

Background: The functional importance of Parkin in the heart is unknown.
Results: Parkin deficiency results in increased susceptibility to myocardial infarction.
Conclusion: Parkin is important in adapting to stress.
Significance: Our studies will advance our knowledge of Parkin in cardiovascular disease.
Keywords: Autophagy, Heart Failure, Mitochondria, Myocardial Infarction, Parkin
Abstract
It is known that loss-of-function mutations in the gene encoding Parkin lead to development of Parkinson disease. Recently, Parkin was found to play an important role in the removal of dysfunctional mitochondria via autophagy in neurons. Although Parkin is expressed in the heart, its functional role in this tissue is largely unexplored. In this study, we have investigated the role of Parkin in the myocardium under normal physiological conditions and in response to myocardial infarction. We found that Parkin-deficient (Parkin−/−) mice had normal cardiac function for up to 12 months of age as determined by echocardiographic analysis. Although ultrastructural analysis revealed that Parkin-deficient hearts had disorganized mitochondrial networks and significantly smaller mitochondria, mitochondrial function was unaffected. However, Parkin−/− mice were much more sensitive to myocardial infarction when compared with wild type mice. Parkin−/− mice had reduced survival and developed larger infarcts when compared with wild type mice after the infarction. Interestingly, Parkin protein levels and mitochondrial autophagy (mitophagy) were rapidly increased in the border zone of the infarct in wild type mice. In contrast, Parkin−/− myocytes had reduced mitophagy and accumulated swollen, dysfunctional mitochondria after the infarction. Overexpression of Parkin in isolated cardiac myocytes also protected against hypoxia-mediated cell death, whereas nonfunctional Parkinson disease-associated mutants ParkinR42P and ParkinG430D had no effect. Our results suggest that Parkin plays a critical role in adapting to stress in the myocardium by promoting removal of damaged mitochondria.
Introduction
Autophagy is important in degrading long-lived proteins and organelles in cells. It occurs constitutively in most cells but can be rapidly increased in response to changes in the intracellular environment (1). Autophagy is very important in the heart, and impaired autophagy is associated with a wide variety of cardiovascular pathologies. For instance, Danon disease is caused by a deficiency in lysosome-associated membrane protein 2 (LAMP2) that results in a fatal cardiomyopathy (2). The LAMP2 deficiency leads to an accumulation of autophagosomes in the heart due to defective fusion between autophagosomes and lysosomes (3). In addition, disruption of autophagy by conditional deletion of Atg5 in the adult heart leads to accumulation of dysfunctional mitochondria and development of cardiac dysfunction (4). Increased autophagy is also commonly observed in acute and chronic myocardial ischemia, heart failure, and dilated cardiomyopathy (5–7). Although the functional role of autophagy is still controversial, most studies suggest that autophagy is a protective response activated by cells in the heart (5, 6, 8, 9).
When mitochondria become dysfunctional, they produce reactive oxygen species that can cause further damage to nearby mitochondria and result in the release of pro-apoptotic proteins. Thus, damaged mitochondria must quickly be removed by autophagy to prevent further damage from occurring in the cell. Exactly how these mitochondria are targeted for removal by autophagosomes in myocytes is still unclear. The E3 ubiquitin ligase Parkin was recently discovered to play an important role in targeting damaged mitochondria for removal via autophagy in neurons (10). Loss-of-function mutations in the gene encoding Parkin were initially identified to be involved in the development of Parkinson disease (11), and recent studies have established a link between mitochondrial dysfunction and Parkinson disease (12). To date, the majority of studies on Parkin have been limited to investigating its role in the brain (10, 13). Although Parkin is highly expressed in the heart (11), its functional importance in this tissue is largely unexplored.
In the present study, we have investigated the functional role of Parkin in the myocardium. We report that Parkin deficiency has no effect on mitochondria or cardiac function in mice under normal conditions, suggesting that Parkin is not required for the base-line turnover of mitochondria. Instead, our studies demonstrate that Parkin plays a critical role in the adaptive response after a myocardial infarction (MI)2 by promoting clearance of damaged mitochondria via autophagy.
EXPERIMENTAL PROCEDURES
Animals
All animal protocols were in accordance with institutional guidelines and approved by the Institutional Animal Care and Use Committee of the University of California San Diego. Adult cardiac myocytes were isolated from 250–300-g male Sprague-Dawley rats or 8–10-week-old male WT or park2 knock-out mice as described previously (14). Park2 knock-out mice were obtained from The Jackson Laboratory (B6.129S4-Park2tm1Shn/J, stock number 006582) and have been described previously (15).
Western Blotting Analysis
Lysates were prepared as described previously (14) in buffer containing 50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1 mm EGTA, 1 mm EDTA, 1% Triton X-100, and Complete protease inhibitor mixture (Roche Applied Bioscience). Protein concentration was determined by Bradford assay using BSA standards. Proteins were separated by SDS-PAGE, transferred to nitrocellulose, and immunoblotted with antibodies against Parkin (Cell Signaling), LC3 (Cell Signaling), GAPDH (Cell Signaling), COXIII (Invitrogen), actin (GeneTex), ubiquitin (Santa Cruz Biotechnology), TOM20 (Santa Cruz Biotechnology), mitofusin 1 (Mfn1) (Santa Cruz Biotechnology), mitofusin 2 (Mfn2) (Sigma-Aldrich), optic atrophy 1 (Opa1) (BD Biosciences), dynamin-like protein 1 (Drp1) (BD Biosciences), or mitochondrial fission 1 (Fis1) (Enzo Life Sciences).
Echocardiography
Echocardiography was performed using a VisualSonics Vevo 770 equipped with an RMV707B 15–45-MHz imaging transducer. Recordings of the parasternal long-axis (B-mode), parasternal short-axis (M-mode), apical four-chamber (pulsed wave Doppler), and aortic arch (pulsed wave Doppler) views were taken. All measurements were collected and analyzed using VisualSonics software. Only data with heart rates >450 beats per minute were accepted.
Mitochondrial Respiration Measurements
The procedures used for mitochondrial isolation and respiration have been described previously (16). Complex I-dependent respiration was measured using 2 mm pyruvate and 2 mm malate as substrates. Complex II-dependent oxygen consumption was measured using 5 μm rotenone with 5 mm succinate as substrate. For measurements of mitochondrial respiration in intact adult mouse cardiomyocytes, cells were resuspended in plating medium (containing 5.56 mm glucose) supplemented with 10 mm pyruvate, and cells were transferred directly into the 37 °C Oxytherm apparatus (Hansatech Instruments) for oxygen consumption measurements. Sequential additions were made of 10 μg/ml oligomycin to inhibit ATP synthase followed by titration with either 200 nm or 400 nm carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone to measure maximal rates of uncoupled mitochondria.
Oxygen consumption of mitochondria isolated from border and remote zones were measured using the Seahorse XF96 analyzer (17). Mitochondria were resuspended in mitochondrial assay solution (1×) composed of 70 mm sucrose, 220 mm mannitol, 10 mm KH2PO4, 5 mm MgCl2, 2 mm HEPES, 1 mm EGTA, and 0.2% (w/v) fatty acid-free BSA, pH 7.2, at 4 °C. Next, 1.5 μg of mitochondria was added to each well while the plate was on ice. The plate was transferred to a centrifuge equipped with a swinging bucket microplate adaptor and was spun at 2000 × g for 20 min at 4 °C. After centrifugation, 10 mm pyruvate plus 1 mm malate or 10 mm succinate plus 2 μm rotenone along with 4 mm ADP were added to the wells. The plate was transferred to the XF96 instrument and allowed to temperature equilibrate for 3 min prior to measuring state 3 respiration.
Mitochondrial Swelling Assay
Sixty micrograms of mitochondria in a volume of 200 μl was added per well in a 96-well plate. Ca2+ was added to a final concentration of 150 μm. Mitochondrial swelling was monitored by measuring absorbance in a plate reader at 520 nm. The amplitude of each curve was obtained by subtracting the end point absorbance from the maximal absorbance obtained from the swelling assay. Maximum rate of change in amplitude (Vmax) was obtained by calculating the slope of the linear segment of the swelling curve.
Myocardial Infarction
Mice were subjected to myocardial infarction by permanently ligating the left anterior descending coronary artery as described previously (18). Briefly, 10–12-week-old WT and Parkin−/− mice were anesthetized with isoflurane, intubated, and ventilated. Pressure-controlled ventilation (Harvard Apparatus) was maintained at 9 cm of H2O. An 8-0 silk suture was placed around the left anterior descending coronary artery and then tightened. The suture was left in place, and the animal was immediately closed up.
Histology
Hearts were arrested in diastole with 200 mm KCl, fixed in 10% formalin for 24 h, and then transferred to 70% ethanol for an additional 20 h before embedding in paraffin. 10-μm-thick heart cross-sections were cut at 300-μm intervals through the left ventricle and stained with Masson's trichrome (Sigma-Aldrich). The percentage of remodeling was determined by dividing the total midline length of all fibrotic remodeled regions by the total midline length of all left ventricle sections (19). The remodeled region was defined as the region in which ≥50% of the total wall thickness had increased collagen deposition. Measurements were performed using ImageJ software.
Transmission Electron Microscopy and Morphometric Analysis
Adult mouse hearts were fixed in 2.5% glutaraldehyde in 0.1 m cacodylate buffer, post-fixed in 1% osmium tetroxide, and then treated with 0.5% tannic acid, 1% sodium sulfate, cleared in 2-hydroxypropyl methacrylate, and embedded in LX112 (Ladd Research, Williston, VT). Sections were mounted on copper slot grids coated with Parlodion and stained with uranyl acetate and lead citrate for examination on a Philips CM100 electron microscope (FEI Co., Hillsboro, OR). Mitochondrial morphometric measurements were performed using Adobe Photoshop CS5 software. A total of 298 mitochondria from WT mice and 272 mitochondria from Parkin−/− mice were analyzed from five electron micrographs taken at 7900× magnification.
Preparation of Parkin Mutants and Adenoviral Constructs
mCherry-Parkin was obtained from Dr. Richard Youle (NINDS, National Institutes of Health) (10). mCherry-ParkinR42P and mCherry-ParkinG430D were generated by site-directed mutagenesis as described previously (20), using mCherry-Parkin as a template. Parkin adenoviruses were generated using the pENTR directional TOPO cloning kit (Invitrogen) followed by recombination into the pAd/CMV/V5-DEST Gateway vector (Invitrogen).
Fluorescence Microscopy
For visualizing Parkin translocation to mitochondria, adult rat cardiomyocytes infected with mCherry-Parkin were fixed with 4% paraformaldehyde (Ted Pella Inc.) in PBS, pH 7.4, permeabilized with 0.2% Triton X-100 in PBS, and blocked in 5% normal goat serum before incubation with anti-COXIV (Invitrogen). Cells were then rinsed and incubated with goat anti-mouse Alexa Fluor 488 secondary antibody (Invitrogen). For induction of autophagy by rotenone, isolated cardiac myocytes from Parkin−/− mice were infected with GFP-LC3 for 24 h before treatment with 40 μm rotenone in DMSO. After 1 h, cells were fixed and stained with anti-COXIV and analyzed by fluorescence microscopy. Fluorescence micrographs were captured using a Carl Zeiss Axio Observer Z1 fitted with a motorized Z-stage and an ApoTome for optical sectioning. Z-stacks were acquired in ApoTome mode using a high-resolution AxioCam MRm digital camera, a 63× Plan-Apochromat (oil immersion) objective, and Zeiss AxioVision 4.8 software (Carl Zeiss). Pseudo-line scans were performed using ImageJ software.
Hypoxia and Cell Death Assay
After isolation, adult rat or mouse cardiomyocytes were allowed to plate on laminin-coated dishes for 2 h before infection with adenoviruses encoding mCherry, mCherry-Parkin, mCherry-Parkin-R42P, and mCherry-Parkin-G430D at a multiplicity of infection of 25 (for mouse) or 50 (for rat) in plating medium plus 2% heat-inactivated serum. Eighteen hours later, myocytes were subjected to hypoxia by incubation in DMEM without glucose (Invitrogen) in hypoxic pouches (GasPak EZ, BD Biosciences) at 37 °C. Cell death was assessed by measuring increased plasma membrane permeability to YoPro-1 (Invitrogen) as described previously (9). Cells were examined by fluorescence microscopy using a Carl Zeiss Axio Observer Z1 at 10× magnification.
Statistical Analyses
All values are expressed as means ± S.E. Statistical analyses were performed using Student's t test or analysis of variance followed by Student-Newman-Keuls multiple comparison test. Survival was assessed using the Kaplan-Meier method and log rank test. p < 0.05 was considered significant.
RESULTS
Parkin-deficient Mice Have Normal Cardiac Function
First, we examined the expression of Parkin in the heart by Western blotting. We found that Parkin is highly expressed in both heart and brain (Fig. 1A). To investigate the functional importance of Parkin in the heart, we characterized the cardiac phenotype in mice deficient for Parkin. Although Parkin−/− mice were smaller than WT mice as reported previously (13), there were no significant differences in heart weight/body weight or lung weight/body weight ratios between WT and Parkin−/− mice (Fig. 1, B–E). To assess the effect of Parkin deficiency on cardiac function, the mice were analyzed by echocardiography. We found that at 3 months of age, WT and Parkin−/− mice had similar fractional shortenings, ejection fractions, and left ventricular internal end diastolic and systolic dimensions (Table 1). To investigate whether Parkin deficiency affected cardiac function in aging mice, we also evaluated cardiac function and structure in 6- and 12-month-old mice. However, no significant differences were observed in cardiac function or in diastolic or systolic dimensions between aged WT and Parkin−/− mice.
FIGURE 1.
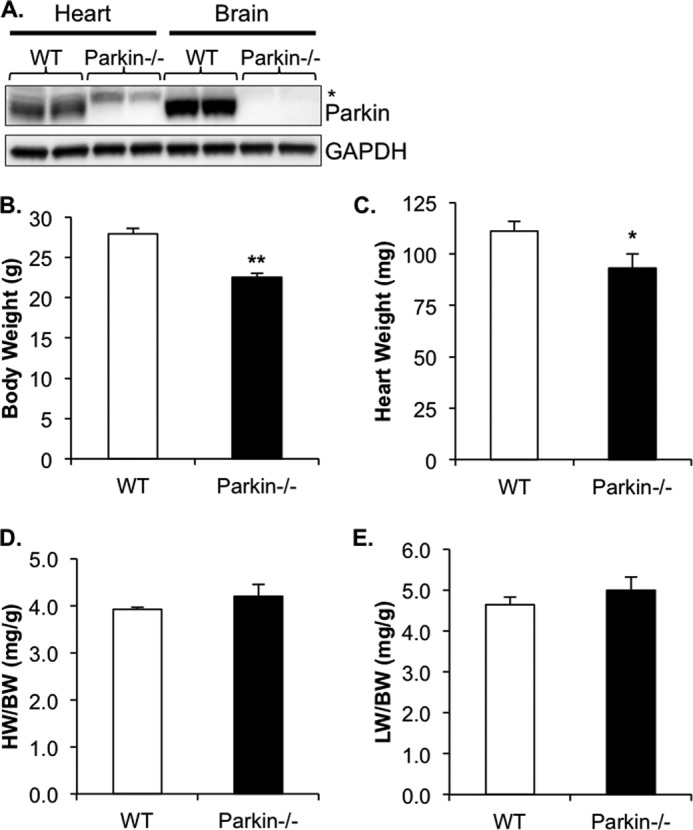
Characterization of Parkin−/− mice. A, Western blot for Parkin in heart and brain tissue (asterisk indicates nonspecific band detected by the Parkin antibody). B–E, WT and Parkin−/− mouse body weight (B), heart weight (C), heart weight/body weight (HW/BW) ratio (D), and lung weight/body weight (LW/BW) ratio (E). Mean ± S.E. (n = 7–11, *, p < 0.05, **, p < 0.01 versus WT).
TABLE 1.
Echocardiographic analysis of WT and Parkin−/− mice
HR, heart rate; BPM, beats per minute; FS, fractional shortening; EF, ejection fraction; IVSd, interventricular septal thickness at diastole; IVSs, interventricular septal thickness at systole; LVIDd, left ventricular internal dimension at diastole; LVIDs, left ventricular internal dimension at systole; LVPWd, left ventricular posterior wall thickness at diastole; LVPWs, left ventricular posterior wall thickness at systole. p > 0.05 for all parameters measured.
| Parameter | Mean value ± S.E. for |
|||||
|---|---|---|---|---|---|---|
| 3 months of age |
6 months of age |
12 months of age |
||||
| WT (n = 17) | Parkin−/− (n = 16) | WT (n = 8) | Parkin−/− (n = 9) | WT (n = 6) | Parkin−/− (n = 6) | |
| HR (BPM) | 582 ± 21 | 603 ± 25 | 524 ± 13 | 521 ± 26 | 567 ± 26 | 574 ± 10 |
| FS (%) | 49.9 ± 2.7 | 49.3 ± 3.6 | 33.6 ± 1.3 | 33.0 ± 2.1 | 48.0 ± 6.0 | 41.5 ± 7.0 |
| EF (%) | 80.5 ± 2.3 | 78.7 ± 3.6 | 63.0 ± 1.9 | 61.7 ± 3.1 | 77.9 ± 5.6 | 69.7 ± 9.1 |
| IVSd (mm) | 0.99 ± 0.06 | 0.96 ± 0.05 | 0.87 ± 0.04 | 0.90 ± 0.04 | 0.95 ± 0.06 | 0.87 ± 0.06 |
| IVSs (mm) | 1.49 ± 0.06 | 1.61 ± 0.10 | 1.24 ± 0.07 | 1.18 ± 0.04 | 1.64 ± 0.16 | 1.44 ± 0.17 |
| LVPWd (mm) | 1.06 ± 0.09 | 1.04 ± 0.08 | 0.89 ± 0.06 | 0.82 ± 0.06 | 1.12 ± 0.07 | 0.94 ± 0.14 |
| LVPWs (mm) | 1.61 ± 0.11 | 1.60 ± 0.11 | 1.27 ± 0.07 | 1.21 ± 0.07 | 1.63 ± 0.05 | 1.32 ± 0.23 |
| LVIDd (mm) | 3.33 ± 0.13 | 3.31 ± 0.13 | 3.83 ± 0.14 | 3.93 ± 0.17 | 3.47 ± 0.17 | 3.89 ± 0.24 |
| LVIDs (mm) | 1.69 ± 0.14 | 1.66 ± 0.17 | 2.56 ± 0.13 | 2.66 ± 0.18 | 1.84 ± 0.26 | 2.33 ± 0.40 |
Parkin-deficient Mitochondria Are Smaller but Have Normal Respiratory Capacity
Parkin has been reported to play an important role in the removal of dysfunctional mitochondria via autophagy (10). Therefore, we investigated whether Parkin deficiency resulted in accumulation of damaged mitochondria. Using mitochondria isolated from WT and Parkin−/− hearts, we assessed mitochondrial respiration by measuring the rate of oxygen consumption. State 3 respiration rates were determined from the maximum rate of ADP-dependent oxygen consumption, whereas state 4 respiration rates were determined after the ADP had been consumed. We found that Parkin−/− mitochondria had similar state 3 and state 4 respiration rates to those of WT mitochondria with substrates for either respiratory complex I (pyruvate/malate) or respiratory complex II (succinate/rotenone) (Fig. 2, A and B). Furthermore, there was no difference in the respiratory control ratio between WT and Parkin−/− mitochondria (Fig. 2C). To ensure that we did not select for functional mitochondria in our isolation procedure, we confirmed our findings in intact cardiac myocytes. Studies measuring maximal mitochondrial respiration in intact isolated adult myocytes confirmed that there was no difference in mitochondrial function between WT and Parkin−/− cells (Fig. 2D). These data suggest that Parkin-deficient cardiac mitochondria are well coupled and capable of efficiently producing ATP. To further confirm that there is not an accumulation of dysfunctional mitochondria resulting in elevated reactive oxygen species production, we examined malonaldehyde content and protein carbonylation as indices of oxidative stress. However, malonaldehyde content and levels of protein carbonyls did not differ between WT and Parkin−/− mice (data not shown).
FIGURE 2.

Mitochondrial respiration is normal in Parkin−/− mouse hearts at 3 months of age. A and B, state 3 and state 4 respiration rates of mitochondria isolated from WT or Parkin−/− mouse hearts with substrates for complex I (pyruvate/malate) (A) or complex II (succinate/rotenone) (B). C, respiratory control (RCR) ratios for complex I and complex II substrates (n = 4). Pyr/Mal, pyruvate/malate; Succ/Rot, rotenone/succinate. D, maximal mitochondrial respiration rates in isolated intact WT and Parkin−/− adult mouse myocytes (n = 3). Mean ± S.E. No significant differences were observed. E, swelling of isolated mitochondria in the presence of 150 μm calcium (n = 3). F, the degree and rate of swelling (amplitude and Vmax, respectively) were not significantly different between WT and Parkin−/− mitochondria. Base-line absorbance values for Parkin−/− mitochondria were significantly higher than WT. Mean ± S.E. (n = 3, *, p < 0.05 versus WT).
Although cardiac mitochondria from Parkin−/− mice appeared normal under base-line conditions, we sought to investigate whether they were more susceptible to mitochondrial permeability transition. To investigate the susceptibility of Parkin-deficient mitochondria to undergo mitochondrial permeability transition, isolated mitochondria were incubated with 150 μm calcium, and swelling was measured as a change in absorbance. We found that there were no differences in the rate or amplitude of swelling between WT and Parkin−/− mitochondria (Fig. 2, E and F). However, we observed that Parkin−/− mitochondria consistently had a higher base-line absorbance value than WT mitochondria (0.712 ± 0.011 for Parkin−/− versus 0.629 ± 0.017 for WT). This suggests that mitochondria from Parkin−/− mice might be smaller than WT mitochondria. To confirm this finding, we examined the mitochondrial ultrastructure of WT and Parkin−/− mouse hearts by transmission electron microscopy and morphometric analysis. As shown in Fig. 3A, mitochondria from WT mice at 12 weeks of age were found to be of homogeneous size and shape and organized into rows perpendicular to the myocardial Z-lines. In contrast, mitochondria in Parkin−/− heart preparations were more disorganized and often found in large clusters with many small, round mitochondria. Morphometric analysis showed that overall, Parkin−/− mitochondria were significantly smaller than WT mitochondria (Fig. 3B). However, there was no evidence of mitochondrial degeneration or damage at this age. The smaller morphology of the Parkin-deficient mitochondria prompted us to investigate the levels of proteins involved in mitochondrial fusion and fission. We hypothesized that changes in these proteins may alter mitochondrial dynamics. However, no changes in the levels of the mitochondrial fusion proteins Mfn1 and Mfn2 or Opa1 were detected (Fig. 3C). There was an increase in Fis1 protein levels, whereas the fission protein Drp1 levels were reduced (Fig. 3D).
FIGURE 3.
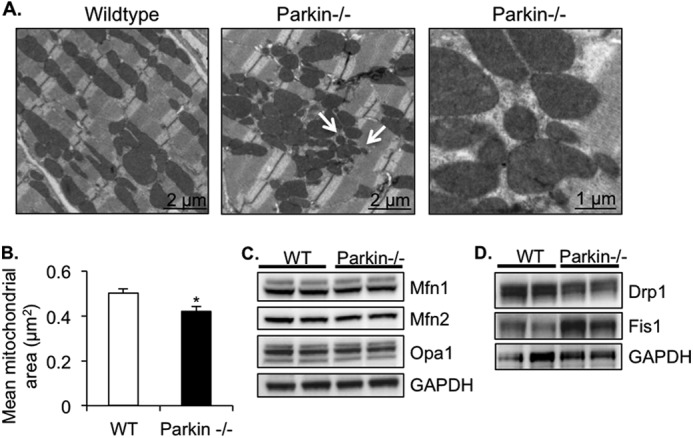
Parkin-deficient hearts have disorganized and smaller mitochondria. A, representative transmission electron micrographs of heart sections from 3-month-old mice. Arrows signify smaller mitochondria. B, quantitation of mean mitochondrial area ± S.E. in WT and Parkin−/− hearts (*, p < 0.05 versus WT). C, Western blot of mitochondrial fusion proteins Mfn1, Mfn2, and Opa1. D, Western blot of mitochondrial fission proteins Drp1 and Fis1.
Increased Sensitivity of Parkin−/− Mice to Myocardial Infarction
Parkin has been reported to be important in removing compromised mitochondria to prevent activation of cell death pathways (10). To investigate whether Parkin−/− mice were more sensitive to cardiac stress, WT and Parkin−/− mice were subjected to MI by permanent ligation of the left anterior descending coronary artery. Interestingly, we discovered that Parkin−/− mice were much more sensitive to MI with ∼60% mortality within the first week when compared with ∼20% for WT mice (Fig. 4A). Histological analysis revealed severe thinning of left ventricular (LV) wall, enlarged LV interior dimensions, and increased remodeling in Parkin−/− hearts when compared with WT (Fig. 4, B–D). In addition, we found that levels of Parkin protein rapidly increased in the border zone after the MI in WT mouse hearts (Fig. 4E). In contrast, Parkin protein levels remained unchanged in the area remote from the infarct (remote zone) (Fig. 4F). We also assessed cardiac function by echocardiography on surviving mice 7 days after MI. Parkin−/− mice had significantly lower fractional shortening and ejection fractions, as well as significantly increased LV end diastolic and systolic dimensions and LV volume when compared with WT (Fig. 5, A–F). These results indicate that Parkin plays an important role in the adaptive response after an MI and that Parkin deficiency results in impaired recovery.
FIGURE 4.

Parkin−/− mice have increased susceptibility to MI. A, Kaplan-Meyer survival curve (p < 0.05). B, representative Masson's trichrome staining of WT and Parkin−/− hearts 7 days after MI. C, representative images of left ventricular remodeling 7 days after MI. D, quantitation of left ventricular remodeling (n = 10, *, p < 0.05 versus WT). E, representative Western blot demonstrating rapid up-regulation of Parkin expression in the border zone of WT mice after MI (Post-MI). Quantitation of Parkin/GAPDH ratio is shown (*, **,***, p < 0.05 versus 0 h, n = 5). AU, arbitrary units. F, representative Western blot of Parkin expression in the remote zone of WT mice after MI (n = 5). Data are mean ± S.E.
FIGURE 5.

Echocardiography of WT and Parkin−/− mice 7 days after MI. A, representative M-mode echocardiograms of WT and Parkin−/− hearts prior to MI and 7 days after MI (Post-MI). B–F, echocardiographic analysis revealed reduced fractional shortening (FS) (B) and ejection fraction (EF) (C), as well as enlarged left ventricular end diastolic dimension (LVEDD) (D), left ventricular end systolic dimension (LVESD) (E), and LV volume (F) in Parkin−/− hearts. Mean ± S.E. (WT, n = 13; Parkin−/−, n = 10, *, p < 0.05, **, p < 0.01 versus WT).
Mitophagy Is Impaired in Parkin−/− Hearts
Studies have shown that changes in the extra- and intracellular environments lead to rapid up-regulation of autophagy in cells (21). To investigate whether autophagy was up-regulated in WT and Parkin−/− hearts following the MI, we assessed the levels of LC3II/LC3I in the border zone of the infarct by Western blotting. The conversion of LC3I to LC3II is indicative of autophagic activity, and the amount of LC3II correlates well with the number of autophagosomes (22). Western blot analysis of endogenous LC3 levels confirmed a significant increase in the LC3II/LC3I ratio in the border zone of the infarct in WT mice following 4 h of MI (Fig. 6A). Although autophagy was also induced in Parkin−/− border zone samples, the magnitude of induction was significantly lower than that of WT at this time point. Interestingly, autophagy was induced to the same extent in both WT and Parkin−/− remote zones (Fig. 6B).
FIGURE 6.

Mitophagy is impaired in Parkin-deficient hearts. A and B, induction of autophagy in the border zone (A) and remote zone (B) 4 h after MI. Western blots and quantitation analyses for LC3II/I levels in the border zone at base-line (Con) in the ventricle and in the border zone 4 h after MI are shown. Mean ± S.E. (*, p < 0.05 versus control, n = 8) AU, arbitrary units; n.s., not significant. C, Western blots for Parkin and LC3 in the mitochondrial fractions of the border zones of WT and Parkin−/− mice 4 h after MI. D, Western blots for ubiquitinated proteins in the cytosolic and mitochondrial fractions of the border zones of WT and Parkin−/− mice 4 h after MI. E, Western blots for Parkin and LC3 in the mitochondrial fractions of the remote zones of WT and Parkin−/− mice 4 h after MI. F, Western blots for ubiquitinated proteins in the cytosolic and mitochondrial fractions of the remote zones of WT and Parkin−/− mice 4 h after MI.
Parkin translocates to damaged mitochondria, where it ubiquitinates a number of substrates (10, 23, 24). The ubiquitination is known to serve as a signal for mitophagy. Analysis of the mitochondria fractions prepared from border zones of WT mice confirmed increased levels of Parkin at the mitochondria after MI (Fig. 6C). Concomitantly, there was an increase in ubiquitination of mitochondrial proteins and binding of LC3II to mitochondria in the border zone of WT mice (Fig. 6, C and D). These are markers of mitophagy. The increase in mitochondrial ubiquitination and LC3II binding was reduced in Parkin−/− border zone mitochondria after MI. Interestingly, there was also an increase in ubiquitination of cytosolic proteins in WT border zone samples, but not in Parkin−/− border zone (Fig. 6D), indicating that Parkin has substrates in the cytosol that are ubiquitinated in response to stress. In contrast, we found a very small increase in mitochondrial Parkin in WT remote zone samples (Fig. 6E). There were no increases in ubiquitination of cytosolic and mitochondrial ubiquitination in either WT or Parkin−/− remote zones after the MI (Fig. 6F). Unexpectedly, there was an increase in mitochondria-associated LC3II levels in both WT and Parkin−/− samples (Fig. 6E). This is indicative of LC3II directly binding to mitochondrial autophagy receptors such as Bnip3 (25) and Nix (26, 27) in the remote zone.
Further examination using transmission electron microscopy confirmed the induction of autophagy and mitophagy in WT myocytes in the border zone after MI (Fig. 7A). In contrast, we did not detect any autophagosomes containing mitochondria in Parkin−/− myocytes. Interestingly, mitochondria in Parkin−/− myocytes in the border zone were swollen and displayed severe cristae remodeling. The accumulation of swollen mitochondria also appeared to contribute to disruption of the contractile elements in the Parkin−/− myocytes (Fig. 7A, white arrows). Additionally, mitochondria isolated from the border zones of Parkin−/− mice subjected to 4 h of MI had significantly lower oxygen consumption rates when compared with mitochondria in the remote zone (Fig. 7, B and C). Although mitochondria from WT border zone had reduced oxygen consumption rates, the decrease was not significant (Fig. 7, B and C).
FIGURE 7.

Accumulation of damaged mitochondria in Parkin-deficient hearts after MI. A, ultrastructural analysis by transmission electron microscopy of WT and Parkin−/− myocytes in the border zone of the infarct 4 h after MI. Black arrows indicate autophagosomes, and white arrows indicate disrupted contractile elements. B and C, oxygen consumption rates (OCR) of mitochondria in the remote zone (RZ) and border zone (BZ) 4 h after MI with pyruvate/malate (B) or succinate/rotenone (C) as substrates. Mean ± S.E. (n = 5 for WT, n = 6 for Parkin−/−).
To further confirm that Parkin−/− myocytes have impaired mitophagy in response to mitochondrial damage, isolated WT and Parkin−/− myocytes were treated with rotenone. Rotenone is a mitochondrial complex I inhibitor and induces translocation of Parkin to mitochondria (28). In WT cells, rotenone treatment caused an increase in mitophagy, as determined by increased co-localization between mitochondria and LC3-GFP-positive autophagosomes (Fig. 8, A and B). In contrast, rotenone did not increase mitophagy in Parkin-deficient myocytes. Consistent with the in vivo data, we found that induction of autophagy was reduced in Parkin-deficient myocytes as rotenone treatment induced fewer LC3-GFP-positive autophagosomes in Parkin−/− cells when compared with WT (Fig. 8C). In addition, isolated Parkin−/− myocytes were more susceptible to hypoxia-induced cell death than WT mouse myocytes (Fig. 8D). The increased sensitivity of Parkin−/− cardiac myocytes to hypoxia was abrogated when Parkin expression was restored in the knock-out cells (Fig. 8D). These data suggest that Parkin deficiency results in defective mitochondrial clearance and subsequent increased susceptibility to stress.
FIGURE 8.

Rotenone treatment fails to induce mitophagy in Parkin−/− myocytes. A, representative images of WT and Parkin−/− myocytes. Cells infected with LC3-GFP were treated with DMSO or 40 μm rotenone for 1 h. After fixation, mitochondria were stained with anti-COXIV. B, quantitation of autophagosomes co-localizing with mitochondria. Mean ± S.E. (n = 3; *, p < 0.05 versus WT control, **, p > 0.05 versus Parkin−/− control). C, quantitation of the mean number of LC3-GFP positive autophagosomes per cell in WT and Parkin−/− myocytes. Mean ± S.E. (n = 3; *, p < 0.05 versus WT control, **, p > 0.05 versus Parkin−/− control). D, adult myocytes from WT or Parkin−/− mice were infected with β-Gal or Parkin for 24 h prior to 4 h of hypoxia and quantitation of cell death. Mean ± S.E. (n = 3, *, p < 0.05 versus Normoxia, **, p < 0.05 versus hypoxia).
Parkin Protects Against Hypoxia-mediated Cell Death
Our in vivo data suggest that Parkin deficiency results in increased susceptibility to tissue damage following stress. Therefore, we investigated whether Parkin overexpression could confer a resistance to stress conditions in vitro. Isolated adult rat cardiac myocytes were infected with adenoviruses encoding empty vector or Parkin prior to hypoxia. We found that by 4 h of hypoxia, there was a significant increase in translocation of Parkin to mitochondria in cardiac myocytes (Fig. 9, A and B). A pseudo-line scan analysis confirmed that mCherry-Parkin co-localized with COXIV-labeled mitochondria in hypoxic cardiac myocytes (Fig. 9C). Moreover, 8 h of hypoxia induced significant cell death in control-infected cardiac myocytes. In contrast, myocytes overexpressing Parkin had significantly reduced cell death (51% ± 6%) when compared with control-infected cells (69% ± 7%) (Fig. 9D). Nonfunctional Parkinson disease-associated mutants of Parkin, ParkinR42P and ParkinG430D, were used as controls. ParkinR42P fails to translocate to mitochondria, whereas ParkinG430D is deficient in ubiquitin ligase activity (29, 30). Overexpression of these mutants in myocytes did not reduce hypoxia-mediated cell death (Fig. 9D), confirming that the protective effect of Parkin is dependent both on the ubiquitin ligase activity of Parkin and on its mitochondrial localization. To investigate whether the protective effect of Parkin was associated with its up-regulation, we analyzed endogenous protein levels of Parkin after exposure to 4 or 8 h of hypoxia. However, we found that hypoxia did not affect endogenous Parkin protein levels in cardiac myocytes (Fig. 9E).
FIGURE 9.

Overexpression of Parkin protects myocytes against hypoxia-mediated cell death. A, representative images of adult rat cardiac myocytes infected with mCherry-Parkin and stained for mitochondrial COXIV. Yellow lines represent line scans. B, quantitation of myocytes with mCherry-Parkin translocation. Mean ± S.E. (*, p < 0.01 versus normoxia, n = 4). C, representative line-scan analyses demonstrating co-localization of Parkin (red line) with mitochondrial COXIV (green line). AU, arbitrary units. D, adult rat cardiomyocytes were infected with adenoviruses encoding mCherry, Parkin, ParkinR42P (R42P), or ParkinG430D (G430D) for 24 h prior to 8 h of hypoxia and quantitation of cell death. Mean ± S.E. (n = 3–5, *, p < 0.05 versus normoxia + mCherry, **, p < 0.05 versus hypoxia + mCherry). E, Western blot analysis for endogenous Parkin in myocytes after exposure to 4 or 8 h of hypoxia.
DISCUSSION
Although Parkin was observed to be highly expressed in the heart over a decade ago (11), its functional role has remained unexplored. Our study reports several new and important findings regarding the role of Parkin in the myocardium. First, Parkin is not essential for the normal turnover of mitochondria via autophagy in the heart. Although loss of Parkin resulted in smaller and more disorganized mitochondria, it did not affect mitochondrial function. Another key finding of this study is that mice lacking Parkin suffer from impaired recovery after MI. Parkin−/− myocytes also have impaired mitophagy and accumulate dysfunctional mitochondria after an infarction. Thus, our data suggest that Parkin plays a critical role in adapting to stress in the myocardium by enhancing mitophagy.
Removal of mitochondria via autophagy is increasingly recognized as an important process to maintain a healthy population of mitochondria in cells. Reduced autophagy results in accumulation of dysfunctional mitochondria and has been linked to aging and development of heart failure (8). For instance, ablation of autophagy in the heart by cardiac specific deletion of Atg5 leads to rapid buildup of dysfunctional mitochondria and development of cardiac dysfunction (4). Similarly, specific deletion of Atg7 in skeletal muscle results in accumulation of impaired mitochondria and increased intracellular reactive oxygen species levels (31). These studies suggest that mitochondria in cardiac and skeletal muscle are frequently removed by autophagy, and a disruption in this process leads to rapid accumulation of dysfunctional mitochondria. Parkin has been identified as an important regulator of mitochondrial autophagy (10), but our studies suggest that Parkin is not critical for the turnover of mitochondria under normal conditions. Because the survival of a cell depends on its ability to clear dysfunctional mitochondria, it is very likely that multiple proteins and pathways can promote mitochondrial removal via autophagy. This redundancy ensures that there is no disruption in the clearance of dysfunctional mitochondria under base-line conditions. Our findings are also consistent with existing patient data. Loss-of-function mutations in the PARK2 gene are associated with early-onset familial Parkinson disease (11), but there are currently no reports that these patients have abnormal heart function. Instead, our findings suggest that these patients might be at increased risk of developing heart failure in response to stress such as myocardial ischemia. Interestingly, studies that were conducted over a decade ago found an increase in mortality of Parkinson disease patients with ischemic heart disease (32, 33). Additional studies are needed to determine whether Parkinson disease patients with cardiovascular disease have increased mortality.
Studies in Drosophila melanogaster initially suggested that Parkin plays a role in regulating mitochondrial function because Parkin gene mutations cause striking changes in both mitochondrial structure and performance (34). However, it is now clear that Parkin has a different role in mammalian cells. Parkin null mice do not suffer from major neurological phenotypes or motor impairments and do not lose dopaminergic neurons of the substantia nigra until they are exposed to stress conditions such as chronic systemic inflammation induced by lipopolysaccharide administration (35). Similarly, our most striking finding in this study was the increased sensitivity of Parkin−/− mice to MI when compared with WT mice. Thus, in mammalian cells, Parkin does not play a critical role in maintaining normal mitochondrial function. Rather, Parkin appears to play an important role in the adaptation to stress. Another striking finding in our study was the ubiquitous deterioration of mitochondria in Parkin−/− myocytes in the border zone after the infarction. In WT myocytes, we detected the presence of some dysfunctional mitochondria after the MI, whereas nearly all mitochondria in Parkin−/− myocytes were swollen and displayed cristae remodeling. Parkin-deficient cardiac mitochondria were normal under base-line conditions but rapidly deteriorated after the MI, suggesting that Parkin might also be involved in maintaining mitochondrial function in response to stress.
In addition, it was recently reported that Parkin-mediated removal of mitochondria plays an important role in cardiac ischemic preconditioning (36). This study found that ischemic preconditioning induces translocation of Parkin to mitochondria and that Parkin-deficient mice are resistant to ischemic preconditioning-induced cardioprotection. Parkin has been reported to ubiquitinate proteins on damaged mitochondria upon translocation (23, 24), and ubiquitination serves as a signal for autophagic degradation of the mitochondria (37). The p62 protein has been reported to first bind ubiquitinated proteins via its ubiquitin-associated domain and then to interact with LC3, leading to autophagosome formation around the mitochondrion (37). Consistent with this, we found that there was an increase in the ubiquitination of mitochondrial proteins and association of LC3 with mitochondria in the border zone of WT hearts after MI. In contrast, the mitochondrial ubiquitination and LC3 binding were reduced in Parkin−/− hearts. This supports the hypothesis that Parkin is important for ubiquitinating proteins on damaged mitochondria. Recent studies suggest a much broader functional role for Parkin than just mitophagy. For instance, Parkin has been reported to regulate fatty acid uptake (38) and mitochondrial biogenesis (39). Our data showing a failure of Parkin−/− hearts to increase cytosolic ubiquitination after MI support the notion that Parkin has a broad range of stress response functions.
The PTEN-induced putative kinase 1 (PINK1) is a serine-threonine kinase that has been reported to be upstream of Parkin. Studies have found that upon collapse of the Δψm, PINK1 accumulates on the outer mitochondrial membrane, which promotes recruitment and activation of Parkin (40). Interestingly, PINK1- and Parkin-deficient mice have very different cardiac phenotypes. PINK1 deficiency results in increased oxidative stress and mitochondrial dysfunction in the myocardium, and these mice develop cardiac dysfunction and hypertrophy by 2 months of age (41). In contrast, we found that Parkin-deficient mice have normal mitochondria and cardiac function in the absence of stress. Thus, although PINK1 and Parkin might function in an overlapping pathway in cells, they clearly have additional and separate functions in the myocardium. PINK1 appears to be important in maintaining normal mitochondrial function in the heart under base-line conditions, whereas Parkin plays an important role in the adaptation to stress by promoting mitophagy. The study by Billia et al. (41) did not investigate whether PINK1 deficiency impaired mitochondrial autophagy in the heart. Thus, additional studies are required to further investigate the role of PINK1 in mitochondrial clearance in the heart.
Our data show that autophagy is rapidly increased in the border zone of the infarct after permanent ligation of the left anterior descending coronary artery and in isolated myocytes treated with rotenone. Unexpectedly, the Parkin−/− myocytes showed reduced formation of autophagosomes in response to MI or rotenone treatment. We recently reported that Bnip3-mediated mitochondrial autophagy in cardiac myocytes involves translocation of Parkin to mitochondria (14). Similar to our findings in the present study, we discovered that induction of autophagy by Bnip3 is reduced in Parkin−/− myocytes. Thus, our data suggest that Parkin is not only responsible for targeting dysfunctional mitochondria for autophagy, but may also participate in initiating the autophagy response. Our data also demonstrate that initiation of autophagy is only impaired in the border zone, where the mitochondrial damage is occurring. Induction of autophagy in the remote zone was not affected in Parkin−/− mice. Interestingly, mitochondria provide an assembly site and lipid source for the nascent autophagosome (42), and it was recently reported that Parkin interacts with the autophagy-promoting protein Ambra1 at mitochondria (43). Ambra1 activates the PI3K complex (Beclin1-Vps34-Vps15) that is required for the formation of new autophagosomes (44). Thus, it is possible that Parkin has much more influence on the regulation of autophagy than previously thought and that perturbation of Parkin function can affect induction of autophagy in response to certain stimuli. Further studies are needed to investigate whether Parkin initiates autophagy at mitochondria via Ambra1 in the heart.
Our data demonstrate clearly that Parkin plays a critical role in the adaptation to stress such as a myocardial infarction. Thus, the prospect of utilizing Parkin as a therapeutic target is very intriguing. Up-regulation of Parkin following MI appears to be a beneficial event that confers resistance to cell death by enhancing mitochondrial autophagy, yet hypoxia alone is not sufficient to up-regulate Parkin protein levels in cardiac myocytes. However, as other studies have discovered, excessive or prolonged activation of autophagy may be detrimental and lead to loss of myocytes (5). It is not surprising that removal of too many mitochondria via autophagy in a contracting myocyte is detrimental because it results in an energy deficiency. Although Parkin represents a great potential therapeutic target to promote clearance of damaged mitochondria and reduce cell death, further studies are necessary to enhance our understanding of mitochondrial autophagy in the myocardium.
This work was supported, in whole or in part, by National Institutes of Health Grants R01HL087023 and R01HL101217 (to A. B. G.).
This article was selected as a Paper of the Week.
- used: MI
- myocardial infarction
- Bnip3
- Bcl-2/adenovirus E1B 19-kDa interacting protein 3
- LC3
- microtubule-associated protein 1 light chain 3
- Drp1
- dynamin-related protein 1
- Fis1
- mitochondrial fission 1
- Mfn1/2
- mitofusin-1/2
- Opa1
- optic atrophy 1
- DMSO
- dimethyl sulfoxide
- PINK1
- PTEN-induced putative kinase 1
- PTEN
- phosphatase and tensin homolog
- LV
- left ventricular.
REFERENCES
- 1. Kanamori H., Takemura G., Maruyama R., Goto K., Tsujimoto A., Ogino A., Li L., Kawamura I., Takeyama T., Kawaguchi T., Nagashima K., Fujiwara T., Fujiwara H., Seishima M., Minatoguchi S. (2009) Functional significance and morphological characterization of starvation-induced autophagy in the adult heart. Am. J. Pathol. 174, 1705–1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nishino I., Fu J., Tanji K., Yamada T., Shimojo S., Koori T., Mora M., Riggs J. E., Oh S. J., Koga Y., Sue C. M., Yamamoto A., Murakami N., Shanske S., Byrne E., Bonilla E., Nonaka I., DiMauro S., Hirano M. (2000) Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 406, 906–910 [DOI] [PubMed] [Google Scholar]
- 3. Tanaka Y., Guhde G., Suter A., Eskelinen E. L., Hartmann D., Lüllmann-Rauch R., Janssen P. M., Blanz J., von Figura K., Saftig P. (2000) Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 406, 902–906 [DOI] [PubMed] [Google Scholar]
- 4. Nakai A., Yamaguchi O., Takeda T., Higuchi Y., Hikoso S., Taniike M., Omiya S., Mizote I., Matsumura Y., Asahi M., Nishida K., Hori M., Mizushima N., Otsu K. (2007) The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 13, 619–624 [DOI] [PubMed] [Google Scholar]
- 5. Matsui Y., Takagi H., Qu X., Abdellatif M., Sakoda H., Asano T., Levine B., Sadoshima J. (2007) Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res. 100, 914–922 [DOI] [PubMed] [Google Scholar]
- 6. Yan L., Vatner D. E., Kim S.-J., Ge H., Masurekar M., Massover W. H., Yang G., Matsui Y., Sadoshima J., Vatner S. F. (2005) Autophagy in chronically ischemic myocardium. Proc. Natl. Acad. Sci. U.S.A. 102, 13807–13812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhu H., Tannous P., Johnstone J. L., Kong Y., Shelton J. M., Richardson J. A., Le V., Levine B., Rothermel B. A., Hill J. A. (2007) Cardiac autophagy is a maladaptive response to hemodynamic stress. J. Clin. Invest. 117, 1782–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gottlieb R. A., Gustafsson A. B. (2011) Mitochondrial turnover in the heart. Biochim. Biophys. Acta 1813, 1295–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hamacher-Brady A., Brady N. R., Logue S. E., Sayen M. R., Jinno M., Kirshenbaum L. A., Gottlieb R. A., Gustafsson A. B. (2007) Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 14, 146–157 [DOI] [PubMed] [Google Scholar]
- 10. Narendra D., Tanaka A., Suen D.-F., Youle R. J. (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kitada T., Asakawa S., Hattori N., Matsumine H., Yamamura Y., Minoshima S., Yokochi M., Mizuno Y., Shimizu N. (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608 [DOI] [PubMed] [Google Scholar]
- 12. Darios F., Corti O., Lücking C. B., Hampe C., Muriel M.-P., Abbas N., Gu W.-J., Hirsch E. C., Rooney T., Ruberg M., Brice A. (2003) Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria-dependent cell death. Hum. Mol. Genet. 12, 517–526 [DOI] [PubMed] [Google Scholar]
- 13. Palacino J. J., Sagi D., Goldberg M. S., Krauss S., Motz C., Wacker M., Klose J., Shen J. (2004) Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J. Biol. Chem. 279, 18614–18622 [DOI] [PubMed] [Google Scholar]
- 14. Lee Y., Lee H.-Y., Hanna R. A., Gustafsson Å. B. (2011) Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 301, H1924–H1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Goldberg M. S., Fleming S. M., Palacino J. J., Cepeda C., Lam H. A., Bhatnagar A., Meloni E. G., Wu N., Ackerson L. C., Klapstein G. J., Gajendiran M., Roth B. L., Chesselet M.-F., Maidment N. T., Levine M. S., Shen J. (2003) Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J. Biol. Chem. 278, 43628–43635 [DOI] [PubMed] [Google Scholar]
- 16. Sayen M. R., Gustafsson A. B., Sussman M. A., Molkentin J. D., Gottlieb R. A. (2003) Calcineurin transgenic mice have mitochondrial dysfunction and elevated superoxide production. Am. J. Physiol. Cell Physiol. 284, C562–C570 [DOI] [PubMed] [Google Scholar]
- 17. Rogers G. W., Brand M. D., Petrosyan S., Ashok D., Elorza A. A., Ferrick D. A., Murphy A. N. (2011) High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS ONE 6, e21746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang C., Zhang X., Ramil J. M., Rikka S., Kim L., Lee Y., Gude N. A., Thistlethwaite P. A., Sussman M. A., Gottlieb R. A., Gustafsson A. B. (2010) Juvenile exposure to anthracyclines impairs cardiac progenitor cell function and vascularization resulting in greater susceptibility to stress-induced myocardial injury in adult mice. Circulation 121, 675–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Takagawa J., Zhang Y., Wong M. L., Sievers R. E., Kapasi N. K., Wang Y., Yeghiazarians Y., Lee R. J., Grossman W., Springer M. L. (2007) Myocardial infarct size measurement in the mouse chronic infarction model: comparison of area- and length-based approaches. J. Appl. Physiol. 102, 2104–2111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kubli D. A., Quinsay M. N., Huang C., Lee Y., Gustafsson A. B. (2008) Bnip3 functions as a mitochondrial sensor of oxidative stress during myocardial ischemia and reperfusion. Am. J. Physiol. Heart Circ. Physiol. 295, H2025–H2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aviv Y., Shaw J., Gang H., Kirshenbaum L. A. (2011) Regulation of autophagy in the heart: “you only live twice”. Antioxid Redox Signal. 14, 2245–2250 [DOI] [PubMed] [Google Scholar]
- 22. Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tanaka A., Cleland M. M., Xu S., Narendra D. P., Suen D.-F., Karbowski M., Youle R. J. (2010) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 191, 1367–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Narendra D., Kane L. A., Hauser D. N., Fearnley I. M., Youle R. J. (2010) p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6, 1090–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hanna R. A., Quinsay M. N., Orogo A. M., Giang K., Rikka S., Gustafsson Å. B. (2012) Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem. 287, 19094–19104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schwarten M., Mohrlüder J., Ma P., Stoldt M., Thielmann Y., Stangler T., Hersch N., Hoffmann B., Merkel R., Willbold D. (2009) Nix directly binds to GABARAP: a possible crosstalk between apoptosis and autophagy. Autophagy 5, 690–698 [DOI] [PubMed] [Google Scholar]
- 27. Novak I., Kirkin V., McEwan D. G., Zhang J., Wild P., Rozenknop A., Rogov V., Löhr F., Popovic D., Occhipinti A., Reichert A. S., Terzic J., Dötsch V., Ney P. A., Dikic I. (2010) Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 11, 45–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tang D., Kang R., Livesey K. M., Kroemer G., Billiar T. R., Van Houten B., Zeh H. J., 3rd, Lotze M. T. (2011) High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab. 13, 701–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee J.-Y., Nagano Y., Taylor J. P., Lim K. L., Yao T.-P. (2010) Disease-causing mutations in Parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J. Cell Biol. 189, 671–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Matsuda N., Sato S., Shiba K., Okatsu K., Saisho K., Gautier C. A., Sou Y.-S., Saiki S., Kawajiri S., Sato F., Kimura M., Komatsu M., Hattori N., Tanaka K. (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 189, 211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu J. J., Quijano C., Chen E., Liu H., Cao L., Fergusson M. M., Rovira I. I., Gutkind S., Daniels M. P., Komatsu M., Finkel T. (2009) Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging 1, 425–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ben-Shlomo Y., Marmot M. G. (1995) Survival and cause of death in a cohort of patients with parkinsonism: possible clues to aetiology? J. Neurol. Neurosurg. Psychiatr. 58, 293–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bennett D. A., Beckett L. A., Murray A. M., Shannon K. M., Goetz C. G., Pilgrim D. M., Evans D. A. (1996) Prevalence of parkinsonian signs and associated mortality in a community population of older people. N. Engl. J. Med. 334, 71–76 [DOI] [PubMed] [Google Scholar]
- 34. Greene J. C., Whitworth A. J., Kuo I., Andrews L. A., Feany M. B., Pallanck L. J. (2003) Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. U.S.A. 100, 4078–4083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Frank-Cannon T. C., Tran T., Ruhn K. A., Martinez T. N., Hong J., Marvin M., Hartley M., Treviño I., O'Brien D. E., Casey B., Goldberg M. S., Tansey M. G. (2008) Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J. Neurosci. 28, 10825–10834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huang C., Andres A. M., Ratliff E. P., Hernandez G., Lee P., Gottlieb R. A. (2011) Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS ONE 6, e20975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pankiv S., Clausen T. H., Lamark T., Brech A., Bruun J. A., Outzen H., Øvervatn A., Bjørkøy G., Johansen T. (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131–24145 [DOI] [PubMed] [Google Scholar]
- 38. Kim K.-Y., Stevens M. V., Akter M. H., Rusk S. E., Huang R. J., Cohen A., Noguchi A., Springer D., Bocharov A. V., Eggerman T. L., Suen D.-F., Youle R. J., Amar M., Remaley A. T., Sack M. N. (2011) Parkin is a lipid-responsive regulator of fat uptake in mice and mutant human cells. J. Clin. Invest. 121, 3701–3712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shin J.-H., Ko H. S., Kang H., Lee Y., Lee Y.-I., Pletinkova O., Troconso J. C., Dawson V. L., Dawson T. M. (2011) PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson's disease. Cell 144, 689–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Narendra D. P., Jin S. M., Tanaka A., Suen D.-F., Gautier C. A., Shen J., Cookson M. R., Youle R. J. (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8, e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Billia F., Hauck L., Konecny F., Rao V., Shen J., Mak T. W. (2011) PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc. Natl. Acad. Sci. U.S.A. 108, 9572–9577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hailey D. W., Rambold A. S., Satpute-Krishnan P., Mitra K., Sougrat R., Kim P. K., Lippincott-Schwartz J. (2010) Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141, 656–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Van Humbeeck C., Cornelissen T., Hofkens H., Mandemakers W., Gevaert K., De Strooper B., Vandenberghe W. (2011) Parkin interacts with Ambra1 to induce mitophagy. J. Neurosci. 31, 10249–10261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fimia G. M., Stoykova A., Romagnoli A., Giunta L., Di Bartolomeo S., Nardacci R., Corazzari M., Fuoco C., Ucar A., Schwartz P., Gruss P., Piacentini M., Chowdhury K., Cecconi F. (2007) Ambra1 regulates autophagy and development of the nervous system. Nature 447, 1121–1125 [DOI] [PubMed] [Google Scholar]