

Increasing evidence indicates that RNA plays an active role in a number of neurodegenerative diseases. The authors recently introduced a theoretical framework, catRAPID, to predict the binding ability of protein and RNA molecules. Here, they use catRAPID to investigate ribonucleoprotein interactions linked to Inherited Intellectual Disability, Amyotrophic Lateral Sclerosis, Creutzfeuld-Jakob, Alzheimer’s, and Parkinson’s diseases. They specifically focus on (1) RNA interactions with fragile X mental retardation protein FMRP; (2) protein sequestration caused by CGG repeats; (3) noncoding transcripts regulated by TAR DNA-binding protein 43 TDP-43; (4) autogenous regulation of TDP-43 and FMRP; (5) iron-mediated expression of amyloid precursor protein APP and α-synuclein; (6) interactions between prions and RNA aptamers. The results are in striking agreement with experimental evidence and provide new insights in processes associated with neuronal function and misfunction.
Keywords: protein–RNA interactions, noncoding RNA, neurodegeneration, autogenous regulation, RNA aptamers
Abstract
Increasing evidence indicates that RNA plays an active role in a number of neurodegenerative diseases. We recently introduced a theoretical framework, catRAPID, to predict the binding ability of protein and RNA molecules. Here, we use catRAPID to investigate ribonucleoprotein interactions linked to inherited intellectual disability, amyotrophic lateral sclerosis, Creutzfeuld-Jakob, Alzheimer’s, and Parkinson’s diseases. We specifically focus on (1) RNA interactions with fragile X mental retardation protein FMRP; (2) protein sequestration caused by CGG repeats; (3) noncoding transcripts regulated by TAR DNA-binding protein 43 TDP-43; (4) autogenous regulation of TDP-43 and FMRP; (5) iron-mediated expression of amyloid precursor protein APP and α-synuclein; (6) interactions between prions and RNA aptamers. Our results are in striking agreement with experimental evidence and provide new insights in processes associated with neuronal function and misfunction.
INTRODUCTION
Although neurodegenerative diseases are traditionally described as protein disorders leading to amyloidosis (Dobson 1999; Rubinsztein 2006), very recent evidence indicates that protein–RNA associations are involved in a number of neuropathies (Anthony and Gallo 2010). In Huntington’s disease, ataxias, and myotonic dystrophy, primary transcripts containing expanded trinucleotide regions form intranuclear foci where proteins are sequestered and inactivated (Kryshtafovych et al. 2005). In Creutzfeldt-Jakob disease, the RNA ability to interact with proteins facilitates conversion of the α-helix-rich prion protein (PrPC) into its infectious β-structure-rich insoluble conformer (PrPSc) (Deleault et al. 2003). In Alzheimer’s disease (AD), translation of aggregation-prone proteins is regulated by iron-dependent ribonucleoprotein interactions (Bartzokis et al. 2000). In many cases, aggregation of RNA-binding proteins (RBP) is triggered by defective transcription and affects processing of RNA molecules, which leads to progressive cell death (Fiesel and Kahle 2011). For a number of RBPs such as TAR DNA-binding protein 43 (TDP-43) and fragile X mental retardation protein (FMRP), regulation of expression levels requires interaction with autogenous mRNA (Johnsen et al. 1982; Schaeffer et al. 2001; Kim et al. 2010; Ayala et al. 2011).
We recently introduced the catRAPID method to predict interactions between protein and RNA molecules (Bellucci et al. 2011). Our approach exploits physico-chemical properties of nucleotide and amino acid chains such as secondary structure, hydrogen bonding, and van der Waals’ propensities to predict protein–RNA associations with a confidence of 80% or higher (Bellucci et al. 2011). Here, we use catRAPID to investigate processes that involve changes in protein and RNA metabolism. We aim to understand the involvement of coding and noncoding RNAs (ncRNAs) in neurodegeneration. Indeed, a number of transcripts have been found to be up- or down-regulated in many neuropathologies, but their mechanisms of action and association with proteins are still uncharacterized (Salta and De Strooper 2012). We are also interested in exploring the ability of proteins to bind to artificial RNA molecules. In fact, the use of RNA aptamers could open up new therapeutic avenues for the cure of neurodegenerative diseases (Lee et al. 2006).
In this study, we introduce an algorithm to estimate the specificity of associations (“interaction strength”) and a method to identify binding regions within polypeptide and nucleotide regions (“interaction fragments”). The new algorithms available at http://tartaglialab.crg.cat were developed to characterize ribonucleoprotein interactions in detail and provide help for future experimental design.
RESULTS
Here we use catRAPID (Bellucci et al. 2011; Agostini et al. 2012) to investigate a number of protein–RNA associations involved in neuronal function and misfunction: (1) protein–RNA interactions associated with fragile X syndrome; (2) protein sequestration in CGG aggregates; (3) the TDP-43 noncoding interactome; (4) FMRP and TDP-43 autogenous regulation; (5) iron-mediated translation of APP and α-synuclein; (6) prion proteins and RNA aptamers.
Protein–RNA interactions associated with fragile X syndrome
Fragile X syndrome (FXS) is the most common inherited form of mental retardation, which affects ∼1:4000 males and 1:7000 females. The fragile X mental retardation-1 gene FMR-1 is located in the X chromosome, and its mutations are responsible for the development of FXS (Pieretti et al. 1991). One of the main mechanisms underlying FXS onset is the repeat codon expansion (>200 CGG repeats) in the 5′ untranslated portion of the gene, which results in hypermethylation and transcriptional silencing of the FMR-1 region. FMRP, the protein codified by FMR-1, is an RNA-binding protein that functions primarily as a regulator of translation in neurons.
Recently, Davidovic et al. (2011) investigated the metabolomic signature associated with FXS. By taking advantage of UV-cross-linking and immunoprecipitation assays (CLIP), the investigators studied FMRP interactions with a number of mRNAs including superoxide dismutase 1 SOD1, Ras-related protein RAB3A, calcium/calmodulin-dependent protein kinase type II α chain CAMK2A, and postsynaptic density protein 95 PSD95 (Davidovic et al. 2011; Subramanian et al. 2011). Using catRAPID we investigate the ability of these four transcripts to interact with FMRP (Methods: Interaction Propensity).
According to our calculations, SOD1, RAB3A, CAMK2A, and PSD95 have high propensity to bind to FMRP (interaction strengths in the range of from 71% to 85%) (Fig. 1A–D; Supplemental Table 1; Methods: Interaction Strength) and interact more strongly than control RNA (RNA interaction strength in the range of from 76% to 99%; Supplemental Fig. 1A–D; Methods: Interaction Strength). In agreement with previous studies (Schaeffer et al. 2001), we find that FMRP has negligible propensity to bind to nucleotides 230–884 of transcript NM_002024.5, which serves as a negative control (Fig. 1E; Supplemental Table 1).
FIGURE 1.

FMRP associations with coding transcripts. Prediction of FMRP association with (A) SOD1, (B) CAMK2A, (C) RAB3A, (D) PSD-95 mRNAs, and (E) negative control RNA. The interaction strength is the propensity to bind with respect to a reference set of protein–RNA associations (black area in the score distribution; Supplemental Table 1).
It has been shown that the RGG box of FMRP interacts with the 5′ of SOD1 (Bechara et al. 2009) and that the APP binds to FMRP through a G-quadruplex motif (Westmark and Malter 2007). To assess whether catRAPID is able to identify regions involved in protein–RNA recognition, we introduced a procedure called “fragmentation” that involves division of polypeptide and nucleotide sequences into fragments and prediction of their interaction propensities (Methods: Interaction Fragments). We find that SOD1 fragments 71–122 nt and 76–127 nt overlap with the experimentally identified region located at nucleotides 86–157 and have the highest propensities to bind to FMRP (Fig. 2A,B; Bechara et al. 2009). As for APP, we predict that FMRP binds to nucleotides 650–751 and 751–852 (Fig. 2C,D; Supplemental Fig. 2A,B), which is in the experimentally validated region encompassing nucleotides 694–846 (nucleotides 699–796 show the highest experimental affinity) (Westmark and Malter 2007). In agreement with experimental evidence, we predict that the FMRP RGG box and the kissing domain KH2 are involved in RNA binding (Fig. 2A,C; Darnell et al. 2001, 2005). Our performances are truly remarkable, especially if we consider that the probability of selecting one experimentally validated interaction by chance is 10−4 (Fig. 2A,C; Methods: Interaction Fragments).
FIGURE 2.

FMRP interactions with SOD1 and APP. Interaction maps of FMRP binding to (A) SOD1 and (C) APP (FMRP secondary structure elements are displayed next to the “protein residue index” axis; blue areas indicate experimentally validated interactions). RNA interaction profiles for FMRP interactions with (B) SOD1 and (D) APP (blue lines indicate experimentally identified binding regions) (Supplemental Table 1).
Our analysis indicates that catRAPID predicts FMRP associations with SOD1, RAB3A, CAMK2A, and PSD95 (Davidovic et al. 2011; Subramanian et al. 2011) and identifies interacting regions within protein (e.g., the RGG box domain) and RNA (e.g., the APP G-quadruplex region) domains (Westmark and Malter 2007; Bechara et al. 2009). Intriguingly, our results suggest that mutations in the RNA-binding domains of FMRP could affect its ability to bind to SOD1 and APP, which could have consequences for the onset of AD and amyotrophic lateral sclerosis (ALS) (Table 1).
TABLE 1.
Open challenges

Protein sequestration in CGG aggregates
Fragile X-associated tremor/ataxia syndrome (FXTAS) is a recently identified neurodegenerative disorder that affects mainly older adult males who carry a pre-mutation codon expansion (55–200 CGG repeats) in the 5′-untranslated region of FMR-1 (Hagerman and Hagerman 2004; Oostra and Willemsen 2009). While FXS is caused by protein deficiency, FXTAS is characterized by an increase up to eightfold in the levels of FMR-1 and displays little effect on FMRP expression (Tassone et al. 2000; Kenneson et al. 2001; Primerano et al. 2002).
Recently, Sellier et al. (2010) investigated the ability of CGG expansions to sequester RBPs. The investigators identified several proteins that colocalize with RNA aggregates: Muscleblind-like MBNL1 and heterogeneous nuclear ribonucleoprotein hnRNP-G colocalize during the initial phases of CGG granule formation, while a number of heterogeneous ribonucleoproteins (hnRNP-A1, hnRNP-A2/B1, hnRNP-C, hnRNP-D, hnRNP-E, and hnRNP-C) coaggregate at late time points in dying cells (Sellier et al. 2010). FMRP, CUGBP1, and PURα do not colocalize with CGG aggregates (Sellier et al. 2010). Interestingly, SRC-associated in mitosis 68 kDa SAM68 is essential for the recruitment of other proteins, but does not interact with CGG repeats, suggesting that other RBPs could mediate its sequestration (Sellier et al. 2010).
According to our calculations, CGG repeats have a strong propensity to sequester MBNL1 and hnRNP-G and all the proteins (hnRNP-A1, hnRNP-A2/B1, hnRNP-C, hnRNP-D, hnRNP-E, and hnRNP-C) that colocalize with CGG repeats (Fig. 3A; Table 1; Methods: Sequestration Propensity). We also find poor sequestration propensity for FMRP and CUGBP1, while we predict that PURα interacts with CGG repeats (Fig. 3A). It should be noted that PURα colocalizes with cytoplasmic CGG repeats in flies (Jin et al. 2007) but not in mammalian cells, where is strictly nuclear (Sellier et al. 2010). Hence, the subcellular localization of PURα might prevent its physical interaction with RNA aggregates.
FIGURE 3.

CGG protein sequestration. Depending on the spatial position with respect to CGG aggregates, proteins are classified as (1) colocalizing (MBNL1 and hnRNP-G, black); (2) colocalizing in the late stage granules (inset); (3) noncolocalizing (FMRP, CUGBP1, and PURα, star), and (4) nonbinding (SAM68, italics). (A) In agreement with experimental results, we observe that colocalizing proteins have strong propensity to be sequestered by CGG repeats (MBNL1, hnRNP-G, and proteins in the inset). SAM68 interacting partners (CIRBP and PTBP2, gray) are found to bind to the CGG repeats. (A,B,D) We predict that SAM68 does not interact with CGG repeats as well as negative controls β-actin 1291–1417 nt and hnRNPA2/B1 1638–1754 nt. (C,E) SAM68 interacts with positive controls β-actin fragment 1398–1504 nt and hnRNPA2/B1 region 435–1173 nt (Supplemental Table 1).
In agreement with experimental evidence (Itoh et al. 2002; Sellier et al. 2010), we predict that SAM68 does not interact with CGG repeats (Fig. 3A) or negative controls β-actin 1291–1417 nt (Fig. 3B) and hnRNPA2/B1 1638–1754 nt (Fig. 3D). In contrast, we find strong interaction propensity for positive controls β-actin fragment 1398–1504 nt (Fig. 3C) and hnRNPA2/B1 region 435–1173 nt (Fig. 3E; Supplemental Table 1), as reported in previous studies (Itoh et al. 2002; Sellier et al. 2010). The fact that SAM68 does not interact with CGG repeats suggests that other proteins could be involved in its sequestration. Using the protein–protein interaction database MINT (http://mint.bio.uniroma2.it/), we retrieved SAM68 protein partners (35 interactions, of which 13 nucleic-acid were binding proteins) and tested their ability to bind to CGG repeats. We find that two proteins, cold-inducible RNA-binding protein CIRBP and polypyrimidine tract-binding protein 2 PTBP2, have sequestration propensities that increase with the length of CGG repeats (Supplemental Fig. 3). We speculate that CIRBP and PTBP2 could be involved in SAM68 sequestration at the early nucleation stages, which represents an intriguing scenario to be further investigated experimentally (Table 1).
The TDP-43 noncoding interactome
TAR DNA-binding protein 43 TDP-43 regulates RNA transcription, splicing, and metabolism (Chen-Plotkin et al. 2010). Although TDP-43 misfolding and aggregation has a major role in the development of ALS and frontotemporal lobar degeneration with ubiquitin-positive inclusions (FTLD-U) (Johnson et al. 2009), TDP-43 pathology has been linked to a number of other neurodegenerative diseases, such as AD and Parkinson’s disease (PD) (Geser et al. 2009; Wilson et al. 2011). Both gain- and loss-of-function are potential disease mechanisms due to TDP-43 nuclear depletion or cytoplasmic aggregation, which affect TDP-43 ability to regulate specific RNAs through direct binding (Lee et al. 2012). Recently, high-throughput RNA sequencing experiments revealed that a group of ncRNAs change their expression levels upon TDP-43 depletion (Polymenidou et al. 2011).
Here, we use catRAPID to predict the interaction potential of TDP-43 with 36 ncRNAs displaying more than twofold change in expression after TDP-43 silencing (Polymenidou et al. 2011). We find that 29 out of 36 associations (i.e., 80% of the entire set) have high propensity to bind to TDP-43 (average interaction strength of 83%), which indicates that a number of interactions are likely to take place (Fig. 4A–D; Table 1; Supplemental Fig. 4). In agreement with previous results (Ayala et al. 2011), we predict that TDP-43 has little propensity to bind to nucleotides 2785–3268 of transcript NM_007375.3, which is a negative control (Fig. 4E; Supplemental Table 1). When compared with a control set of RNA molecules, 84% of ncRNAs (30 out of 36 transcripts) are predicted to have significantly higher propensity to bind to TDP-43, which indicates great specificity for these interactions (Methods: Interaction Strength; Supplemental Fig. 5; Strong et al. 2007).
FIGURE 4.

TDP-43 associations with ncRNAs. Predictions of TDP-43 interactions with (A) FR033920; (B) FR311881; (C) FR330477; (D) FR143870, and (E) negative control RNA (Supplemental Material). (A,B) Natural antisense transcripts FR033920 and FR311881 regulate docosahexaenoic acid levels and dopamine transport, relatively. (C,D) vault RNAs FR330477 and FR143870 could be implicated in redox regulatory networks (Table 1).
Two of the most interacting targets are natural antisense transcripts participating in the regulation of gene expression (Supplemental Table 1): RevΔ5ase (FR033920/LIT3360, interaction strength = 99%), which modulates docosahexaenoic acid levels preventing cytoplasmic accumulation of TDP-43 (Tremblay et al. 2011) and Otx2OS (FR311881/LIT3411, interaction strength = 99%), which regulates the dopamine transport system impaired in PD/ALS patients (Buttarelli et al. 2006). Other ncRNAs predicted to highly interact with TDP-43 are vault RNAs (FR330477/LIT2028, interaction strength = 90%, and FR143870/LIT2029, interaction strength = 92%) (Supplemental Table 1; Kedersha and Rome 1986). The interaction between TDP-43 and vault RNAs might be implicated in redox regulatory networks, as suggested by preliminary experimental evidence (Iwashita et al. 2010; Fiesel et al. 2011). Although controversial hypotheses exist on the mechanisms by which TDP-43 contributes to neurodegeneration, catRAPID could help in the identification of protein–RNA interactions dysregulated in disease. The analysis of TDP-43 noncoding interactome shows that catRAPID is extremely powerful to investigate protein–ncRNA associations inferred from changes in regulatory networks (Table 1).
FMRP and TDP-43 autogenous regulation
Autogenous regulation of gene expression involves interaction between protein and RNA encoded by the same gene (Lee et al. 2012). Regulation of expression levels through autogenous associations has been reported for a number of RNA processing factors, including serine–arginine rich proteins SC35 and SF2, and heterogeneous ribonucleoprotein members PTB and hnRNP-L (Johnsen et al. 1982; Kim et al. 2010). Here, we focus on the autogenous regulation of FMRP and TDP-43, because impairment of this feedback mechanism could have consequences for the onset of FXS and ALS.
Coimmunoprecipitation assays indicate that a region located at position 1557–1658 nt in the 3′ UTR of FMR-1 is crucial for autogenous interaction (Schaeffer et al. 2001). Using catRAPID to compute the interaction scores between protein and RNA fragments (Methods: Interaction Fragments), we predict that FMR-1 region 1587–1660 nt, overlapping with the experimental region 1557–1658 nt, has high propensity to interact with the RGG domain of FMRP (Fig. 5A–C; Supplemental Fig. 6A; Schaeffer et al. 2001). As for TDP-43, CLIP experiments performed in HEK293 cells indicate that the protein self-associates at the 3′ UTR of its pre-mRNA TARDBP (Ayala et al. 2011). catRAPID correctly identifies the two experimentally validated RNA regions located at positions 301–744 nt and 745–1060 nt (Fig. 5D–F; Supplemental Fig. 6B; Ayala et al. 2011). The algorithm also identifies two RNA recognition motifs (RRM1 and RRM2) and the glycine-rich C-terminal domain of TDP-43 (Fig. 5D; Lagier-Tourenne and Cleveland 2009). Our predictions indicate that the C-terminal region spanning residues 321–366 of TDP-43 is directly involved in autogenous regulation, in agreement with previous experimental observations (Ayala et al. 2011). We speculate that many other RNA-binding proteins have evolved similar mechanisms of autogenous regulation.
FIGURE 5.

FMRP and TDP-43 self-regulation. (A) FMRP-FMR-1 and (D) TDP-43-TARDBP interaction maps (blue areas indicate experimentally validated interactions, protein secondary structure elements are displayed next to the “protein residue index” axis); (B) FMR-1 and (E) TARDBP RNA interaction profiles (blue lines indicate experimentally identified binding regions; Methods: Interaction Fragments); interaction strengths with (C) nucleotides 1557–1658 of FMR-1 and (F) nucleotides 495–555 of TARDBP (Supplemental Table 1).
Iron-mediated expression of APP and α-synuclein
Iron regulatory protein 1, IRP-1, is a cytosolic iron sensor that binds to an iron-sulfur cluster and acts as aconitase when cellular iron levels are high (Philpott et al. 1994). If iron levels are low, IRP-1 binds to stem–loop structures called Iron-Responsive Elements (IREs, conserved nucleotides 5′CAGU/AGN3′) in target mRNA species, and regulates uptake, storage, and transport of iron ions. Binding of IRP-1 to an iron–sulfur cluster impairs its ability to interact with RNA. An electrophoretic motility shift assay (REMSA) has been used to probe the interaction between IRP-1 and the iron-responsive element (IRE-Type II) within the 5′ UTR of APP mRNA (Rogers et al. 2002). Importantly, IRE-Type II sequence shows high similarity with the IRE in the mRNA of ferritin H-subunit (Supplemental Fig. 7) that is regulated by IRP-induced translational repression in case of iron deficiency.
We use catRAPID to predict the interaction potential of IRP-1 with both ferritin and APP transcripts (Methods: Interaction Fragments) (Rogers et al. 2002). Our method correctly predicts that the 5′ UTRs of ferritin (Supplemental Fig. 8) and APP (Fig. 6A,B) interact with IRP-1 in several regions of the protein sequence, which is consistent with experimental evidence (Rogers et al. 2002; Walden et al. 2006). As for the APP transcript, nucleotides 25–76 show the highest interaction propensity and overlap with the experimental segment at nucleotides 40–95 (Fig. 6A,B; Supplemental Fig. 8). We note that the IRE-binding surface of IRP-1 does not consist of canonical RNA-binding motifs such as those of other ribonucleoproteins, and that IRE binding is mediated by residues from each of the four IRP-1 domains (Walden et al. 2006). Recently, Cho et al. (2010) showed that disruption of the CAGAGC motif leads to strong reduction in the IRP-1 propensity to bind to APP IRE (Supplemental Table 1; Supplemental Fig. 9A). We use catRAPID to carry out an exhaustive analysis of all the mutations in the CAGAGC site (4095 sequence variants) and discovered that 87% of the mutations strongly reduce IRP-1 binding ability (Supplemental Fig. 9A–C). Intriguingly, shuffling the IRE sequence is predicted to abrogate IRP-1 interactions in 95 out 100 cases (Supplemental Fig. 9B,C). The same effect is observed for TDP-43 interactions with ncRNAs, where shuffling reduces the interaction propensity in about 80 out of 100 cases (Supplemental Fig. 9D–F).
FIGURE 6.

IRP-1 interactions with APP and α-synuclein mRNA. (A) Interaction map of IRP-1 with APP (secondary structure elements are displayed at the “protein residue index” axis; blue areas indicate experimentally validated interactions). RNA-interaction profiles for IRP-1 associations with (B) APP and (C) α-synuclein mRNA (blue lines indicate experimentally identified binding regions) (Supplemental Table 1). (D) Interaction strength for IRP-1 domain 4 region (amino acids 661–889) and putative IRE fragment in α-synuclein transcript (nucleotides 190–252) (Supplemental Table 1).
Interestingly, one RNA stem–loop within the 5′ UTR of human α-synuclein transcript has been predicted to be structurally related to the IRE element present in ferritin mRNA (Supplemental Fig. 7; Olivares et al. 2009). According to our calculations, the IRE-containing fragment of α-synuclein mRNA (nucleotides 190–252) has the highest propensity to bind to IRP-1 (Fig. 6C; Supplemental Fig. 8B). Moreover, we predict that IRP-1 domain 4, containing the largest number of binding sites (Walden et al. 2006), interacts with the IRE-containing fragment (interaction strength = 85%) (Fig. 6D). Our predictions suggest that IRP-1 could be involved in regulating α-synuclein, which might have implications for the alteration of iron levels found in PD (Table 1).
Prions and RNA aptamers
Mammalian prions (PrP) are infectious agents causing neurodegenerative diseases (Prusiner 1998). To date, prion infectivity is attributed to conversion of the soluble PrPc into an aggregation-prone structural isoform PrPSc (Pan et al. 1993). The exact physiological function of PrPc remains elusive; however, there is an increasing understanding of the molecular mechanisms underlying PrPc pathological conversion and its interactions with other biological macromolecules. Among these, cellular adhesion molecules, nucleic acids, basal membrane molecules, and sulfated glycans have been reported to interact with PrPc and to induce or modulate conversion into β-sheet-rich structures that shares many features with infectious PrPSc (Silva et al. 2011). It has been proposed that PrPc could undergo specific structural rearrangements modulated by binding with specific nucleic acids molecules, such as highly structured RNAs (Deleault et al. 2003) or RNA aptamers (Mercey et al. 2006).
Several studies showed that residues 90–141 are crucial for the conversion from natural to pathological form (Tartaglia et al. 2005, 2008). Proske et al. (2002) discovered an RNA aptamer, DP7, which binds with great affinity to an epitope located within residues 90–141 of hamster PrP (highly conserved in mouse and human sequences) (Supplemental Table 1). Here we estimate the binding ability of DP7 to hamster PrP using catRAPID (Methods: Interaction Fragments). Based on our calculations, the fragment located at residues 104–155 shows the highest propensities to bind to DP7 and has the largest overlap with the experimental region spanning residues 90–141 (Fig. 7A; Proske et al. 2002). In agreement with experimental evidence, we predict that full-length hamster PrP and DP7 aptamer are highly interacting (interaction strength = 85%) (Fig. 7B; Proske et al. 2002). We also report very high interaction propensity between residues 90 and 141 of hamster PrP and DP7 aptamer (interaction strength = 99%; RNA interaction strength = 100%), which is consistent with experimental findings (Methods: Interaction Score) (Fig. 7C; Proske et al. 2002). In agreement with Proske et al. (2002), we predict high interaction propensities between DP7 and mouse and human PrP (Supplemental Fig. 10A–D).
FIGURE 7.

Prion and aptamers. (A) Protein interaction profile for the association of hamster prion protein (PrP) with RNA aptamer DP7. Interaction strength of DP7 interactions with (B) full-length PrP, and (C) PrP fragment 90–141 (highlighted); (D) Prediction of PrP90–141 binding specificity for DP7 (control set of 1000 DP7 single point mutations; Supplemental Table 1; Methods: Interaction Strength).
The analysis of 100 single point mutations on the DP7 aptamer reveals that 73% of variants are predicted to reduce the ability to bind to hamster PrP (Fig. 7D), while the remaining 27% increases the interaction propensity (interaction strength in the range from 99% to 100%), thus indicating that catRAPID is able to capture the specificity of DP7 for PrP (Methods: Interaction Score). In conclusion, our results suggest that catRAPID represents a valuable tool for the in silico screening of RNA aptamers (Table 1).
DISCUSSION
We used our computational approach catRAPID to investigate protein–RNA interactions linked to neurodegenerative diseases. As summarized in Table 1, we were interested in understanding the involvement of ribonucleoprotein associations in a number of regulatory processes as well as disorders such as PD and AD. We studied the metabolic signature associated with FXS and analyzed FMRP associations with SOD1 and APP, highlighting possible links with ALS and AD (Davidovic et al. 2011). We investigated the X-chromosome disorder FXTAS, which is caused by CGG expansions in the FMR-1 untranslated region (Sellier et al. 2010) and characterized key players involved in protein sequestration. We predicted TDP-43 interactions with several ncRNAs displaying changes of expression levels upon TDP-43 depletion (Polymenidou et al. 2011) and identified a set of vault-associated and natural antisense transcripts that could be linked to clinical manifestations of TDP-43 proteinopathy (Chen-Plotkin et al. 2010). We also studied the ability of FMRP and TDP-43 to regulate their own expression levels through autogenous interactions, characterizing their binding sites in great detail (Schaeffer et al. 2001; Ayala et al. 2011). We analyzed the interaction between IRP-1 and APP (Cho et al. 2010) and predicted the interaction between IRP-1 and an IRE-like region of α-synuclein mRNA, which represents a link to the iron-pathway deregulation associated with PD (Olivares et al. 2009). Finally, we investigated the ability of RNA aptamers to bind to aggregation-prone regions of prions (Proske et al. 2002), which shows that our theoretical framework could be useful for the in silico screening of RNA-based therapeuticals (Table 1).
In this work we introduced the interaction strength algorithm to measure the robustness of our predictions with respect to an “ensemble” of negative controls (e.g., Fig. 5F). In each case studied, we observed that experimentally validated associations have significantly higher interaction propensities than control sets (e.g., Fig. 4). Moreover, to assess catRAPID’s performances on negative controls, we studied a number of noninteracting pairs present in literature (Schaeffer et al. 2001; Itoh et al. 2002; Ayala et al. 2011) (Supplemental Table 1). We found strong agreement between experimental results and our calculations (Figs. 1E, 3B,D, 5E, 7D), which indicates that the use of control sets is very important to achieve accurate predictions. As shown in Figures 1B,D, 6B,C, 7A, the interaction fragments algorithm is able to identify binding regions in both protein and RNA molecules.
We propose catRAPID for predictions of protein–RNA associations, to flag putative interactions and select candidates for experimental studies (Table 1). Our method allows processing of a large amount of protein–RNA pairs and can lead to finding previously unknown interactions. Due to the vastly increased analysis throughput, even whole protein–RNA networks could be soon investigated without the need to focus on small subsets. Our methodology provides a significant amount of new information on protein–RNA associations, discovery of which would not be possible with a purely experimental workflow due to the sheer volume. Most importantly, our approach works on the intersection of protein and RNA biology and will help to bridge the gap between the two disciplines.
MATERIALS AND METHODS
Interaction propensity
We use the catRAPID method to predict protein–RNA interactions (Bellucci et al. 2011). In catRAPID, the contributions of secondary structure, hydrogen bonding, and van der Waals’ are combined together into the “interaction profile”:
| (1) |
The “interaction propensity” π is defined as the inner product between the protein propensity profile 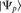 and the RNA propensity profile
and the RNA propensity profile 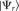 weighted by the “interaction matrix” I:
weighted by the “interaction matrix” I:
| (2) |
The interaction matrix I as well as the parameters αS, αH, and αW were derived using a Montecarlo procedure under the condition that interaction propensities π take maximal values for associations in the positive training set and minimal values for associations in the negative training set:
| (3) |
The catRAPID method was trained to predict interaction propensities of protein–RNA pairs in the range of from 50 to 750 amino acids and 50 to 1500 nt. The algorithm to compute the interaction propensity with respect to the negative training set (discriminative power) is available at www.tartaglialab.crg.cat/catrapid.html.
Interaction strength
The concept of interaction strength is introduced to compare the interaction propensity of a protein–RNA pair with a reference set that has little propensity to bind (random associations between polypeptide and nucleotide sequences). For each protein–RNA pair under investigation, we use a “reference set” of 102 protein and 102 RNA molecules. To assess the strength of a particular association, we compute its interaction propensity π and compare with the interaction propensities  of the reference set (total of 104 nonredundant protein–RNA pairs). Using the interaction propensity distribution of the reference set, we generate the “interaction score”:
of the reference set (total of 104 nonredundant protein–RNA pairs). Using the interaction propensity distribution of the reference set, we generate the “interaction score”:
 |
(4) |
The number of interactions is Λ = 104. From the distribution of interaction propensities we compute the “interaction strength”:
 |
(5) |
Reference sequences have the same lengths as the pair of interest to guarantee that the interaction strength does not depend on protein and RNA lengths. The “protein interaction strength” and the “RNA interaction strength” are special cases of the interaction strength in which only the protein or the RNA sequence is randomized to generate a reference set. For instance, the RNA interaction strength used for the analysis of the TDP43 interactome is the RNA-binding ability of a protein with respect to a pool of 100 proteins. The algorithm to compute the interaction strength is available at http://tartaglialab.crg.cat/catrapid.strength.html.
Interaction fragments
In some cases, protein or RNA sequences exceed the size compatible with our computational requirements, and catRAPID could not be used to calculate the interaction propensity. To overcome this limitation, we developed a procedure called “fragmentation,” which involves division of polypeptide and nucleotide sequences into fragments, followed by prediction of the interaction propensities. The analysis of fragments is particularly useful to identify regions involved in the binding (e.g., self-interactions of TDP-43 and FMRP). The fragmentation approach is based on the division of protein and RNA sequences into overlapping segments:
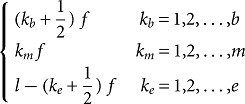 |
(6) |
Where kb, km, and ke indicate the position of fragments, f is their length and l is the overall sequence length. The number of total fragments is b + m + e = t ≤ 100 (limited by catRAPID sequence restrictions). The maximum number of protein–RNA interactions is 104, which implies that the ability to identify an experimentally validated interaction by chance is 10−4. The list of all the protein–RNA fragment associations is called “interaction map.” “Protein and RNA interaction profiles” are bidimensional projections of the interaction map onto the protein or RNA positions, respectively. A variant of the fragmentation algorithm developed to analyze protein interactions with long transcripts has been described in a recent paper (Agostini et al. 2012). The algorithms to compute interaction fragments are available at http://tartaglialab.crg.cat/catrapid.fragments.html.
CGG sequestration propensity
The CGG sequestration propensity is calculated as the interaction strength multiplied by the protein concentration:
 |
(7) |
Where 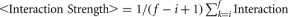 Strength (k) is the average interaction strength for CGG repeats ranging from i = 20 to f = 200 and α = 1/log(Abundance)max is the normalization factor. Protein abundances are retrieved from the database http://pax-db.org.
Strength (k) is the average interaction strength for CGG repeats ranging from i = 20 to f = 200 and α = 1/log(Abundance)max is the normalization factor. Protein abundances are retrieved from the database http://pax-db.org.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Professor R. Guigo’, Dr. B. Lehner, and Viola Tartaglia for stimulating discussions. Our work was supported by the Spanish Ministry of Economy and Competitiveness (SAF2011-26211). We are grateful to the Programa de Ayudas FPI del Ministerio de Economia y Competitividad—BES-2012-052457. This project was supported by a grant from “la Caixa” to Petr Klus.
REFERENCES
- Agostini F, Cirillo D, Bolognesi B, Tartaglia GG 2012. X-inactivation: Quantitative predictions of protein interactions in the Xist network. Nucleic Acids Res 10.1093/nar/gks968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony K, Gallo J-M 2010. Aberrant RNA processing events in neurological disorders. Brain Res 1338: 67–77 [DOI] [PubMed] [Google Scholar]
- Ayala YM, De Conti L, Avendaño-Vázquez SE, Dhir A, Romano M, D’Ambrogio A, Tollervey J, Ule J, Baralle M, Buratti E, et al. 2011. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J 30: 277–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartzokis G, Sultzer D, Cummings J, Holt LE, Hance DB, Henderson VW, Mintz J 2000. In vivo evaluation of brain iron in Alzheimer disease using magnetic resonance imaging. Arch Gen Psychiatry 57: 47–53 [DOI] [PubMed] [Google Scholar]
- Bechara EG, Didiot MC, Melko M, Davidovic L, Bensaid M, Martin P, Castets M, Pognonec P, Khandjian EW, Moine H, et al. 2009. A novel function for fragile X mental retardation protein in translational activation. PLoS Biol 7: e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellucci M, Agostini F, Masin M, Tartaglia GG 2011. Predicting protein associations with long noncoding RNAs. Nat Methods 8: 444–445 [DOI] [PubMed] [Google Scholar]
- Buttarelli FR, Circella A, Pellicano C, Pontieri FE 2006. Dopamine transporter immunoreactivity in peripheral blood mononuclear cells in amyotrophic lateral sclerosis. Eur J Neurol 13: 416–418 [DOI] [PubMed] [Google Scholar]
- Chen-Plotkin AS, Lee VM-Y, Trojanowski JQ 2010. TAR DNA-binding protein 43 in neurodegenerative disease. Nat Rev Neurol 6: 211–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H-H, Cahill CM, Vanderburg CR, Scherzer CR, Wang B, Huang X, Rogers JT 2010. Selective translational control of the Alzheimer amyloid precursor protein transcript by iron regulatory protein-1. J Biol Chem 285: 31217–31232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Jensen KB, Jin P, Brown V, Warren ST, Darnell RB 2001. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell 107: 489–499 [DOI] [PubMed] [Google Scholar]
- Darnell JC, Fraser CE, Mostovetsky O, Stefani G, Jones TA, Eddy SR, Darnell RB 2005. Kissing complex RNAs mediate interaction between the Fragile-X mental retardation protein KH2 domain and brain polyribosomes. Genes Dev 19: 903–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidovic L, Navratil V, Bonaccorso CM, Catania MV, Bardoni B, Dumas M-E 2011. A metabolomic and systems biology perspective on the brain of the fragile X syndrome mouse model. Genome Res 21: 2190–2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleault NR, Lucassen RW, Supattapone S 2003. RNA molecules stimulate prion protein conversion. Nature 425: 717–720 [DOI] [PubMed] [Google Scholar]
- Dobson CM 1999. Protein misfolding, evolution and disease. Trends Biochem Sci 24: 329–332 [DOI] [PubMed] [Google Scholar]
- Fiesel FC, Kahle PJ 2011. TDP-43 and FUS/TLS: Cellular functions and implications for neurodegeneration. FEBS J 278: 3550–3568 [DOI] [PubMed] [Google Scholar]
- Fiesel FC, Schurr C, Weber SS, Kahle PJ 2011. TDP-43 knockdown impairs neurite outgrowth dependent on its target histone deacetylase 6. Mol Neurodegener 6: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geser F, Martinez-Lage M, Kwong LK, Lee VM-Y, Trojanowski JQ 2009. Amyotrophic lateral sclerosis, frontotemporal dementia and beyond: The TDP-43 diseases. J Neurol 256: 1205–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman PJ, Hagerman RJ 2004. The fragile-X premutation: A maturing perspective. Am J Hum Genet 74: 805–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh M, Haga I, Li Q-H, Fujisawa J 2002. Identification of cellular mRNA targets for RNA-binding protein Sam68. Nucleic Acids Res 30: 5452–5464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwashita K, Ikeda R, Takeda Y, Sumizawa T, Furukawa T, Yamaguchi T, Akiyama S, Yamada K 2010. Major vault protein forms complexes with hypoxia-inducible factor (HIF)-1α and reduces HIF-1α level in ACHN human renal adenocarcinoma cells. Cancer Sci 101: 920–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin P, Duan R, Qurashi A, Qin Y, Tian D, Rosser TC, Liu H, Feng Y, Warren ST 2007. Pur α binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of Fragile X Tremor/Ataxia Syndrome. Neuron 55: 556–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsen M, Christensen T, Dennis PP, Fiil NP 1982. Autogenous control: Ribosomal protein L10-L12 complex binds to the leader sequence of its mRNA. EMBO J 1: 999–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BS, Snead D, Lee JJ, McCaffery JM, Shorter J, Gitler AD 2009. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem 284: 20329–20339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha NL, Rome LH 1986. Isolation and characterization of a novel ribonucleoprotein particle: Large structures contain a single species of small RNA. J Cell Biol 103: 699–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenneson A, Zhang F, Hagedorn CH, Warren ST 2001. Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum Mol Genet 10: 1449–1454 [DOI] [PubMed] [Google Scholar]
- Kim HD, Kim T-S, Joo YJ, Shin H-S, Kim S-H, Jang C-Y, Lee CE, Kim J 2010. RpS3 translation is repressed by interaction with its own mRNA. J Cell Biochem 110: 294–303 [DOI] [PubMed] [Google Scholar]
- Kryshtafovych A, Venclovas C, Fidelis K, Moult J 2005. Progress over the first decade of CASP experiments. Proteins 61 (Suppl 7): 225–236 [DOI] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Cleveland DW 2009. Rethinking ALS: The FUS about TDP-43. Cell 136: 1001–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JF, Stovall GM, Ellington AD 2006. Aptamer therapeutics advance. Curr Opin Chem Biol 10: 282–289 [DOI] [PubMed] [Google Scholar]
- Lee EB, Lee VM-Y, Trojanowski JQ 2012. Gains or losses: Molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci 13: 38–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercey R, Lantier I, Maurel M-C, Grosclaude J, Lantier F, Marc D 2006. Fast, reversible interaction of prion protein with RNA aptamers containing specific sequence patterns. Arch Virol 151: 2197–2214 [DOI] [PubMed] [Google Scholar]
- Olivares D, Huang X, Branden L, Greig NH, Rogers JT 2009. Physiological and pathological role of α-synuclein in Parkinson’s disease through iron mediated oxidative stress; The role of a putative iron-responsive element. Int J Mol Sci 10: 1226–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oostra BA, Willemsen R 2009. FMR1: A gene with three faces. Biochim Biophys Acta 1790: 467–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick RJ, Cohen FE 1993. Conversion of α-helices into β-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci 90: 10962–10966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpott CC, Klausner RD, Rouault TA 1994. The bifunctional iron-responsive element binding protein/cytosolic aconitase: The role of active-site residues in ligand binding and regulation. Proc Natl Acad Sci 91: 7321–7325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT, Nelson DL 1991. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66: 817–822 [DOI] [PubMed] [Google Scholar]
- Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling S-C, Sun E, Wancewicz E, Mazur C, et al. 2011. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci 14: 459–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Primerano B, Tassone F, Hagerman RJ, Hagerman P, Amaldi F, Bagni C 2002. Reduced FMR1 mRNA translation efficiency in fragile X patients with premutations. RNA 8: 1482–1488 [PMC free article] [PubMed] [Google Scholar]
- Proske D, Gilch S, Wopfner F, Schätzl HM, Winnacker E-L, Famulok M 2002. Prion-protein-specific aptamer reduces PrPSc formation. Chembiochem 3: 717–725 [DOI] [PubMed] [Google Scholar]
- Prusiner SB 1998. Prions. Proc Natl Acad Sci 95: 13363–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JT, Randall JD, Cahill CM, Eder PS, Huang X, Gunshin H, Leiter L, McPhee J, Sarang SS, Utsuki T, et al. 2002. An iron-responsive element type II in the 5′-untranslated region of the alzheimer’s amyloid precursor protein transcript. J Biol Chem 277: 45518–45528 [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC 2006. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443: 780–786 [DOI] [PubMed] [Google Scholar]
- Salta E, De Strooper B 2012. Non-coding RNAs with essential roles in neurodegenerative disorders. Lancet Neurol 11: 189–200 [DOI] [PubMed] [Google Scholar]
- Schaeffer C, Bardoni B, Mandel J-L, Ehresmann B, Ehresmann C, Moine H 2001. The fragile X mental retardation protein binds specifically to its mRNA via a purine quartet motif. EMBO J 20: 4803–4813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C, Rau F, Liu Y, Tassone F, Hukema RK, Gattoni R, Schneider A, Richard S, Willemsen R, Elliott DJ, et al. 2010. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J 29: 1248–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva JL, Vieira TCRG, Gomes MPB, Rangel LP, Scapin SMN, Cordeiro Y 2011. Experimental approaches to the interaction of the prion protein with nucleic acids and glycosaminoglycans: Modulators of the pathogenic conversion. Methods 53: 306–317 [DOI] [PubMed] [Google Scholar]
- Strong MJ, Volkening K, Hammond R, Yang W, Strong W, Leystra-Lantz C, Shoesmith C 2007. TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol Cell Neurosci 35: 320–327 [DOI] [PubMed] [Google Scholar]
- Subramanian M, Rage F, Tabet R, Flatter E, Mandel J-L, Moine H 2011. G-quadruplex RNA structure as a signal for neurite mRNA targeting. EMBO Rep 12: 697–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia GG, Cavalli A, Pellarin R, Caflisch A 2005. Prediction of aggregation rate and aggregation-prone segments in polypeptide sequences. Protein Sci 14: 2723–2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia GG, Pawar AP, Campioni S, Dobson CM, Chiti F, Vendruscolo M 2008. Prediction of aggregation-prone regions in structured proteins. J Mol Biol 380: 425–436 [DOI] [PubMed] [Google Scholar]
- Tassone F, Hagerman RJ, Loesch DZ, Lachiewicz A, Taylor AK, Hagerman PJ 2000. Fragile X males with unmethylated, full mutation trinucleotide repeat expansions have elevated levels of FMR1 messenger RNA. Am J Med Genet 94: 232–236 [DOI] [PubMed] [Google Scholar]
- Tremblay C, St-Amour I, Schneider J, Bennett DA, Calon F 2011. Accumulation of transactive response DNA binding protein 43 in mild cognitive impairment and Alzheimer disease. J Neuropathol Exp Neurol 70: 788–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walden WE, Selezneva AI, Dupuy J, Volbeda A, Fontecilla-Camps JC, Theil EC, Volz K 2006. Structure of dual function iron regulatory protein 1 complexed with ferritin IRE-RNA. Science 314: 1903–1908 [DOI] [PubMed] [Google Scholar]
- Westmark CJ, Malter JS 2007. FMRP mediates mGluR5-dependent translation of amyloid precursor protein. PLoS Biol 5: e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AC, Dugger BN, Dickson DW, Wang D-S 2011. TDP-43 in aging and Alzheimer’s disease - a review. Int J Clin Exp Pathol 4: 147–155 [PMC free article] [PubMed] [Google Scholar]