of plasma clot lysis experiment: A405 is plotted versus time. Curves are shown for PAI-1 with 82.5 (b), 41.25 (c), 20.6 (d), 10.3 (e), 5.16 (f), and 0 μm (g) AZ3976 and for tPA only (a), without added PAI-1. B, the IC50 determination. The clot lysis time was defined as the time to halve the amplitude on the negative slope minus the time to halve the amplitude on the positive slope where the maximal effect (100%) was the time without added PAI-1 and minimal effect (0%) the time without added compound. The line depicts the best fit using Grafit IC50 fit. The mean of four experiments is plotted ± S.E.
Isothermal Calorimetry
Titration experiments were conducted at 35 °C (Fig. 3) to increase the binding enthalpy and thus the sensitivity of the measurements as well as to getting close to physiological conditions. As shown in Table 2 and Fig. 3, the titration of PAI-1 with AZ3976 revealed a KD of 0.38 μm with a binding stoichiometry (n value) of 0.30. The low stoichiometry suggested that AZ3976 only bound to a subpopulation in the preparation of active PAI-1. Because the specific activity in that preparation of active PAI-1 was ∼75%, the remaining 25% represents a different, inactive form of the protein. Given the presence of at least two different populations in that preparation, as well as the inherent instability of active PAI-1 at elevated temperatures, additional experiments were performed using latent PAI-1.
FIGURE 3.

Titration of glycosylated active and latent PAI-1 by isothermal calorimetry with AZ3976. Titration of 45 μm active PAI-1 with specific activity ∼75%, or 50 μm glycosylated latent PAI-1 in 50 mm sodium phosphate (pH 7.4), 100 mm NaCl, and 2% DMSO was achieved by injecting 20 × 0.5 μl and 20 × 1 μl aliquots, respectively, of 1 mm AZ3976 at 35 °C with 90 s waiting time between subsequent injections. The line depicts the best fit according to a single site binding model used to extract the thermodynamic properties of the interactions, which are shown in Table 2.
TABLE 2.
Thermodynamic parameters for AZ3976 interaction with active and latent PAI-1 determined by ITC at 35 °C
The parameters were extracted from the complete titration of active PAI-1 (specific activity ∼75%) or latent PAI-1, as shown in Fig. 3, by the best fit according to a single site binding model, yielding the binding enthalpy (ΔH), stoichiometry (n), entropy (−TΔS), and the association constant (Ka), from which the dissociation constant (KD = 1/Ka) as well as the binding enthalpy (ΔG) were calculated. Errors represent the errors from the data fit applying the Levenberg-Marquardt algorithm. For experimental conditions, see legend to Fig. 3.
| KD | ΔG | ΔH | −TΔS | n value | |
|---|---|---|---|---|---|
| μm | kcal/mol | kcal/mol | kcal/mol | ||
| Active PAI-1 | 0.38 ± 0.05 | −9.08 | −12.53 ± 0.12 | 3.45 | 0.30 ± 0.01 |
| Latent PAI-1 | 0.29 ± 0.05 | −9.22 | −14.99 ± 0.07 | 5.77 | 0.93 ± 0.01 |
The ITC data showed a KD of 0.29 μm with a binding stoichiometry of 0.93 for the complex formation of AZ3976 with glycosylated latent PAI-1 (Fig. 3). The thermodynamic parameters indicate that the binding of AZ3976 is purely enthalpy-driven and partially offset by an entropic penalty. These data point to good hydrogen bonding and specific interactions with the target and further suggest that conformational changes in the protein and/or the ligand lead to a reduction in overall ligand binding affinity. Despite similar affinities observed in the active and latent PAI-1 preparations, there are significant differences in the thermodynamic parameters, namely the binding enthalpy and entropy, see Table 2. The smaller binding enthalpy observed with the inactive fraction present in the active PAI-1 solution (ΔΔH = 2.46 kcal/mol) is balanced by a similar increase in entropy (Δ(−TΔS) = 2.32 kcal/mol), thus leaving the affinity almost unchanged. Thus, the data would suggest that the subpopulation of inactive PAI-1 in the active PAI-1 preparation is not completely identical with isolated latent PAI-1, i.e. there may be several ligand binding competent forms in the active PAI-1 preparation.
Taken together, the ITC data showed that although AZ3976 did not bind to active PAI-1, it bound with submicromolar affinity to latent PAI-1. Also, the thermodynamic signature revealed that AZ3976 binding to latent PAI-1 is largely enthalpy-driven and that the conformational flexibility of latent PAI-1 is probably restricted upon ligand binding.
X-ray Structure of Latent PAI-1 in Complex with AZ3976
Motivated by the ITC data, latent PAI-1 was co-crystallized with AZ3976, and the 2.4 Å structure was solved by molecular replacement using the structure of latent PAI-1 (PDB code 1DVN) (18). Although the overall fold of PAI-1 in the AZ3976 complex is similar to latent PAI-1, there are significant differences (Fig. 4). The largest deviations are observed in β-sheet C (residues Pro-180–Pro-200) and helices D and E (using nomenclature as defined by Stein and Chotia (50)).
FIGURE 4.

The structure of latent PAI-1 in complex with AZ3976. A, comparison of the latent PAI-1·AZ3976 holostructure with the latent PAI-1 apoform (PDB code 1DVN) (18). PAI-1 is colored by sequence from blue (N-terminal) to red (C-terminal). The inhibitor is shown in stick representation. The structure of the latent PAI-1 apostructure is shown as a gray ribbon representation. B, comparison of the AZ3976 binding site in ligand bound and free latent PAI-1. C, AZ3976 binding site in latent PAI-1. The Fo − Fc map calculated using the refined AZ3979-bound structure of latent PAI-1 is contoured at 3σ. D–F show the surface of the ligand binding site in three different forms of PAI-1. D, latent ligand complexed (this work); E, latent (PDB code 1DVN) (18); and F, active (PDB code 1DVM) (18). The magenta-colored surface describes the solvent accessible area in the region of AZ3979 binding. The nomenclature for the secondary structure is adopted from Stein and Chotia (50).
The Fo − Fc difference map displayed a strong feature where AZ3976 could unambiguously be modeled into the electron density in a low energy conformation. The ligand is bound in a cavity defined by α-helices A, B, D, and E, β-strands 1A and 2A, and the loops connecting α-helices B and D and connecting α-helix E and β-strand 1A, shown in detail in Fig. 4C.
The ligand binding pocket (Fig. 4, B and C) displays a close geometric fit to the ligand with good complementarities of polar and non-polar areas of the ligand. The side chain hydroxyl group of Tyr-37 in α-helix A forms a hydrogen bond to the carbamate carbonyl group. Asp-95 provides for three hydrogen bonds, with two via the side chain carboxyl group to nitrogens in the ligand and one via the main chain nitrogen to the carbonyl group in the pyrimidinone ring. Also, the side chain of Arg-76 forms a hydrogen bond to the pyridine nitrogen. In addition, π-stacking interaction between Tyr-79 and the pyridopyrimidinone bicyclic ring in the ligand is observed.
The ligand binding pocket identified here is poorly conserved across species. For example, five of the residues within 5 Å from the bound AZ3976 differ between human and rat (data not shown). Indeed, the inhibitory effect of AZ3976 appears to be species dependent as 100 μm of AZ3976 showed no effect on rat PAI-1 in the chromogenic assay.
SPR Measurements
A set of SPR experiments were designed to investigate how a compound with demonstrated ability to bind to latent, but not active, PAI-1 can inhibit this serpin in functional assays. First, as a control experiment, it was established that the initial binding rate of complex formation of active PAI-1 to captured tPA was linearly dependent on the PAI-1 concentration under the conditions of the experiment (data not shown). With latent PAI-1, no binding was observed, as shown previously (45). A similar control experiment was used with a concentration series of active PAI-1 injected over 900 RU immobilized VN (data not shown), and the conditions for linearity were found to be fulfilled also under these conditions. No binding of 10 nm latent PAI-1 to the VN surface was observed. Moreover, injection of tPA onto active PAI-1 captured on immobilized VN caused PAI-1 to dissociate.
In the presence of 100 μm AZ3976, but without preincubation, neither complex formation of active PAI-1 with tPA, nor the binding of active PAI-1 to VN was inhibited (data not shown). However, after gradually increasing preincubation time of active PAI-1 (10 nm) with AZ3976 (10 μm), active PAI-1 progressively lost its binding ability for VN (Fig. 5A). AZ3976 was found not to affect the stability of the complex between PAI-1 and VN, as injection of the compound for several hours onto PAI-1 bound to immobilized VN was indistinguishable from injection of buffer only (data not shown). Moreover, the rate of dissociation of PAI-1 from VN by addition of tPA was not either affected by the presence of AZ3976 (Fig. 5A). These data agree with the chromogenic screening data, where 100 μm AZ3976 did not inhibit the PAI-1·VN activity.
FIGURE 5.
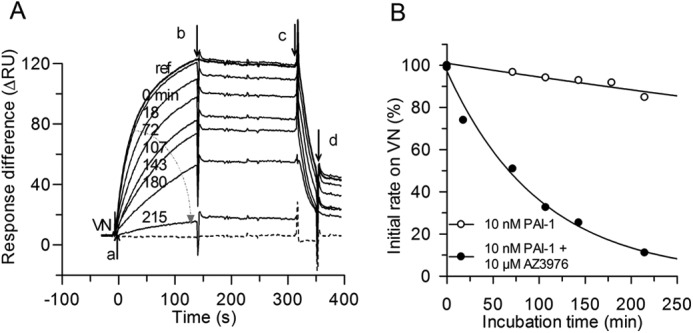
Interaction of PAI-1 with immobilized VN and effect of AZ3976. A, sensorgram overlay of the association and dissociation in real time after injection of 10 nm PAI-1 with 10 μm AZ3976 (a), incubated for increasing time intervals (arrow from 0 to 215 min) at 20 °C >900 RU immobilized VN, at change to buffer in the presence of 10 μm AZ3976 (b), and at injection of 20 μl of 25 nm tPA in the presence of 10 μm AZ3976 (c), followed by running buffer (d). The sensorgram of buffer only (dashed line) and reference, 10 nm PAI-1 in buffer without preincubation (marked ref) are included, whereas the sensorgrams of PAI-1 incubated in buffer are not shown for clarity. The ΔRU between the channel with immobilized VN, flow channel 2, and the channel without VN, flow channel 1, is shown. B, data are from the experiment shown in A. Initial binding rate to VN, measured after 10 s, as a function of the incubation time for 10 nm PAI-1 in buffer and for 10 nm PAI-1 with AZ3976 are shown. The lines are fitted to the data assuming single exponential decay, k = 0.0007 (buffer) and 0.01 min−1 (AZ3976), respectively.
The evaluation of the initial binding rate of PAI-1 to VN after different incubation times of active PAI-1 with 10 μm AZ3976 showed a mono exponential loss in binding of PAI-1 to VN (Fig. 5B). Because only active PAI-1 and not latent PAI-1 can bind to VN, this indicated loss of active PAI-1 after incubation with AZ3976. Additional experiments were therefore performed to investigate the binding of PAI-1 to captured tPA after different incubation times of active PAI-1 with AZ3976. Also, in this experimental set up, active PAI-1 lost the propensity to form an irreversible complex with tPA (Fig. 6A). For example, the t½ for decay of 10 nm active PAI-1 at 20 °C decreased from 15 h in buffer (k = 0.0008 min−1), to 40 min in the presence of 20 μm AZ3976 (Fig. 6B), a 23-fold apparent increase in the rate of transition from active PAI-1 to an “inactive” PAI-1 form. In Fig. 6B, it is shown that the rate constants calculated for the exponential decay in PAI-1 activity induced by the compound (Fig. 6A) was linearly dependent on the concentration of AZ3976, with a second order rate constant of 14 m−1·s−1 at 20 °C (slope in Fig. 6B). Note that the same rate constants were found both from the binding experiments with captured tPA and with immobilized VN (Fig. 6B), indicating that the same mechanism is at play.
FIGURE 6.
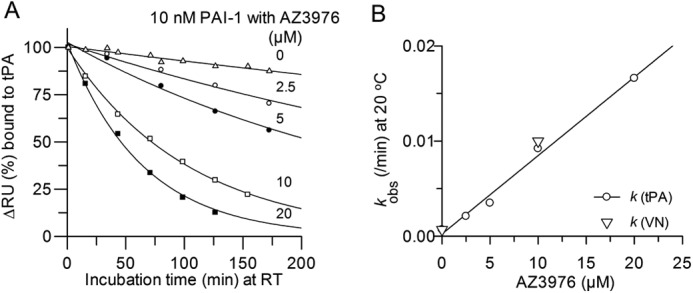
Interaction of PAI-1 with captured tPA and effect of incubation time with AZ3976. A, initial rate, measured as ΔRU, the change in RU 10 s after injection over tPA, as a function of the incubation time (min) of 10 nm PAI-1 with AZ3976 at different concentrations at 20 °C. Lines are drawn by fitting a single exponential decay to the data. B, rate constants from the data in A (circles) for single exponential decay of PAI-1, or from the data in Fig. 5 (triangles) from binding to VN, versus the concentration of AZ3976. The line was drawn by a linear fit with a slope of 14 m−1·s−1.
The above-described experiments have shown a transition of active PAI-1 to inactive PAI-1 after incubation with AZ3976. To investigate whether the inactive PAI-1 species induced by AZ3976 is identical to latent PAI-1, H4B3, a monoclonal antibody specific for non-glycosylated latent PAI-1, was used. By flowing active non-glycosylated PAI-1 over captured H4B3 some binding was found (Fig. 7A), but this is expected because the active PAI-1 batch did contain ∼25% inactive/latent PAI-1. Incubation with AZ3976 dramatically increased the binding to H4B3, concomitant with the decrease in activity of the binding to tPA, run in a parallel channel on the same chip. In the control experiment with 100% latent PAI-1, similar binding to H4B3 was found both with and without AZ3976 (Fig. 7B).
FIGURE 7.
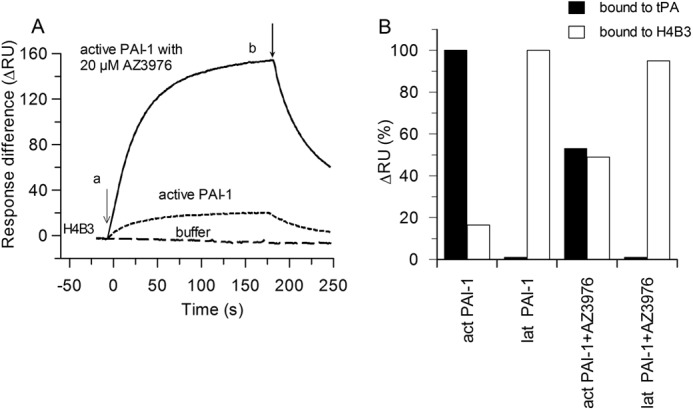
Decay of PAI-1 activity and concomitant generation of latent PAI-1. A, sensorgram overlay of injection onto the monoclonal antibody H4B3 (specific for non-glycosylated latent PAI-1) captured on immobilized RamFc at 10 nm non-glycosylated active PAI-1 in buffer (dotted line) or with 20 μm AZ3976 for 1 h (solid line), or buffer alone (dashed line) at 20 °C (a). b, flow change to running buffer with 20 μm AZ3976. The ΔRU between the channel with H4B3 and the channel with the reference antibody is shown. B, values in % of ΔRU for 10 nm PAI-1, measured from the sensorgram 70 s after injection, which represent active PAI-1 bound to captured tPA and latent PAI-1 bound to H4B3, without AZ3976 and with 20 μm AZ3976 incubated with PAI-1 for 1 h at 20 °C.
To extend the study and to monitor directly target engagement of AZ3976 with either glycosylated active or latent PAI-1, SPR experiments were conducted using immobilized proteins. The sensorgrams (Fig. 8) indicated reversible binding of AZ3976 with submicromolar affinity to both latent PAI-1 and to the fraction of inactive PAI-1 present in the active PAI-1 batch, following a simple 1:1 binding model. This is consistent with the ITC data. Also, the Rmax values that relate to the stoichiometry in the ITC experiments show a similar pattern, with active PAI-1 having only ∼23% ligand binding competence that appears to originate from an inactive/latent form of the protein in the preparation of active PAI-1. In contrast, latent PAI-1 appears to have a close to 100% ligand binding competence, taking into account the potential inaccessibility of the ligand binding site and/or denaturation processes due to the immobilization procedure. Furthermore, as shown in Table 3, the affinities of AZ3976 binding to either forms of latent PAI-1 are statistically indistinguishable from the ITC results.
FIGURE 8.

SPR sensorgrams for the interaction of AZ3976 with immobilized glycosylated latent and active PAI-1. Concentrations ranged from 20 μm to 27.4 nm following a 3-fold dilution scheme. The SPR experiments were run in PBSTD buffer at 25 °C, using a flow rate of 30 μl/min. The normalized responses were obtained by dividing the observed response levels with the predicted maximum response levels for a 1:1 binding interaction. The kinetic analysis was performed by fitting the data to a 1:1 binding model (fitted model traces as red dotted line). A, sensorgrams of AZ3976 binding to latent PAI. The normalized responses indicate a ligand binding activity of ∼90%. B, sensorgrams of AZ3976 binding to active PAI. The normalized responses indicate a ligand binding activity of ∼30%.
TABLE 3.
Kinetic and equilibrium binding parameters for AZ3976 interaction with active and latent PAI-1 determined by SPR at 25 °C
The kinetic rate constants kon and koff as well as the dissociation rate constant KD were extracted by kinetic analysis of the SPR sensorgrams by applying a simple 1:1 binding model. The data represent the average of four independent measurements, and errors represent the errors from the data fit applying the Levenberg-Marquardt algorithm. The parameter Rmax describes the normalized maximal analyte responses under steady state conditions. For experimental conditions, see legend to Fig. 8.
| KD | kon | koff | Rmax | |
|---|---|---|---|---|
| μm | m−1 s−1 | s−1 | ||
| Active PAI-1 | 0.24 ± 0.03 | 8.94 ± 2.91 × 104 | 0.021 ± 0.005 | 0.23 ± 0.02 |
| Latent PAI-1 | 0.19 ± 0.01 | 9.01 ± 2.98 × 104 | 0.017 ± 0.005 | 0.87 ± 0.04 |
DISCUSSION
The small molecule PAI-1 inhibitor AZ3976 was identified in a high throughput screening campaign. AZ3976 is a neutral molecule with overall drug-like properties, and it displayed profibrinolytic activities in a human plasma clot lysis assay. SPR experiments support that AZ3976 accelerates latency transition.
Remarkably, AZ3976 did not bind to active PAI-1 but bound reversibly to latent PAI-1 with submicromolar affinity, as shown both by ITC and SPR. The structure presented in this report is the first x-ray structure of latent PAI-1 in complex with a small molecule. AZ3976 binds in a deep pocket with the entry located between α-hD and β-s2A. This part of PAI-1 includes what is referred to as the flexible joint region (i.e. α-hD, α-hF, and β-s1A) because it participates in structural rearrangements going from active PAI-1 to the irreversible PAI-1·tPA complex (51). The presence of five hydrogen bonds indicates specific interactions between PAI-1 and AZ3976. Overall, the x-ray structural information agrees well with the results and interpretations of the ITC data, i.e. that ligand binding is largely enthalpy-driven and that the conformational flexibility of latent PAI-1 is probably restricted upon ligand binding.
The binding site for AZ3976 is >20 Å apart from the glycosylation sites in PAI-1, Asn-209, and Asn-265 (Fig. 9) (8). This is consistent with the finding that AZ3976 enhanced latency transition both of active glycosylated (Fig. 6) and non-glycosylated human PAI-1 (Fig. 7).
FIGURE 9.

Overview of inhibitor binding to PAI-1. A, the structure of active PAI-1 in complex with the somatomedin B domain of VN (PDB code 1OC0). PAI-1 is shown in a gray ribbon representation, except for the RCL, which is colored red (gap due to missing residues in structure). Somatomedin B is shown as a ribbon diagram in magenta. Additional residues implicated in VN binding are shown in stick representation colored in magenta and yellow. Residues implicated in binding of the small molecule PAI-1 inhibitor PAI-039 are shown in cyan and yellow, i.e. residues shown in yellow are implicated in both PAI-039 and VN binding. Glycosylation sites are shown in blue stick representation, and additional residues mentioned in the discussion are shown in green. B, the structure described in this work. The overall structure is colored the same way as in A, highlighting the different topology of the central β-sheet in the two structures. AZ3976 is shown in an orange stick representation and residues within 5 Å of the inhibitor are shown as cyan sticks.
The structural basis for unique binding of AZ3976 to latent PAI-1 over active PAI-1 may be related to intramolecular interactions in the two PAI-1 forms (Fig. 9). It should be noted that also latent PAI-1 needs to undergo conformational changes to accommodate AZ3976 binding (Fig. 4B and compare Fig. 4, D and E), mainly by departing α-hD from β-s2A. However, the changes required seem to be more extensive for active than for latent PAI-1 (Fig. 4, D–F). In active PAI-1 (PDB code 1DVM) (18), there is a hydrogen bond between the side chains of Thr-93 in β-s2A and Tyr-79 in α-hD, and the side chain of Asn-87 in the loop between α-hD and β-s2A forms hydrogen bonds to the peptide backbone of Leu-82 in α-hD and to Glu-90 and Ile-91 in β-s2A. None of these “tethering” interactions between α-hD and β-s2A are present in latent PAI-1 (PDB code 1DVN) (18). In latent PAI-1, Tyr-79 needs to switch to a different side chain rotamer to allow AZ3976 binding (Fig. 4B). Another difference between latent PAI-1 and the latent PAI-1 AZ3976 complex is the N-terminal part of α-hA where the Tyr-7 backbone conformation disrupts the α-helix in the complex structure, as a way to allow movement of α-hD necessary for ligand binding. It is uncertain whether the observed conformation of latent PAI-1 in complex with AZ3976 is present as an equilibrium state in the absence of ligand or whether it is induced by the ligand. However, the direct binding SPR experiments favor the former scenario because a simple 1:1 binding model fits the data (Fig. 8).
Structural data show that AZ3976 binds latent PAI-1 and VN binds active PAI-1 in partly overlapping regions (see Fig. 9). There is also evidence that VN has additional binding interactions with active PAI-1 outside the somatomedin domain B involving residues in α-hD (Lys-69, Arg-76, Tyr-79, and Lys-80) and α-hE (Arg-115 and Arg-118) (26, 27). Some of these residues are the same as those interacting with AZ3976 in the present structure, i.e. residues Arg-76, Tyr-79, and Arg-118. VN protects PAI-1 from the neutralizing effect by AZ3976. For example, AZ3976 did neither inhibit the PAI-1·VN complex in the chromogenic assay nor disrupt the PAI-1·VN complex in the SPR experiments with immobilized VN. The protective effects of VN may be due to direct steric effects, but prevention of the formation of the structural state to which AZ3976 binds might be important as well.
The region hosting the AZ3976 binding site has been implicated in binding to other small molecules, mainly through modeling and mutagenesis studies. Notably, these agents appear to act via several different mechanisms. For example, the small molecule AR-H029953xx has been suggested to bind near residues Arg-76 in α-hD and Arg-118 in the loop between α-hF and β-s1A, and it inhibited PAI-1 by preventing complex formation between PAI-1 and tPA (52). For another small molecule, PAI-039, e.g. Arg-76 and Tyr-79 in α-hD and Thr-93, Thr-94, and Asp-95 in β-s2A have been suggested to be involved in binding (see Fig. 9A) (53). Notably, all of these residues are within 5 Å from the AZ3976 ligand in the present structure (Fig. 9B). PAI-039 inhibited PAI-1 mainly by inducing substrate PAI-1 (53), where hydrolysis of the PAI-1·tPA complex becomes favored over formation of the irreversible PAI-1·tPA complex. Several other small molecules have been determined to bind in the same, or similar, region of PAI-1, e.g. ANS, bis-ANS, and XR5118 (51). Among the small molecule inhibitors, AZ3976 is unique because it (a) binds preferentially to latent PAI-1 over active PAI-1 and (b) inhibits PAI-1 by accelerating latency transition. In summary, the region centered on α-helices D and E and β-strands 1A and 2A, both in active and latent PAI-1, represents a “flexible joint hot spot” for interactions with large and small molecules in a region of great plasticity. Depending on the specific interactions between the inhibitory molecule and PAI-1, several different inhibitory mechanisms are possible.
The mechanistic path by which PAI-1 inhibits tPA (and uPA) is complex and requires acrobatic conformational flexibility of the protein (54, 55). This provides both a opportunity and a challenge for drug design; an opportunity because there are many conformational states of PAI-1 to which a molecule may interfere to achieve desired inhibition; a challenge because there are experimental difficulties to measure binding of an inhibitor to an isolated relevant PAI-1 form and to understand the mode of action and structure activity relationship.
A simplified mechanistic scheme for PAI-1 latency conversion, partially adapted from earlier literature (20, 56), is shown in Fig. 10. In this model, PAI-1 is hypothesized to be in a state of dynamic equilibrium between the active structure A, and an intermediate conformation, prelatent fraction, where prelatent fraction can be irreversibly converted into latent PAI-1, L. In active PAI-1, RCL is a flexible solvent-exposed loop, whereas in latent PAI-1, the RCL has inserted as a β-strand in β-sheet A (see Fig. 9). The current work supports that one or several intermediate states do exist and that these states are amenable to small molecule intervention to prevent the inhibitory action of PAI-1.
FIGURE 10.
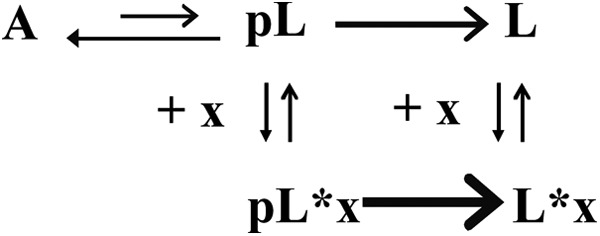
Mechanistic scheme of PAI-1 in presence of latency accelerating inhibitor. A represents active PAI-1, pL represents prelatent PAI-1, L represents latent PAI-1, and x indicates inhibitor.
The SPR competition data presented in this study show that the incubation time-dependent loss of active PAI-1 binding to tPA and VN is paralleled with increased inactive PAI-1 binding to the antibody H4B3 that is specific for latent PAI-1. The most plausible conclusion is that AZ3976 accelerates active PAI-1 conversion to the latent state. As shown, the t½ for decay of 10 nm active PAI-1 into inactive, latent PAI-1 at 20 °C decreased from 15 h in buffer to 40 min in the presence of 20 μm AZ3976 (Fig. 6). Previously, the activation energy for the spontaneous transition has been shown to be Ea = 92 ± 6 kJ in the same buffer as used in this study, with a t½ at 25 °C of 8.2 ± 0.3 h (22). This value is consistent with a t½ of 15 h at 20 °C in this report. Moreover, the rate constant for the exponential decay in PAI-1 activity induced by the compound is linearly dependent on the concentration of AZ3976, suggesting that the complex formation with a prelatent fraction is rate-limiting. The SPR data are thus consistent with the hypothesis that AZ3976 binds to a prelatent conformation of PAI-1 and decreases the transition state energy for the conversion of active to latent PAI-1.
Although this is the first report of a small molecule inactivating PAI-1 by accelerating latency transition, both antibodies (11, 13) and a peptide (57) have been shown to inhibit PAI-1 through a comparable mode of action. One of these antibodies is MA-159M12, a monoclonal antibody against rat PAI-1, which decreased the half-life of active PAI-1 ∼7-fold (11). Alanine-scanning mutagenesis experiments showed that the N-terminal region of α-hA constitutes major part of the epitope for MA-159M12 (11). Interestingly, this epitope is adjacent to α-hD, which is involved in binding of AZ3976, allowing speculations that the kinetic stability of active PAI-1 is sensitive to its structural integrity in this region. MA-33B8 is a monoclonal antibody against human PAI-1, which also inhibits PAI-1 by accelerating latency transition (13). MA-33B8 did bind both to latent PAI-1 and to the complex between tPA and PAI-1 at a faster rate compared with active PAI-1, and it was suggested that the antibody binding epitope was more exposed in a prelatent conformation of PAI-1 (e.g. prelatent PAI-1 in Fig. 10) (13). Subsequent mutagenesis studies indicated that the epitope for MA-33B8 includes the loops between α-hD and β-s2A and between β-s2C and β-s3C and also includes the residues proceeding β-s3A (10, 56). Additional characterization of this antibody using SPR experiments indicated about 100-fold greater affinity of MA-33B8 for latent PAI-1 compared with active PAI-1, a finding that was substantiated by an analysis of the active and latent x-ray structures indicating that the epitope is more compact in latent compared with active PAI-1 (56). Note the similarity to AZ3976, which, according to ITC and SPR measurements, binds latent PAI-1 with high affinity (KD 0.19–0.29 μm), whereas the affinity to active PAI-1 was negligible. Both for MA-33B8 and AZ3976, the binding sites were apparently more accessible in latent PAI-1 than in active PAI-1. Another monoclonal antibody H4B3 also speeds up formation of latent PAI-1 (58). This antibody has been described to depend on partial detachment of s1C for binding a prelatent form of PAI-1, and it was shown to bind strongly to latent PAI-1 but not active PAI-1 (58). Even though the binding epitope of H4B3 is far from the regions of binding for AZ3976, MA-159M12, and MA-33B8, it is uncertain whether the prelatent forms that they are hypothesized to bind to are the same or different. Paionin-4-D1D2 is a 31-residue peptide fused to domains 1 and 2 of the phage coat protein g3p, which also stimulates latency transition (57). Based on site-directed mutagenesis, the binding site mainly involved the loop between α-hD and β-s2A (overlapping binding site with that of MA-33B8). Furthermore, paionin-4-D1D2 displayed similar submicromolar binding affinities both for active and latent human PAI-1, and VN was shown to protect active PAI-1 from binding the peptide (57).
Taken together, AZ3976 shares several important features with both the antibodies and the peptide reported to accelerate PAI-1 latency transition. First, the regions in PAI-1 to which they bind all participate in the structural rearrangements required for the transition of active PAI-1 to its latent form. Second, they bind to latent PAI-1 with similar or higher affinity compared with active PAI-1. From this, it may be hypothesized that latent PAI-1 better mimics one or more prelatent forms than active PAI-1, and therefore latent PAI-1 could represent a valuable “surrogate” for such prelatent structures.
AZ3976 was not tested in vivo because it did not inhibit rat PAI-1 in vitro and was unstable in rat plasma. Thus, in vivo efficacy of a small molecule that accelerates PAI-1 latency transition remains to be established. Interestingly, however, the latency inducing antibody MA-33B8 has been shown to have a profibrinolytic effect in vivo (40). However, there are significant differences between the MA-33B8 antibody and the small molecule AZ3976. For example, MA-33B8 decreased the half-life of active PAI-1 ∼4000-fold (13) compared with ∼20-fold for AZ3976, although direct comparison of figures may be misleading as the values depend on the experimental conditions. Furthermore, MA33B8 induced latency transition also in the presence of VN (13), whereas 100 μm AZ3976 did not inhibit the PAI-1·VN complex. Still, a potential benefit with a small molecule is that it can act on intracellular PAI-1, e.g. PAI-1 in the platelets, smooth muscle cells, endothelial cells, and adipocytes. The main part of active PAI-1 in blood is stored in the α-granules in platelets (30). This PAI-1 is not in complex with VN (28–30) and is released upon platelet activation (28, 59). Blood clots contain large amounts of PAI-1 that may originate from the α-granules of activated platelets and arterial clots contain 2- to 3-fold more PAI-1 than venous clots and are more resistant to thrombolysis (60, 61). Thus, a chronic administration of a small molecule with the same mode of action as AZ3976 could potentially inactivate PAI-1 stored in platelets before it becomes excreted. Because molecules like AZ3976 would be expected to be less effective in inhibiting already circulating PAI-1, which is complexed to VN (21), this approach would be predicted to be more advantageous in long term prevention therapy than in acute treatment of thrombosis and fibrosis.
In summary, the small molecule PAI-1 inhibitor AZ3976 displayed profibrinolytic activities in a human plasma clot lysis assay, and biophysical characterization support a mode of action with induced accelerated latency transition. This mechanism and the presented x-ray data outline an exciting opportunity for improved structure-aided drug design endeavors in the hunt for therapeutically useful small molecule PAI-1 inhibitors.
All authors are, or were, employees of AstraZeneca.
The atomic coordinates and structure factors (code 4AQH) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PAI-1
- plasminogen activator inhibitor type 1
- DMSO
- dimethyl sulfoxide
- EDC
- N-ethyl-N′-(3-dimethyl aminopropyl)-carbodiimide hydrochloride
- IC50
- concentration giving 50% inhibition
- ITC
- isothermal calorimetry
- NHS
- N-hydroxy-succinimide
- RamFc
- rabbit anti-mouse Fc
- RCL
- reactive center loop
- RU
- resonance units
- SPR
- surface plasmon resonance
- tPA
- tissue-type plasminogen activator
- uPA
- urokinase-type plasminogen activator
- VN
- vitronectin
- PDB
- Protein Data Bank.
REFERENCES
- 1. Maksimenko A. V. (2002) Macromolecular ensembles of external and internal fibrinolysis: ways to enhance the thrombolytic effect (a review). Pharmaceut. Chem. J. 36, 348–355 [Google Scholar]
- 2. Dupont D. M., Madsen J. B., Kristensen T., Bodker J. S., Blouse G. E., Wind T., Andreasen P. A. (2009) Biochemical properties of plasminogen activator inhibitor-1. Front. Biosci. 14, 1337–1361 [DOI] [PubMed] [Google Scholar]
- 3. Cesari M., Pahor M., Incalzi R. A. (2010) Plasminogen activator inhibitor-1 (PAI-1): a key factor linking fibrinolysis and age-related subclinical and clinical conditions. Cardiovasc. Ther. 28, e72-e91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rijken D. C., Lijnen H. R. (2009) New insights into the molecular mechanisms of the fibrinolytic system. J. Thromb. Haemost. 7, 4–13 [DOI] [PubMed] [Google Scholar]
- 5. Lin Z., Jiang L., Yuan C., Jensen J. K., Zhang X., Luo Z., Furie B. C., Furie B., Andreasen P. A., Huang M. (2011) Structural basis for recognition of urokinase-type plasminogen activator by plasminogen activator inhibitor-1. J. Biol. Chem. 286, 7027–7032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jensen J. K., Thompson L. C., Bucci J. C., Nissen P., Gettins P. G., Peterson C. B., Andreasen P. A., Morth J. P. (2011) Crystal structure of plasminogen activator inhibitor-1 in an active conformation with normal thermodynamic stability. J. Biol. Chem. 286, 29709–29717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Whisstock J. C., Silverman G. A., Bird P. I., Bottomley S. P., Kaiserman D., Luke C. J., Pak S. C., Reichhart J. M., Huntington J. A. (2010) Serpins flex their muscle: II. Structural insights into target peptidase recognition, polymerization, and transport functions. J. Biol. Chem. 285, 24307–24312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xue Y., Björquist P., Inghardt T., Linschoten M., Musil D., Sjölin L., Deinum J. (1998) Interfering with the inhibitory mechanism of serpins: crystal structure of a complex formed between cleaved plasminogen activator inhibitor type 1 and a reactive-centre loop peptide. Structure 6, 627–636 [DOI] [PubMed] [Google Scholar]
- 9. Mottonen J., Strand A., Symersky J., Sweet R. M., Danley D. E., Geoghegan K. F., Gerard R. D., Goldsmith E. J. (1992) Structural basis of latency in plasminogen activator inhibitor-1. Nature 355, 270–273 [DOI] [PubMed] [Google Scholar]
- 10. Naessens D., Gils A., Compernolle G., Declerck P. J. (2003) Elucidation of the epitope of a latency-inducing antibody: identification of a new molecular target for PAI-1 inhibition. Thromb. Haemost. 90, 52–58 [PubMed] [Google Scholar]
- 11. Ngo T. H., Zhou Y., Stassen J. M., Declerck P. J. (2002) Importance of N-terminal residues in plasminogen activator inhibitor 1 on its antibody induced latency transition. Thromb. Haemost. 88, 288–293 [PubMed] [Google Scholar]
- 12. Strömqvist M., Andersson J. O., Boström S., Deinum J., Ehnebom J., Enquist K., Johansson T., Hansson L. (1994) Separation of active and inactive forms of recombinant human plasminogen activator inhibitor type 1 (PAI-1) expressed in Chinese hamster ovary cells: comparison with native human PAI-1. Protein Expr. Purif. 5, 309–316 [DOI] [PubMed] [Google Scholar]
- 13. Verhamme I., Kvassman J. O., Day D., Debrock S., Vleugels N., Declerck P. J., Shore J. D. (1999) Accelerated conversion of human plasminogen activator inhibitor-1 to its latent form by antibody binding. J. Biol. Chem. 274, 17511–17517 [DOI] [PubMed] [Google Scholar]
- 14. Ehnebom J., Björquist P., Andersson J., Johansson T., Deinum J. (1997) Detergent Tween 80 modifies the specific activity of Pal-1. Fibrinolysis Proteol. 11, 165–170 [Google Scholar]
- 15. Ehnebom J., Pusa S., Bjorquist P., Deinum J. (1997) Comparison of chromogenic substrates for tissue plasminogen activator and the effects on the stability of plasminogen activator inhibitor type-1. Fibrinolysis Proteol. 11, 287–293 [Google Scholar]
- 16. Levin E. G., Santell L. (1987) Conversion of the active to latent plasminogen activator inhibitor from human endothelial cells. Blood 70, 1090–1098 [PubMed] [Google Scholar]
- 17. Lindahl T. L., Sigurdardottir O., Wiman B. (1989) Stability of plasminogen activator inhibitor 1 (PAI-1). Thromb. Haemost. 62, 748–751 [PubMed] [Google Scholar]
- 18. Stout T. J., Graham H., Buckley D. I., Matthews D. J. (2000) Structures of active and latent PAI-1: a possible stabilizing role for chloride ions. Biochemistry 39, 8460–8469 [DOI] [PubMed] [Google Scholar]
- 19. Willems P. K., Rabijns A., Aertgeerts K., Vleugels N., Knockaert I., De Bondt H. L., De Ranter C. J., Declerck P. J. (1999) Plasminogen activator inhibitor 1 (PAI-1) in its active conformation: crystallization and preliminary X-ray diffraction data. Acta Crystallogr. D 55, 574–576 [DOI] [PubMed] [Google Scholar]
- 20. Nar H., Bauer M., Stassen J. M., Lang D., Gils A., Declerck P. J. (2000) Plasminogen activator inhibitor 1. Structure of the native serpin, comparison to its other conformers and implications for serpin inactivation. J. Mol. Biol. 297, 683–695 [DOI] [PubMed] [Google Scholar]
- 21. Sigurdardóttir O., Wiman B. (1990) Complex formation between plasminogen activator inhibitor 1 and vitronectin in purified systems and in plasma. Biochim. Biophys. Acta 1035, 56–61 [DOI] [PubMed] [Google Scholar]
- 22. Ehnebom J., Björquist P., Sigurdardottir O., Deinum J. (2000) Characterization of the interaction of plasminogen activator inhibitor type 1 with vitronectin by surface plasmon resonance. Fibrinolysis Proteol. 14, 47–57 [Google Scholar]
- 23. Zhou A., Huntington J. A., Pannu N. S., Carrell R. W., Read R. J. (2003) How vitronectin binds PAI-1 to modulate fibrinolysis and cell migration. Nat. Struct. Biol. 10, 541–544 [DOI] [PubMed] [Google Scholar]
- 24. Kluft C., Verheijen J. H. (1990) Leiden Fibrinolysis Working Party: Blood collection and handling procedures for assessment of tissue-type plasminogen activator (t-PA) and plasminogen activator inhibitor-1 (PAI-1). Fibrinolysis 4, Suppl 2, 155–161 [Google Scholar]
- 25. Blouse G. E., Dupont D. M., Schar C. R., Jensen J. K., Minor K. H., Anagli J. Y., Gårdsvoll H., Ploug M., Peterson C. B., Andreasen P. A. (2009) Interactions of plasminogen activator inhibitor-1 with vitronectin involve an extensive binding surface and induce mutual conformational rearrangements. Biochemistry 48, 1723–1735 [DOI] [PubMed] [Google Scholar]
- 26. Schar C. R., Blouse G. E., Minor K. H., Peterson C. B. (2008) A deletion mutant of vitronectin lacking the somatomedin B domain exhibits residual plasminogen activator inhibitor-1-binding activity. J. Biol. Chem. 283, 10297–10309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schar C. R., Jensen J. K., Christensen A., Blouse G. E., Andreasen P. A., Peterson C. B. (2008) Characterization of a site on PAI-1 that binds to vitronectin outside of the somatomedin B domain. J. Biol. Chem. 283, 28487–28496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Robbie L. A., Booth N. A., Brown A. J., Bennett B. (1996) Inhibitors of fibrinolysis are elevated in atherosclerotic plaque. Arterioscl. Thromb. Vasc. Biol. 16, 539–545 [DOI] [PubMed] [Google Scholar]
- 29. Lang I. M., Schleef R. R. (1996) Calcium-dependent stabilization of type I plasminogen activator inhibitor within platelet α-granules. J. Biol. Chem. 271, 2754–2761 [DOI] [PubMed] [Google Scholar]
- 30. Nordenhem A., Wiman B. (1997) Plasminogen activator inhibitor-1 (PAI-1) content in platelets from healthy individuals genotyped for the 4G/5G polymorphism in the PAI-1 gene. Scand. J. Clin. Lab. Invest. 57, 453–461 [DOI] [PubMed] [Google Scholar]
- 31. Brogren H., Wallmark K., Deinum J., Karlsson L., Jern S. (2011) Platelets retain high levels of active plasminogen activator inhibitor 1. PLoS ONE 6, e26762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hrafnkelsdottir T., Gudnason T., Wall U., Jern C., Jern S. (2004) Regulation of local availability of active tissue-type plasminogen activator in vivo in man. J. Thromb. Haemost. 2, 1960–1968 [DOI] [PubMed] [Google Scholar]
- 33. Cale J. M., Li S. H., Warnock M., Su E. J., North P. R., Sanders K. L., Puscau M. M., Emal C. D., Lawrence D. A. (2010) Characterization of a novel class of polyphenolic inhibitors of plasminogen activator inhibitor-1. J. Biol. Chem. 285, 7892–7902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Crandall D. L., Quinet E. M., El Ayachi S., Hreha A. L., Leik C. E., Savio D. A., Juhan-Vague I., Alessi M. (2006) Modulation of adipose tissue development by pharmacological inhibition of PAI-1. Arterioscl. Thromb. Vasc. Biol. 26, 2209–2215 [DOI] [PubMed] [Google Scholar]
- 35. Elokdah H., Abou-Gharbia M., Hennan J. K., McFarlane G., Mugford C. P., Krishnamurthy G., Crandall D. L. (2004) Tiplaxtinin, a novel, orally efficacious inhibitor of plasminogen activator inhibitor-1: design, synthesis, and preclinical characterization. J. Med. Chem. 47, 3491–3494 [DOI] [PubMed] [Google Scholar]
- 36. Falk K., Björquist P., Strömqvist M., Holmdahl L. (2001) Reduction of experimental adhesion formation by inhibition of plasminogen activator inhibitor type 1. Brit. J. Surg. 88, 286–289 [DOI] [PubMed] [Google Scholar]
- 37. Izuhara Y., Takahashi S., Nangaku M., Takizawa S., Ishida H., Kurokawa K., van Ypersele de Strihou C., Hirayama N., Miyata T. (2008) Inhibition of plasminogen activator inhibitor-1: its mechanism and effectiveness on coagulation and fibrosis. Arterioscl. Thromb. Vasc. Biol. 28, 672–677 [DOI] [PubMed] [Google Scholar]
- 38. Izuhara Y., Yamaoka N., Kodama H., Dan T., Takizawa S., Hirayama N., Meguro K., van Ypersele de Strihou C., Miyata T. (2010) A novel inhibitor of plasminogen activator inhibitor-1 provides antithrombotic benefits devoid of bleeding effect in nonhuman primates. J. Cereb. Blood Flow Metab. 30, 904–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Levi M., Biemond B. J., van Zonneveld A. J., ten Cate J. W., Pannekoek H. (1992) Inhibition of plasminogen activator inhibitor-1 activity results in promotion of endogenous thrombolysis and inhibition of thrombus extension in models of experimental thrombosis. Circulation 85, 305–312 [DOI] [PubMed] [Google Scholar]
- 40. Montes R., Declerck P. J., Calvo A., Montes M., Hermida J., Muñoz M. C., Rocha E. (2000) Prevention of renal fibrin deposition in endotoxin-induced DIC through inhibition of PAI-1. Thromb. Haemost. 84, 65–70 [PubMed] [Google Scholar]
- 41. Pandya V., Jain M., Chakrabarti G., Soni H., Parmar B., Chaugule B., Patel J., Joshi J., Joshi N., Rath A., Raviya M., Shaikh M., Sairam K. V., Patel H., Patel P. (2011) Discovery of inhibitors of plasminogen activator inhibitor-1: structure-activity study of 5-nitro-2-phenoxybenzoic acid derivatives. Bioorg. Med. Chem. Lett. 21, 5701–5706 [DOI] [PubMed] [Google Scholar]
- 42. van Giezen J. J., Wahlund G., Nerme, Abrahamsson T. (1997) The Fab-fragment of a PAI-1 inhibiting antibody reduces thrombus size and restores blood flow in a rat model of arterial thrombosis. Thromb. Haemost. 77, 964–969 [PubMed] [Google Scholar]
- 43. Lucking A. J., Visvanathan A., Philippou H., Fraser S., Grant P. J., Connolly T. M., Gardell S. J., Feuerstein G. Z., Fox K. A., Booth N. A., Newby D. E. (2010) Effect of the small molecule plasminogen activator inhibitor-1 (PAI-1) inhibitor, PAI-749, in clinical models of fibrinolysis. J. Thromb. Haemost. 8, 1333–1339 [DOI] [PubMed] [Google Scholar]
- 44. Lucking A. J., Chelliah R., Trotman A. D., Connolly T. M., Feuerstein G. Z., Fox K. A., Boon N. A., Badimon J. J., Newby D. E. (2010) Characterisation and reproducibility of a human ex vivo model of thrombosis. Thromb. Res. 126, 431–435 [DOI] [PubMed] [Google Scholar]
- 45. Björquist P., Brohlin M., Ehnebom J., Ericsson M., Kristiansen C., Pohl G., Deinum J. (1994) Plasminogen activator inhibitor type-1 interacts exclusively with the proteinase domain of tissue plasminogen activator. Biochim. Biophys. Acta 1209, 191–202 [DOI] [PubMed] [Google Scholar]
- 46. Gardell S. J., Krueger J. A., Antrilli T. A., Elokdah H., Mayer S., Orcutt S. J., Crandall D. L., Vlasuk G. P. (2007) Neutralization of plasminogen activator inhibitor I (PAI-1) by the synthetic antagonist PAI-749 via a dual mechanism of action. Mol. Pharmacol. 72, 897–906 [DOI] [PubMed] [Google Scholar]
- 47. Leslie A. (1999) Integration of macromolecular diffraction data. Acta Crystallogr. D 55, 1696–1702 [DOI] [PubMed] [Google Scholar]
- 48. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 49. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stein P., Chothia C. (1991) Serpin tertiary structure transformation. J. Mol. Biol. 221, 615–621 [DOI] [PubMed] [Google Scholar]
- 51. Egelund R., Einholm A. P., Pedersen K. E., Nielsen R. W., Christensen A., Deinum J., Andreasen P. A. (2001) A regulatory hydrophobic area in the flexible joint region of plasminogen activator inhibitor-1, defined with fluorescent activity-neutralizing ligands. Ligand-induced serpin polymerization. J. Biol. Chem. 276, 13077–13086 [DOI] [PubMed] [Google Scholar]
- 52. Björquist P., Ehnebom J., Inghardt T., Hansson L., Lindberg M., Linschoten M., Strömqvist M., Deinum J. (1998) Identification of the binding site for a low-molecular-weight inhibitor of plasminogen activator inhibitor type 1 by site-directed mutagenesis. Biochemistry 37, 1227–1234 [DOI] [PubMed] [Google Scholar]
- 53. Gorlatova N. V., Cale J. M., Elokdah H., Li D., Fan K., Warnock M., Crandall D. L., Lawrence D. A. (2007) Mechanism of inactivation of plasminogen activator inhibitor-1 by a small molecule inhibitor. J. Biol. Chem. 282, 9288–9296 [DOI] [PubMed] [Google Scholar]
- 54. Björquist P., Ehnebom J., Deinum J. (1999) Protein movement during complex-formation between tissue plasminogen activator and plasminogen activator inhibitor-1. Biochim. Biophys. Acta 1431, 24–29 [DOI] [PubMed] [Google Scholar]
- 55. Huntington J. A. (2006) Shape-shifting serpins–advantages of a mobile mechanism. Trends Biochem. Sci. 31, 427–435 [DOI] [PubMed] [Google Scholar]
- 56. Gorlatova N. V., Elokdah H., Fan K., Crandall D. L., Lawrence D. A. (2003) Mapping of a conformational epitope on plasminogen activator inhibitor-1 by random mutagenesis. Implications for serpin function. J. Biol. Chem. 278, 16329–16335 [DOI] [PubMed] [Google Scholar]
- 57. Mathiasen L., Dupont D. M., Christensen A., Blouse G. E., Jensen J. K., Gils A., Declerck P. J., Wind T., Andreasen P. A. (2008) A peptide accelerating the conversion of plasminogen activator inhibitor-1 to an inactive latent state. Mol. Pharmacol. 74, 641–653 [DOI] [PubMed] [Google Scholar]
- 58. Dupont D. M., Blouse G. E., Hansen M., Mathiasen L., Kjelgaard S., Jensen J. K., Christensen A., Gils A., Declerck P. J., Andreasen P. A., Wind T. (2006) Evidence for a pre-latent form of the serpin plasminogen activator inhibitor-1 with a detached β-strand 1C. J. Biol. Chem. 281, 36071–36081 [DOI] [PubMed] [Google Scholar]
- 59. Roessler F. C., Ohlrich M., Marxsen J. H., Stellmacher F., Sprenger A., Dempfle C. E., Seidel G. (2011) The platelet-rich plasma clot: a standardized in vitro clot formation protocol for investigations of sonothrombolysis under physiological flows. Blood Coagul. Fibrinolysis 22, 407–415 [DOI] [PubMed] [Google Scholar]
- 60. Booth N. A., Robbie L. A., Croll A. M., Bennett B. (1992) Lysis of platelet-rich thrombi: the role of PAI-1. Ann. N.Y. Acad. Sci. 667, 70–80 [DOI] [PubMed] [Google Scholar]
- 61. Potter van Loon B. J., Rijken D. C., Brommer E. J., van der Maas A. P. (1992) The amount of plasminogen, tissue-type plasminogen activator and plasminogen activator inhibitor type 1 in human thrombi and the relation to ex vivo lysibility. Thromb. Haemost. 67, 101–105 [PubMed] [Google Scholar]