

This article examines how tRNALys3 that serves as a primer for HIV-1 genome replication is targeted to the viral RNA during virion assembly. Evidence is presented that lysyl-tRNA synthetase may carry out this task, by specifically interacting with a genome region near the primer binding site (PBS) which mimics the anti-codon of tRNALys, a process that may increase the efficiency of the final tRNALys3 annealing to viral RNA.
Keywords: tRNA anti-codon-like element, lysyl-tRNA synthetase, reverse transcription, primer placement
Abstract
The primer for initiating reverse transcription in human immunodeficiency virus type 1 (HIV-1) is tRNALys3. Host cell tRNALys is selectively packaged into HIV-1 through a specific interaction between the major tRNALys-binding protein, human lysyl-tRNA synthetase (hLysRS), and the viral proteins Gag and GagPol. Annealing of the tRNA primer onto the complementary primer-binding site (PBS) in viral RNA is mediated by the nucleocapsid domain of Gag. The mechanism by which tRNALys3 is targeted to the PBS and released from hLysRS prior to annealing is unknown. Here, we show that hLysRS specifically binds to a tRNA anti-codon-like element (TLE) in the HIV-1 genome, which mimics the anti-codon loop of tRNALys and is located proximal to the PBS. Mutation of the U-rich sequence within the TLE attenuates binding of hLysRS in vitro and reduces the amount of annealed tRNALys3 in virions. Thus, LysRS binds specifically to the TLE, which is part of a larger LysRS binding domain in the viral RNA that includes elements of the Psi packaging signal. Our results suggest that HIV-1 uses molecular mimicry of the anti-codon of tRNALys to increase the efficiency of tRNALys3 annealing to viral RNA.
INTRODUCTION
Transfer RNAs are the adaptor molecules in protein synthesis, binding trinucleotide codons through specific base-pairing interactions and translating them into the amino acid sequence of proteins. They accomplish this by delivering specific amino acids, which are covalently attached to their 3′ ends by aminoacyl-tRNA synthetases. In addition to their central role in protein biosynthesis, tRNAs and tRNA-like elements are used for a variety of cellular roles and are exploited by viruses in their replication (Dreher 2009). For example, plant viruses contain tRNA anti-codon-like elements (TLEs) at their 3′ ends that are bound by aminoacyl-tRNA synthetases and aminoacylated (Dreher 2009; Hammond et al. 2009). The solution structure of a portion of the 3′ UTR of turnip crinkle virus has structural features that resemble those of a tRNA (Hammond et al. 2009), the pea enation mosaic virus has been predicted to contain a T-shaped tRNA-like structure (Gao et al. 2012), and the intergenic region of cricket paralysis virus also mimics the tRNA structure to enhance its own translation (Costantino et al. 2008).
Many retroelements and all retroviruses, including human immunodeficiency virus type 1 (HIV-1) initiate reverse transcription of their viral RNA genome (vRNA) from the 3′ end of a host cellular tRNA (Coffin et al. 1997). Human tRNALys3 is the primer for HIV-1 and is selectively packaged into the virus along with the other major tRNALys isoacceptors in the cell, tRNALys1 and tRNALys2 (Jiang et al. 1993; Pavon-Eternod et al. 2010). Upon virus entry into target cells, the single-stranded vRNA is reverse transcribed into double-stranded proviral DNA, which is integrated into host DNA. Following export from the nucleus, newly synthesized vRNA, viral precursor proteins Gag and GagPol, and other cellular factors co-opted by the virus assemble at a cytoplasmic site prior to viral budding. While the processing of the major HIV-1 precursor proteins Gag and GagPol into mature viral proteins occurs during and after viral budding from the cell, annealing of tRNALys3 to vRNA occurs in the absence of precursor protein processing (Huang et al. 1997). The selective incorporation of primer tRNALys3 is required for optimizing both tRNALys3 annealing to the viral RNA primer binding site (PBS) and viral infectivity of the HIV-1 population (Gabor et al. 2002). The cytoplasmic nucleoprotein complex into which tRNALys3 is recruited prior to PBS annealing also includes human lysyl-tRNA synthetase (hLysRS), Gag, GagPol, and vRNA (Kleiman et al. 2010). The selective incorporation of tRNALys isoacceptors appears to be due to a specific interaction between Gag and hLysRS (Javanbakht et al. 2003; Kovaleski et al. 2007). GagPol is also required for binding to tRNALys and facilitating its incorporation (Khorchid et al. 2000; Kobbi et al. 2011), and an interaction between hLysRS and Pol has been proposed (Saadatmand et al. 2008; Kobbi et al. 2011).
Human tRNALys and hLysRS are both present in 20–25 molecules per virion and are likely packaged into virions as a complex (Huang et al. 1994; Cen et al. 2002). The relative level of packaged tRNALys is linked to both packaged LysRS and virus infectivity; that is, knockdown of cytoplasmic hLysRS reduces the amount of tRNALys in the virus and subsequently reduces virus infectivity, whereas overexpression of hLysRS causes the opposite effect (Guo et al. 2003, 2005). The selective incorporation of tRNALys into virions may also reflect a higher concentration of tRNALys at the cellular site of HIV-1 assembly or alterations to the tRNA pool induced by HIV-1 infection (van Weringh et al. 2011; Li et al. 2012).
Although the ∼9.4-kB vRNA contains secondary structural elements whose functions are, in most cases, still unknown (Watts et al. 2009), the 5′ untranslated region (UTR) is especially rich in complex secondary structures with known functions in many steps of the virus life cycle (Coffin et al. 1997; Bolinger and Boris-Lawrie 2009). For example, Gag facilitates tRNALys3 primer annealing onto a highly conserved sequence in the 5′ UTR (Feng et al. 1999; Roldan et al. 2005; Jones et al. 2011). The longest motif in this sequence, the 18-nt PBS is complementary to the 3′ 18-nt of tRNALys3. Additional interactions occur between vRNA and complementary sequences in the variable arm and anti-codon stem–loop of tRNALys3 (Isel et al. 1993; Arts et al. 1996; Beerens et al. 2001; Iwatani et al. 2003; Wilkinson et al. 2008).
An essential component of the translation machinery, hLysRS binds to tRNALys through specific interactions with its anti-codon binding domain (Stello et al. 1999) and less specific interactions via an N-terminal extension (Francin et al. 2002; Francin and Mirande 2006). Mammalian LysRS binds its tRNA substrates with high affinity (Francin et al. 2002), and the mechanism of tRNALys3 release from hLysRS and targeting to the PBS during formation of the annealing complex is presently unknown. This is an important question since only a limited number of Gag-GagPol complexes are expected to be bound to hLysRS/tRNALys3, and only two copies of vRNA are packaged per virion. Thus, a mechanism for increasing the likelihood that the tRNALys-containing complexes bind proximal to the PBS may be beneficial to the virus. In this work, we provide evidence that a tRNALys anti-codon-like sequence in the vRNA located near the PBS can interact with hLysRS, and we discuss possible roles for tRNA mimicry in initiation complex formation and HIV-1 replication.
RESULTS
HIV-1 genome contains a tRNALys anti-codon-like element
Using a recent secondary structural model of the HIV-1 vRNA (Watts et al. 2009), we searched for tRNALys3-like secondary structural elements proximal to the PBS. A hairpin containing a loop sequence resembling the anti-codon loop of tRNALys3 was identified directly upstream of the PBS (Fig. 1A, TLE, boxed). We examined the conservation of the tRNA anti-codon-like element across ∼2170 HIV-1 isolates (Fig. 2; Methods in Supplemental Material). The center U nucleotide in the loop, corresponding to U35 in tRNALys3, is 95% conserved. Anti-codon positions U35 and U36 are identity elements critical for tRNALys3 binding and aminoacylation by hLysRS in vitro (Stello et al. 1999) and in vivo (Javanbakht et al. 2002). Point mutations at position 35 have the most severe effects on hLysRS catalytic efficiency (ranging from ∼150-fold to >3000-fold reduction of kcat/KM) (Stello et al. 1999). Thus, the high conservation of the central U in the TLE loop is consistent with a favorable hLysRS binding site.
FIGURE 1.

(A) Predicted secondary structure of a 329-nt RNA derived from the HIV-1 NL4-3 5′ UTR with the TLE sequence boxed and PBS circled and in bold. Although only the TLE loop sequence resembles the anti-codon domain of tRNALys3, we have included the flanking 3-bp stem domain in our definition of the TLE to emphasize the presentation of anti-codon-like nucelotide in an exposed loop. The NL4-3 “PBS/TLE” RNA used in this study (105 nt) contains nt 125–223 plus three additional 5′-G-C-3′ pairs to stabilize the terminal RNA stem and aid in vitro transcription. The 330-nt 5′ UTR RNA used in this study contains the 329-nt sequence shown with an additional 5′-G residue for in vitro transcription. The ΔTARpolyA mutation is a deletion of nt 1–124, the ΔSL3 mutation is a deletion of nt 300–329, and the ΔSL2,3 mutation is a deletion of nt 284–329. (B) The sequence of a 23-mer derived from the vRNA (TLE23) is shown next to the anti-codon stem–loop of tRNALys3 for comparison. This construct contains two additional nonviral 5′-C-G-3′ pairs to stabilize the terminal RNA stem. (C) Predicted secondary structure of the MAL construct derived from nt 123–218 of the HIV-1 MAL isolate is shown, with the PBS sequence circled and in bold. The A-rich stem–loop examined in Puglisi and Puglisi (1998) includes nt 156–172.
FIGURE 2.
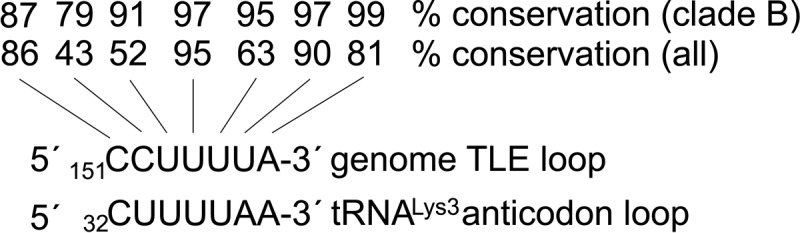
The anti-codon loop of human tRNALys3 is shown aligned with the loop sequence of the TLE (nt 151–157). The % conservation (all) and % conservation of clade B viruses were determined from alignment of ∼2170 and 736 HIV-1 sequences, respectively, from the HIV sequence database (http://www.hiv.lanl.gov/).
To further examine the conservation of the viral genome TLE loop sequences across HIV-1 isolates, we divided sequences by virus subtype or clade (described in Supplemental Material, Methods). Most of the sequences originate from clades B and C, which represent the majority of infections in the Americas, Europe, Asia, and South and East Africa (Coffin et al. 1997). Alignment of TLE loop nucleotides from clades B and C demonstrates that in these viruses, the TLE is U-rich (Supplemental Fig. S1) and is highly conserved, especially among clade B viruses (Fig. 2). In contrast, viruses from other clades—including clades A and G and recombinant forms of these clades—contain a central U flanked by two C nucleotides (Supplemental Fig. S1). This variation contributes to the lower conservation in the nucleotides surrounding the highly conserved U154 when all clades are included in the alignment (Fig. 2). Structure-probing studies of HXB2 (a clade B isolate) and MAL (a mosaic of clades A, D, and I) concluded that the two viruses have different PBS stem folds (Goldschmidt et al. 2004). In HXB2, the U-rich anti-codon-like element is an exposed 7-nt loop (Fig. 1A), as found in previous structure probing of the clade B HIV-1 LAI strain (Damgaard et al. 1998). In the MAL isolate, an alternative 6-nt A-rich apical loop is formed (Fig. 1C). The CUC triplet corresponding to the clade B UUU triplet is located in the base-paired stem region (Goldschmidt et al. 2004). In this work, we examine TLE sequences from a clade B virus, NL4-3, as well as an RNA derived from the MAL isolate (Fig. 1).
hLysRS specifically binds viral TLE-containing RNAs
Electrophoretic mobility shift assays (EMSAs) were carried out to determine if hLysRS could bind to a 105-nt segment of the HIV-1 vRNA containing the putative TLE and the PBS (Fig. 1A, nt 123–223). The PBS/TLE RNA bound to hLysRS but not to human prolyl-tRNA synthetase (hProRS) (Fig. 3A). As mammalian LysRS contains a 65-aa N-terminal extension that confers nonspecific RNA-binding (Francin and Mirande 2006), we also tested binding to hLysRSΔN65 and found that it also binds the PBS/TLE (Fig. 3B). Since the truncated protein has been shown to be more sensitive to specific RNA binding interactions (Francin and Mirande 2006), all in vitro studies described below were performed with hLysRSΔN65. Additionally, a large fraction of hLysRS in virions is truncated (Cen et al. 2001).
FIGURE 3.
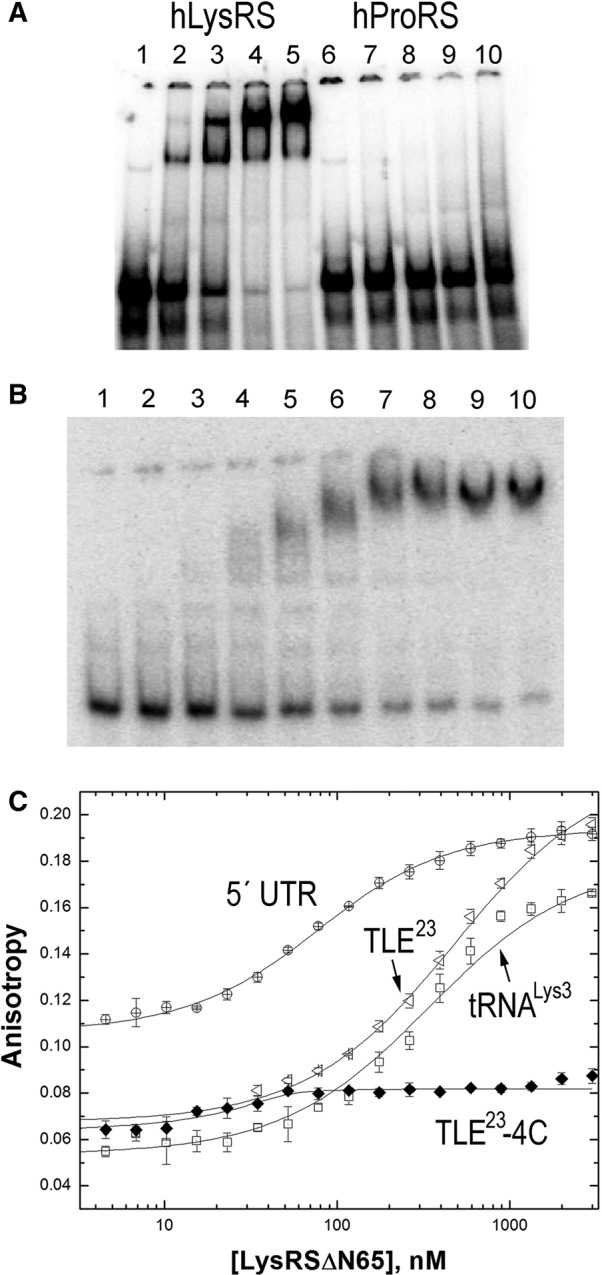
Binding studies investigating the specificity of hLysRS/TLE interaction. (A) Electrophoretic mobility shift assays performed with 25 nM 105-nt 32P-labeled HXB2 PBS/TLE RNA and 0, 0.50, 1.0, 2.0, or 3.0 μM hLysRS (lanes 1–5) or hProRS (lanes 6–10). (B) Electrophoretic mobility shift assays were performed with 30 nM PBS/TLE and 0, 0.12, 0.18, 0.26, 0.40, 0.59, 0.89, 1.3, 2.0, or 3.0 μM hLysRSΔN65 (lanes 1–10). (C) FA binding assay wherein varying amounts of hLysRSΔN65 were incubated with 20–50 nM fluorescently labeled TLE23, TLE23-4C, 330-nt 5′ UTR, or tRNALys3. The averages of three experiments with standard deviations are shown.
To establish whether hLysRSΔN65 could also bind to the isolated TLE stem–loop, binding to a 23-nt TLE RNA (TLE23) (Fig. 1B) was tested using fluorescence anisotropy (FA). TLE23 is derived from the HIV-1 genome (nt 146–164) with two additional C-G closing base pairs to stabilize the stem–loop. Initial studies performed at 15 mM NaCl, 37.5 mM KCl, and 4 mM MgCl2 were complicated by the dependence of the binding affinity (Kd) on fluorescent RNA concentration (Supplemental Fig. S2A), suggesting that the measured binding affinities (Supplemental Table S1) were underestimated under these conditions. Therefore, FA binding experiments were performed under more stringent conditions (i.e., 15 mM NaCl, 35 mM KCl, 10 mM MgCl2) so that the binding affinities are weak enough to measure accurately without the dependence of Kd on the fluorescent RNA concentration (Supplemental Fig. S2B). Under these conditions, hLysRSΔN65 bound the TLE23 (Fig. 3C) with an affinity (Kd = 436 nM), similar to that of in vitro transcribed tRNALys3 (Kd = 407 nM) or a minihelix derived from the anti-codon stem–loop of tRNALys3 (anti-codonLys3, Kd = 268 nM) (Table 1). Human TrpRS and ProRS did not show any significant binding to the TLE23 even under the less stringent low Mg2+ conditions (Supplemental Fig. S3). Mutation of the U-rich TLE loop sequence to AAAA (TLE23-4A) or CCCC (TLE23-4C) resulted in a large reduction in binding affinity to hLysRSΔN65 (Kd > 3 μM) (Fig. 3C; Table 1), suggesting that binding is mediated through the TLE23 U-loop residues. Under conditions of lower ionic strength (Supplemental Table S1), hLysRSΔN65 bound TLE23-4A and TLE23-4C ∼30- and ∼100-fold more weakly than the WT TLE23, respectively.
TABLE 1.
Apparent equilibrium dissociation constants (Kd) for hLysRSΔN65 binding to tRNALys3 and HIV-1 derived RNAs determined by FA measurements

Binding of hLysRSΔN65 to longer RNAs was also tested to assess the role of sequences surrounding the TLE stem–loop to LysRS interaction. As shown in Figure 3C, a 330-nt RNA consisting of most of the 5′ UTR of the HIV-1 vRNA (Fig. 1A) binds to hLysRSΔN65 with ∼6.5-fold greater affinity than the TLE23 and tRNALys3 (Kd = 65.2 nM) (Table 1). By comparison, mutation of the U-rich motif in the 5′ UTR by changing all four Us to Cs (5′ UTR-4C) reduced binding by approximately threefold (Kd = 172 nM) (Table 1), suggesting that binding to the 5′ UTR is not mediated only by the anti-codon-like U-rich sequence. The 5′ UTR construct consists of TAR, polyA, PBS, SL1, SL2, and SL3 stem–loops (Fig. 1A). To determine the contribution of other 5′ UTR elements to high-affinity hLysRSΔN65 binding, 5′ UTR deletion variants ΔTARpolyA, ΔTARpolyA ΔSL3, and ΔTARpolyA ΔSL2 + 3 were constructed. ΔTARpolyA bound hLysRSΔN65 with an affinity only slightly lower (Kd = 104 nM) than the 5′ UTR (Table 1), suggesting that the presence of the TAR and polyA stem–loops does not greatly enhance binding to hLysRSΔN65. The additional deletion of SL3 also showed little effect (ΔTARpolyA ΔSL3, Kd = 85.4 nM). In contrast, deletion of both SL2 and SL3 reduced binding approximately fourfold (Kd = 281 nM). Binding to PBS/TLE—which lacks the TAR and polyA stem–loops in addition to SL1, SL2, and SL3—is characterized by an even weaker affinity (Kd = 383 nM). However, the ΔTARpolyA ΔSL2,3 variant containing the U153-156C quadruple point mutation bound to hLysRSΔN65 with an affinity (Kd = 231 nM) similar to that of ΔTARpolyA ΔSL2,3 (281 nm) and 5′ UTR-4C (172 nM). The nonadditivity of these multiple changes on hLysRSΔN65 binding suggests that the interaction between hLysRSΔN65 and the TLE motif is context-dependent and that there may be direct or indirect coupling between these sites. Taken together, these data suggest that the TLE loop is part of a larger structure representing a hLysRS binding domain (LysRS-BD) in the viral RNA. Both SL1 and SL2 (Fig. 1A) in the viral LysRS-BD are required for the highest affinity binding, and the U-rich TLE motif generally contributes less to hLysRS binding to larger 5′ UTR-derived RNAs than to shorter RNAs such as TLE23.
The secondary structure of the NL4-3 vRNA differs from that of the MAL RNA (Goldschmidt et al. 2004). The latter contains sequences in its 5′ UTR most similar to HIV-1 clade A viruses (Gao et al. 1998). The region proximal to the PBS in the MAL isolate folds into an A-rich stem–loop (nt 160–170) (Fig. 1C) which adopts a tRNA-like U-turn motif (Puglisi and Puglisi 1998). To test binding of this region of the MAL isolate to hLysRSΔN65, we prepared an RNA derived from nt 123–218 of the MAL genome (Fig. 1C). MAL123-218 also binds to hLysRSΔN65 with a Kd of 313 nM, which is similar to that observed for the corresponding region of the NL4-3 genome (PBS/TLE, Kd = 383 nM). This finding shows that hLysRSΔN65 binds to tRNA anti-codon-like motifs in both HIV-1 isolate 5′ UTRs despite their differences in sequence and secondary structure and suggests that direct binding to these RNAs may depend more on their three-dimensional structures.
HIV-1 genome-derived RNAs compete for hLysRS/tRNALys3 binding
In the cell, hLysRS interaction with the HIV-1 genome would occur in the presence of tRNALys3. Thus, competition binding studies were carried out to determine the extent to which the viral TLE-containing sequences can displace bound tRNALys3-derived sequences from hLysRS. When hLysRSΔN65 is prebound to anti-codonLys3 (Fig. 1B, right) prior to TLE23 addition, the wild-type (WT) TLE23 (IC50 = 980 nM) (Table 2) can effectively compete for binding to anti-codonLys3, whereas TLE23-4A cannot (Fig. 4). The tRNALys3 transcript competes better than the TLE23 (IC50 = 257 nM). Interestingly, the 330-nt 5′ UTR RNA competes even more effectively (IC50 = 90 nM) (Table 2) than tRNALys3, likely due to additional favorable contacts between LysRS-BD and hLysRSΔN65. Competition for tRNALys3 binding was also tested (Supplemental Fig. S4; Table 3), and a similar result was obtained. When hLysRSΔN65 was prebound to tRNALys3 and competed with an ∼180-nt RNA derived from the Rous sarcoma virus genome (RSV RNA), competition was not observed, but a 105-nt PBS/TLE RNA (nt 127–221) containing the TLE competed modestly for tRNALys3, with an IC50 of 808 nM. As for the anti-codonLys3 competition assay, the 5′ UTR and tRNALys3 competed similarly for the tRNALys3/hLysRSΔN65 complex (Supplemental Fig. S4), with IC50 values of ∼200 nM each (Table 3).
TABLE 2.
Relative binding affinities of hLysRSΔN65 to HIV-1 genome-derived RNAs based on anti-codonLys3 competition binding assays

FIGURE 4.

FA competition binding experiments. hLysRSΔN65 (200 nM) was prebound to 40 nM fluorescently labeled anti-codonLys3, followed by titration with the following unlabeled competitor RNAs: 330-nt 5′ UTR, in vitro transcribed human tRNALys3, TLE23, and TLE23-4A.
TABLE 3.
Relative binding affinities of hLysRSΔN65 to HIV-1 genome-derived RNAs based on tRNALys3 competition binding assays
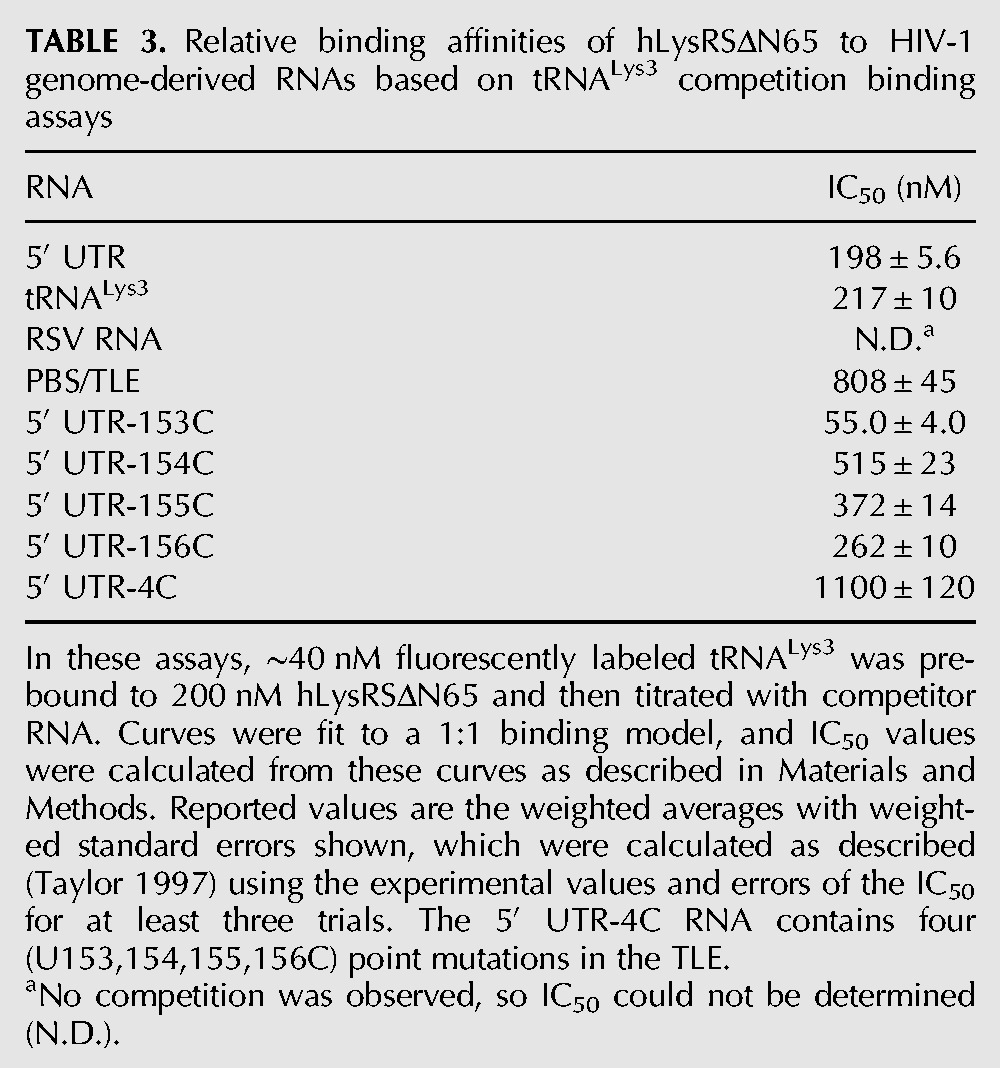
The importance of each U for hLysRSΔN65 binding was tested to determine if binding specificity to the TLE resembled the previously established binding specificity for tRNALys3. In these assays, hLysRSΔN65 was prebound to tRNALys3 and titrated with competitor RNAs. The results obtained paralleled the effects of the corresponding point mutations on aminoacylation of tRNALys3 by hLysRS (Stello et al. 1999). The binding affinity decreased in the order U153C > WT ∼ U156C > U155C > U154C > U153-156C (Table 3). U153 corresponds to the wobble base, which is C34 in tRNALys1,2. Since hLysRS also aminoacylates this tRNA isoacceptor, mutation to C is not expected to negatively affect hLysRSΔN65 binding. In fact, increased binding of the genomic RNA to hLysRSΔN65 is observed, as indicated by the approximately fourfold lower IC50 value obtained with this variant (55.0 nM) (Table 3). Increased IC50 values (lower affinity) are observed for the U154C (515 nM) and U155C (372 nM) variants, which correspond to critical tRNALys3 recognition elements U35 and U36, respectively. The U156C mutation did not alter binding relative to the WT sequence (IC50= 262 nM). Finally, mutation of all four U residues in the U-rich TLE sequence (nt 153–156) to CCCC increased the IC50 from 198 nM for WT to 1100 nM, suggesting that the U-rich motif enhances the competition of the 5′ UTR for tRNALys3/hLysRSΔN65 interaction. Compared to tRNALys3 aminoacylation data—153-fold and 11-fold decreases in kcat/KM for tRNALys3 U35C and U36C variants, respectively (Stello et al. 1999)—the U154C and U155C 5′ UTR are only modestly—approximately twofold—defective in competition binding. However, the reductions in kcat/KM for aminoacylation for these mutations were primarily due to decreased kcat (Stello et al. 1999). In fact, the KM values for tRNALys3 WT, U35C, and U36C were 3.4, 7.8, and 3.7 μM, respectively, which are in good agreement with the tRNALys3 competition binding data (Table 3; Stello et al. 1999).
Wild-type TLE is required for efficient tRNALys3 primer annealing
To address the function of the TLE/hLysRS interaction in HIV-1 replication, 293T cells were transfected with either WT BH10 virus or virus containing a CCC mutation in the U154-156 positions of the TLE. Viral production was similar for both WT and mutant virus (CAp24/ml culture medium), and precursor protein processing and amounts of reverse transcriptase (RTp66/p51) and hLysRS in cells were also unaffected (Supplemental Fig. S5). These findings indicate that the CCC mutation does not affect protein production or packaging of hLysRS into virions.
Recent data suggest that tRNALys3 annealing may be a two-step process, with the initial annealing step promoted by Gag prior to protein processing, followed by final primer/template remodeling by mature nucleocapsid protein (Guo et al. 2009). In protease-negative virions, only Gag-facilitated tRNALys3 annealing occurs, as no mature nucleocapsid protein is present. Therefore, vRNA was isolated from either WT or protease-negative (Pr−) viruses, either lacking or containing the CCC mutation. Hybridization with probes specific for either vRNA or tRNALys3 indicated that the tRNALys3/genomic RNA ratios were similar in all four types of viruses, showing that tRNALys3 packaging was unaffected, a key control (Fig. 5A,B, “tRNALys3/Genome RNA”). The effect of the mutations on reverse transcription initiation from the tRNALys3 primer was assayed using a previously published strategy (Guo et al. 2009). In this assay, total vRNA is used as the source of primer tRNA annealed to genomic RNA. An in vitro reverse transcription reaction is performed in the presence of ddATP, resulting in a 6-nt extension product. Images of these 6-nt extension products are shown in Figure 5A and B with values of the quantified bands indicated (“Relative tRNALys3 initiation”). Quantification of these results shows that the CCC mutation reduces initiation by 68% and 92% in WT (Fig. 5A) and Pr− viruses (Fig. 5B), respectively. This is consistent with a reduction in tRNALys3 annealing resulting from the CCC mutation. Moreover, as the defect in annealing is more severe in Pr− viruses, the results reinforce nucleocapsid protein's role in repositioning or remodeling the primer/template complex prior to initiation by reverse transcriptase.
FIGURE 5.

Effect of TLE loop mutations on the incorporation of tRNALys3 into HIV-1, the initiation of reverse transcription, and viral infectivity. WT and protease-negative (Pr−) virus were produced from transfected 293T cells. (A) Results of WT virus with or without the U154–156C mutation. The tRNALys3:vRNA ratio (top) was determined by hybridizing RNA with specific DNA probes. In the initiation assay (bottom), total vRNA was used as the source of primer/template. A denaturing polyacrylamide gel shows the results of 6-nt primer extension assays performed with WT virus (BH10[UUU]) or virus containing a triple mutation in the TLE (BH10[CCC]). (B) The results of tRNA packaging and primer extension assays similar to those described in A, performed with Pr− virus. (C) Single-round viral infectivity of TZM-bl cells transfected with WT or CCC mutant virus, normalized to BH10. TZM-bl cells contain an integrated copy of the luciferase gene behind the HIV-1 promoter, and infectivity was measured by using the luciferase activity in the lysates of cells 48 h post-transfection.
To assess the effect of the TLE mutation on viral infection, single-round infectivity of WT and CCC mutant viruses was measured using a luciferase reporter system, in which TMZ-bl cells are used that contain a luciferase gene under the control of an HIV-1 promoter (Wei et al. 2002). The CCC mutation causes a 40% reduction in infectivity (Fig. 5C), which is less than the expected reduction based on the primer extension assay. This may, in part, be due to the lack of a strict correlation between the number of cDNA transcripts produced and the number integrated into the host genome. The fact that the TLE is not absolutely essential for tRNALys3 placement and viral infectivity is, however, consistent with the significant contributions of sequence elements outside the U-rich loop to hLysRS binding.
Thus, the reduced ability of the CCC mutation to bind LysRS (Table 3) does not result in less viral production (Supplemental Fig. S5) but does result in reduced infectivity. This is not surprising since virus production from cells transfected with viral DNA does not depend upon tRNALys3-primed reverse transcription to produce new proviral DNA whose integration into the host cell’s genome will lead to the production of viral proteins. On the other hand, the viruses produced from such transfected cells might be expected to be less infectious (Fig. 5C), i.e., produce less viral DNA and proteins upon a new round of infection, if the CCC mutant results in less tRNALys3 annealed to the viral RNA.
DISCUSSION
The data presented here show that the HIV-1 vRNA contains a tRNA anti-codon-like element proximal to the PBS that is part of a larger binding domain for hLysRS. The TLE confers specific binding to hLysRS (Fig. 3A), but additional sequence elements, including SL1 and SL2, are involved in high-affinity binding (Table 1). In the isolated TLE stem–loop, the U-rich motif is essential for hLysRSΔN65 binding; however, in larger RNAs containing the TLE stem–loop, the contribution of the U-rich motif is less important. WT TLE-containing sequences effectively compete for hLysRSΔN65/tRNALys3 interaction in vitro, and mutation of the U-rich loop, which mimics the tRNALys3 anti-codon loop, reduces tRNALys3 annealing in virions. The presence of a LysRS-BD in the 5′ UTR may also promote binding of Gag/hLysRS/tRNALys3 complexes over those that do not contain hLysRS. Thus, these findings are consistent with a model in which HIV-1 uses molecular mimicry of tRNALys to increase the efficiency of tRNA primer annealing by providing a site of preferential hLysRS binding (Fig. 6). The simplified model in Figure 6 suggests that binding of hLysRS to the TLE may promote tRNALys3 annealing through release of tRNALys3 from hLysRS; however, the annealing of tRNALys3 to viral RNA takes place within a Gag/GagPol nucleoprotein complex that also promotes this process (Saadatmand and Kleiman 2012). Within this complex, hLysRS has been reported to have multiple interactions with sequences within both Gag (Javanbakht et al. 2003; Kovaleski et al. 2007) and Pol (Saadatmand et al. 2008), and we report here an additional interaction with the TLE in the viral RNA. While this interaction may promote tRNALys3 annealing to the nearby PBS sequence by facilitating the release of tRNALys3 from LysRS, it might also help promote the localization of the hLysRS/tRNALys3-containing nucleoprotein complex to the site of annealing within the large vRNA genome. Thus, mutation of the U-rich sequence within the TLE reduces annealing by either reducing recruitment of the hLysRS/tRNALys3 complex or by weakening the competition of the TLE for hLysRS/tRNALys3 and thereby preventing the subsequent release of tRNALys3 for annealing and priming.
FIGURE 6.
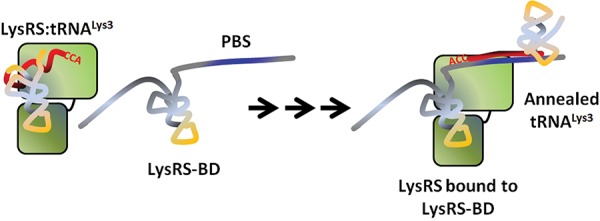
Hypothetical model for TLE-assisted tRNALys3 primer placement. The tRNALys3 primer shares 18 nt of complementarity (red) to the HIV-1 vRNA and must be annealed for reverse transcription to begin from the primer’s CCA-3′OH end. Human LysRS and tRNALys3 are packaged as a complex along with genomic RNA, which has a TLE (yellow) upstream of the primer binding site (PBS) (blue). Although the full extent to which the TLE-containing viral RNA mimics the structure of a tRNA is unknown, the TLE is part of a LysRS binding domain (LysRS-BD) that effectively competes for binding to hLysRS (green, two domains representing catalytic and anti-codon-binding domains). This competition may facilitate release of bound tRNALys3 from the synthetase, which can then be annealed to the vRNA. Arrows indicate intermediate steps in the annealing pathway, which requires viral proteins that facilitate tRNALys3 annealing such as Gag and GagPol (omitted for clarity).
The clade B virus TLE studied here mimics tRNALys3 more closely than the putative TLE sequence from other virus clades (Supplemental Fig. S1). Previously published chemical and enzymatic probing of the initiation complex of the MAL isolate has shown that an alternate secondary structure is preferred in which a U-rich loop is not exposed (Fig. 1C; Isel et al. 1995; Goldschmidt et al. 2004). Instead, in these isolates an A-rich stem–loop is present with a tRNA anti-codon-like U-turn structure (Puglisi and Puglisi 1998; Goldschmidt et al. 2004), and this loop is hypothesized to interact with tRNALys3 via its complementarity to the U-rich anti-codon sequence (Isel et al. 1995; Puglisi and Puglisi 1998). Extensive tRNA/genome interactions outside the PBS have been proposed based on structure probing studies of the MAL isolate (Goldschmidt et al. 2004). Despite the differences in predicted secondary structure between MAL and NL4-3 5′ UTRs, both contain an anti-codon-like stem–loop upstream of the PBS (Goldschmidt et al. 2004), and both bind to hLysRSΔN65. Additional studies are needed to establish whether the tRNA anti-codon-like stem–loops in different virus clades share a common function in promoting efficient tRNA primer annealing. Interestingly, in vitro selection for RNA aptamers that bind to hLysRSΔN65 has revealed that high-affinity hLysRS-binding RNAs fall primarily into two classes with consensus motifs containing either a U-rich or A-rich loop (A Curtright, W Wang, and K Musier-Forsyth, in prep.).
HIV-1 may take advantage of tRNA mimicry for an additional purpose. In Dicistroviridae viruses (Costantino et al. 2008; Zhu et al. 2011), tRNA mimicry is critical for internal ribosome entry site (IRES) function. Although many retroviruses contain an IRES (Balvay et al. 2009), the presence of an IRES in HIV-1 is somewhat controversial (Miele et al. 1996; Brasey et al. 2003) as the IRES appears to be active only under certain physiological conditions, such as G2/M arrest (Brasey et al. 2003) or stress (Gendron et al. 2011), and there is also evidence that ribosomal scanning occurs in part of the 5′ UTR (Berkhout et al. 2011). In recent studies (Brasey et al. 2003; Gendron et al. 2011), IRES activity mapped to a region in the 5′ UTR overlapping with the TLE described herein. Taken together, these findings suggest that the TLE might be part of an IRES that aids in ribosome recruitment via tRNA mimicry. Recently, glycyl-tRNA synthetase (GlyRS) was shown to bind to the poliovirus IRES to activate translation initiation (Andreev et al. 2012). Thus, a common strategy employed by viruses may be to recruit aminoacyl-tRNA synthetases, which are well-established to have moonlighting roles outside of protein translation (Nechushtan et al. 2009; Kim et al. 2011).
NMR studies using the hLysRS anti-codon binding domain support a direct interaction between the HIV-1 TLE loop and LysRS (S Liu, CP Jones, K Musier-Forsyth, and P Tsang, in prep.). Additional biophysical studies are needed to determine the extent to which the LysRS-BD in the 5′ UTR imitates other structural features of tRNALys3. Binding studies comparing the 5′ UTR 330-mer to deletion constructs indicated that the TARpolyA and SL3 domains are dispensable but that the SL1/SL2 region of the 5′ UTR contributes to high-affinity LysRS binding, possibly by stabilizing a tRNA-like structure or by directly interacting with LysRS. Mutating the anti-codon-like sequence of the TLE in the context of the 5′ UTR was not sufficient to abolish LysRS interaction in vitro or viral infectivity in cell-based assays, consistent with the involvement of other vRNA sequences. Testing a true “LysRS-BD-minus” mutant virus in cell-based assays may be difficult due to the critical role that these sequence elements play in other steps of the virus life cycle—SL1 contains the dimerization initiation site, SL2 contains the splice donor site, and SL2 and SL3 are part of the Psi packaging signal (Coffin et al. 1997; De Guzman et al. 1998; Amarasinghe et al. 2000).
Previous studies identified a nonanucleotide sequence located in the U3 region at the 3′ end of the HIV-1 vRNA complementary to nt 38–46 of tRNALys3 (Brule et al. 2000; Song et al. 2009). Recently, Bambara and coworkers discovered a much longer sequence element that resembles the tRNALys3 gene embedded in the 3′ end of the HIV-1 vRNA and closely related lentiviral genomes (Piekna-Przybylska et al. 2010). This sequence includes the previously identified nonanucleotide sequence, has extensive complementarity to tRNALys3, and even contains a vestigial intron inserted in approximately the position expected for a tRNA intron. In vitro studies showed that including this extended sequence in the acceptor template further enhances the efficiency of minus-strand transfer beyond that observed with the 9-nt segment (Piekna-Przybylska et al. 2010). Thus, functionally important sequence elements with extensive complementarity to tRNALys3 are present in the 5′ and 3′ UTR of HIV-1 genomic RNA (i.e., the 5′ PBS region and 3′ tRNALys3-like gene). The identification of the anti-codon-like TLE reported here provides a third example of a functionally relevant tRNALys-derived sequence that is present in the HIV-1 genome.
MATERIALS AND METHODS
Preparation of proteins and nucleic acids
Human LysRS (Shiba et al. 1997), LysRSΔN65 (Shiba et al. 1997), TrpRS (Wakasugi et al. 2002), and ProRS (Heacock et al. 1996) were prepared as previously described. Each protein contains a His6 tag and was purified using Ni2+-NTA resin (Qiagen), Co2+ Talon resin (Clontech), or His-select resin (Sigma). Protein concentrations were determined using the Bradford assay with BSA as a standard (Bio-Rad). Active site titrations were used for assaying TrpRS and ProRS enzyme activities (Fersht et al. 1975). Enzyme activity was additionally tested using aminoacylation assays with in vitro transcribed human tRNALys3 in the case of hLysRS and hLysRSΔN65 and human tRNAPro in the case of ProRS (Heacock et al. 1996; Shiba et al. 1997).
Short RNAs (TLE23, TLE23-4A, TLE23-4C, and anti-codonLys3) were purchased from Dharmacon. All other RNAs—330-nt HIV-1 5′ UTR variants, 206-nt ΔTARpolyA, 186-nt ΔTARpolyA ΔSL3, 169-nt ΔTARpolyA ΔSL2+3, 105-nt TARpolyA, 105-nt NL4-3 PBS/TLE, 105-nt HXB2 PBS/TLE, 98-nt Psi RNA, 76-nt human tRNALys3, 180-nt RSV RNA, and 98-nt MAL123–218 RNA—were prepared by in vitro transcription using T7 RNA polymerase (Milligan et al. 1987) and purified using denaturing (8 M urea) polyacrylamide gel electrophoresis. Extinction coefficients at 260 nm used for the UTR-derived RNAs are as follows: 5′ UTR WT and U-to-C mutants, 304 × 104 M−1 cm−1; NL4-3 and HXB2 PBS/TLE, 95.5 × 104 M−1 cm−1; ΔTARpolyA, 190 × 104 M−1 cm−1; ΔTARpolyA ΔSL3, 162 × 104 M−1 cm−1; ΔTARpolyA ΔSL2+3, 146 × 104 M−1 cm−1; ΔTARpolyA ΔSL2+3-4C, 146 × 104 M-1 cm−1; MAL123–218, 87 × 104 M−1 cm−1. The 76-nt tRNALys3 used in this study was prepared as described previously (Shiba et al. 1997). The gene encoding the 330-nt HIV-1 5′ UTR RNA was amplified from pMSMΔEnv (McBride and Panganiban 1996) using PCR and cloned into PstI and BamHI restriction sites of pET15b (Novagen) behind a T7 promoter. The resulting plasmid (pHIV330) was digested with FokI to generate the DNA template for in vitro transcription reactions. The sequence of the RNA resulting from in vitro transcription is identical to the first 329 nt of the HIV-1 vRNA with an additional G placed at the 5′ end to improve transcription efficiency. From this clone, genes encoding other genomic RNAs were made using standard PCR and Quikchange mutagenesis procedures (Stratagene). The plasmid pMAL123–218 encoding the MAL123–218 RNA was cloned from pJCB (Paillart et al. 1994), a gift of Dr. Roland Marquet (Université de Strasbourg). This RNA is derived from MAL nt 123–218 with two additional G residues placed at the 5′ end for efficient in vitro transcription. Prior to use, the 96- to 330-nt RNAs were folded by heating to 85°C for 2.5 min, 50°C for 8 min, adding MgCl2 to 10 mM, heating at 37°C for 10 min, and cooling on ice for at least 30 min. Shorter RNAs (tRNALys3, anti-codonLys3, and TLE23) were folded by heating for 2 min at 80°C, 2 min at 60°C, adding MgCl2 to 10 mM, and cooling on ice for at least 30 min.
Radiolabeled RNAs used in EMSAs were prepared by treating ∼150 pmol RNA with calf intestine phosphatase and 5′ end-labeling with γ-32P-ATP using T4 polynucleotide kinase (New England Biolabs). 32P-labeled RNAs were purified using G-25 spin columns (Roche), aliquoted, and stored at −20°C in diethylpyrocarbonate (DEPC)-treated H2O.
Fluorescently labeled RNAs were prepared using previously described methods (Pagano et al. 2007) with some modifications. In 100 mM sodium acetate, pH 5.1, 5 nmol RNA was treated with 10-fold excess sodium periodate in a 50-μL reaction volume for 1.5 h at room temperature in the dark. The reaction was quenched with 5 μL glycerol and ethanol precipitated. The pellet from the ethanol precipitation was resuspended in 50 μL 100 mM sodium acetate, pH 5.1, with 1 mM fluorescein-5-semithiocarbazide and incubated at 4°C in the dark for 18–24 h. The mixture was then eluted through a G-25 spin column (Roche) to remove free dye and checked for purity using 1% agarose or denaturing polyacrylamide gel electrophoresis (8% or 12% gels, depending on RNA length). Labeling efficiency was determined by measuring the UV absorbances at 260 nm and 495 nm at pH 8 and using the following extinction coefficients (and those above): fluorescein, 8.5 × 104 M−1 cm−1 (pH 8); anti-codonLys3, 32.8 × 104 M−1 cm−1; tRNALys3, 60.4 × 104 M−1 cm−1; TLE23, 22.1 × 104 M−1 cm−1; TLE23-4A, 23.0 × 104 M−1 cm−1; TLE23-4C, 21.0 × 104 M−1 cm−1. Labeling efficiencies were typically between 60% and 80%. RNAs were aliquoted after labeling and stored in DEPC-treated H2O in amber tubes at −20°C.
Electrophoretic mobility shift assays
Prior to use, RNAs were refolded as described above. 32P-labeled RNA substrate (25 nM) was mixed with varying amounts of protein (final concentrations in Fig. 3 legend) in 10 mM Tris-HCl, pH 7.4, 75 mM KCl, 10 mM MgCl2, and 5% glycerol. Binding reactions (10 μL) were incubated for 30 min at room temperature and mixed with 4 μL loading dye (50% glycerol, xylene cyanol, bromophenol blue) prior to electrophoresis on a 6% native polyacrylamide gel at 4°C. Gels were dried and exposed overnight, and band intensities were visualized and quantified using a Typhoon Trio phosphorimager (GE Healthcare).
Fluorescence anisotropy equilibrium binding measurements
Reaction mixtures contained 20 mM Tris-HCl, pH 7.4, 35 mM KCl, 10 mM MgCl2, 15 mM NaCl, 20–50 nM labeled RNA, and various protein concentrations. Reactions were incubated for 30 min in amber tubes at room temperature. Aliquots (20 μL) were read in 384-well plates using a SpectraMax M5 plate reader system (Molecular Devices) by exciting at 485 nm and measuring emission at 530 nm. FA binding curves were fit to a 1:1 binding model (Stewart-Maynard et al. 2008).
For competition assays, either 40 nM fluorescently labeled tRNALys3 or anti-codonLys3 was incubated with 200 nM hLysRSΔN65 in 10 mM Tris-HCl, pH 7.4, 37.5 mM KCl, 5 mM MgCl2, and 15 mM NaCl. Under these conditions, the difference between the anisotropy of the complex and the anisotropy of the free RNA is large enough that competition binding curves show significant and reproducible changes upon titration with competitor. Conditions in competition assays were identical to binding conditions. After incubation of hLysRSΔN65 with the fluorescently labeled RNA for 30 min at room temperature, competitor RNA was added, and the mixture was incubated 30 min at room temperature. To establish points for normalization, the anisotropy of the free RNA in the absence of hLysRSΔN65 (rfree) and the anisotropy of the complex without competitor (rbound) were also measured.
Competition binding curves were normalized by using the following equation: (r – rfree)/(rbound – rfree). This normalization sets rbound to “100% RNA bound” and rfree to “0% RNA bound.” For determination of IC50, competition binding curves were fit to the same 1:1 binding model used above, as previously described (Stewart-Maynard et al. 2008).
Cells, transfections, and virus purification
A description of the plasmids used is given in the Supplemental Material, Methods. HEK-293T cells were grown in complete Dulbecco’s modified Eagle’s medium plus 10% fetal bovine serum, 100 units of penicillin, and 100 μg of streptomycin/ml. For the production of viruses, HEK-293T cells were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Supernatant was collected 48 h post-transfection. Viruses were pelleted from culture medium by centrifugation in a Beckman SW41 rotor at 35,000 rpm for 1 h through a 20% sucrose cushion. The pellet of purified virus was resuspended in 1× TNE (20 mM Tris, pH 7.8, 100 mM NaCl, 1 mM EDTA).
Viral RNA isolation
Total vRNA was extracted from viral pellets using the guanidinium isothiocyanate procedure as previously described (Huang et al. 1994) and was dissolved in RNAse free, DEPC-treated dH2O.
Real-time PCR quantification of viral genomic RNA and tRNALys3
RT-PCR was performed upon vRNA and tRNALys3 using SuperScript One-Step RT/PCR with Platinum Taq (Invitrogen Life Technologies). Primers for quantifying genomic RNA encompassed nt 710–910: forward, 5′-GAGATGGGTGCGAGAGCGTCAGTA-3′; reverse, 5′-GCTCCCTGCTTGCCCATACTATATGT-3′. Primers for quantifying tRNALys3: forward, 5′-GTCGGTAGAGCATCAGACTT-3′; reverse, 5′-CGCCCGAACAGGGACTT-3′.
tRNALys3-primed initiation of reverse transcription
Total RNA isolated from virus produced in transfected 293T cells was used as the source of primer tRNA annealed in vivo to vRNA in an in vitro reverse transcription reaction, as previously described (Huang et al. 1996). Briefly, total vRNA was incubated at 37°C for 15 min in 20 μL of RT buffer (50 mM Tris-HCl [pH 7.5], 60 mM KCl, 3 mM MgCl2, and 10 mM dithiothreitol) containing 50 ng purified HIV RT, 10 units of RNasin, and various deoxynucleotide triphosphates (dNTPs). To measure the ability of annealed tRNALys3 to be extended by six deoxyribonucleotides, the RT reaction mixture contained 200 μM dCTP, 200 μM dTTP, 5 μCi of [α]-32P dGTP (0.16 μM), and 50 μM ddATP. Reaction products were resolved by polyacrylamide gel electrophoresis using a 6% polyacrylamide gel containing 7 M urea (Huang et al. 1996).
Virus infectivity assay
Single-cycle infectivity levels were determined by challenging 105 TZM-bl indicator cells (Wei et al. 2002) with equal amounts of viruses (corresponding to 20 ng of CAp24) that had been produced from HEK-293T cells transfected with wild-type or mutant HIV-1 DNA. The induced expression of luciferase activity in cell lysates was measured after 48 h. TZM-bl indicator cells were generated from the stably transfected CD4+ Hela cell line, JC53, by introducing separate integrated copies of the luciferase and β–galactosidase genes under control of the HIV-1 promoter. Infection was measured by the induction of luciferase activity by newly synthesized HIV-1 proteins and was measured using a Lumat LB9507 luminometer (EG&G Berthold).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Medha Raina for performing initial gel shift experiments and Dr. Roland Marquet (Université de Strasbourg) for the plasmid pJCB. We also thank Tiffiny Rye-McCurdy, Joseph Webb, and Maheen Masood for assistance with protein purification and Roopa Comandur for assistance with MAL RNA preparation. This work was supported by NIH grant RO1 AI077387 (to L.K. and K.M.-F.). C.P.J. was supported by NIH Training Grant T32 GM008512 and a Center for RNA Biology Fellowship from Ohio State University.
REFERENCES
- Amarasinghe GK, De Guzman RN, Turner RB, Summers MF 2000. NMR structure of stem-loop SL2 of the HIV-1 Ψ RNA packaging signal reveals a novel A-U-A base-triple platform. J Mol Biol 299: 145–156 [DOI] [PubMed] [Google Scholar]
- Andreev DE, Hirnet J, Terenin IM, Dmitriev SE, Niepmann M, Shatsky IN 2012. Glycyl-tRNA synthetase specifically binds to the poliovirus IRES to activate translation initiation. Nucleic Acids Res 40: 5602–5614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arts EJ, Stetor SR, Li X, Rausch JW, Howard KJ, Ehresmann B, North TW, Wohrl BM, Goody RS, Wainberg MA, et al. 1996. Initiation of (−) strand DNA synthesis from tRNA3Lys on lentiviral RNAs: Implications of specific HIV-1 RNA-tRNA3Lys interactions inhibiting primer utilization by retroviral reverse transcriptases. Proc Natl Acad Sci 93: 10063–10068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balvay L, Soto Rifo R, Ricci EP, Decimo D, Ohlmann T 2009. Structural and functional diversity of viral IRESes. Biochim Biophys Acta 1789: 542–557 [DOI] [PubMed] [Google Scholar]
- Beerens N, Groot F, Berkhout B 2001. Initiation of HIV-1 reverse transcription is regulated by a primer activation signal. J Biol Chem 276: 31247–31256 [DOI] [PubMed] [Google Scholar]
- Berkhout B, Arts K, Abbink TE 2011. Ribosomal scanning on the 5′-untranslated region of the human immunodeficiency virus RNA genome. Nucleic Acids Res 39: 5232–5244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolinger C, Boris-Lawrie K 2009. Mechanisms employed by retroviruses to exploit host factors for translational control of a complicated proteome. Retrovirology 6: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasey A, Lopez-Lastra M, Ohlmann T, Beerens N, Berkhout B, Darlix JL, Sonenberg N 2003. The leader of human immunodeficiency virus type 1 genomic RNA harbors an internal ribosome entry segment that is active during the G2/M phase of the cell cycle. J Virol 77: 3939–3949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brule F, Bec G, Keith G, Le Grice SF, Roques BP, Ehresmann B, Ehresmann C, Marquet R 2000. In vitro evidence for the interaction of tRNA3Lys with U3 during the first strand transfer of HIV-1 reverse transcription. Nucleic Acids Res 28: 634–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cen S, Khorchid A, Javanbakht H, Gabor J, Stello T, Shiba K, Musier-Forsyth K, Kleiman L 2001. Incorporation of lysyl-tRNA synthetase into human immunodeficiency virus type 1. J Virol 75: 5043–5048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cen S, Javanbakht H, Kim S, Shiba K, Craven R, Rein A, Ewalt K, Schimmel P, Musier-Forsyth K, Kleiman L 2002. Retrovirus-specific packaging of aminoacyl-tRNA synthetases with cognate primer tRNAs. J Virol 76: 13111–13115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffin JM, Hughes SH, Varmus H 1997. Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- Costantino DA, Pfingsten JS, Rambo RP, Kieft JS 2008. tRNA-mRNA mimicry drives translation initiation from a viral IRES. Nat Struct Mol Biol 15: 57–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damgaard CK, Dyhr-Mikkelsen H, Kjems J 1998. Mapping the RNA binding sites for human immunodeficiency virus type-1 gag and NC proteins within the complete HIV-1 and -2 untranslated leader regions. Nucleic Acids Res 26: 3667–3676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Guzman RN, Wu ZR, Stalling CC, Pappalardo L, Borer PN, Summers MF 1998. Structure of the HIV-1 nucleocapsid protein bound to the SL3 Ψ-RNA recognition element. Science 279: 384–388 [DOI] [PubMed] [Google Scholar]
- Dreher TW 2009. Role of tRNA-like structures in controlling plant virus replication. Virus Res 139: 217–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng YX, Campbell S, Harvin D, Ehresmann B, Ehresmann C, Rein A 1999. The human immunodeficiency virus type 1 Gag polyprotein has nucleic acid chaperone activity: Possible role in dimerization of genomic RNA and placement of tRNA on the primer binding site. J Virol 73: 4251–4256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fersht AR, Ashford JS, Bruton CJ, Jakes R, Koch GL, Hartley BS 1975. Active site titration and aminoacyl adenylate binding stoichiometry of aminoacyl-tRNA synthetases. Biochemistry 14: 1–4 [DOI] [PubMed] [Google Scholar]
- Francin M, Mirande M 2006. Identity elements for specific aminoacylation of a tRNA by mammalian lysyl-tRNA synthetase bearing a nonspecific tRNA-interacting factor. Biochemistry 45: 10153–10160 [DOI] [PubMed] [Google Scholar]
- Francin M, Kaminska M, Kerjan P, Mirande M 2002. The N-terminal domain of mammalian lysyl-tRNA synthetase is a functional tRNA-binding domain. J Biol Chem 277: 1762–1769 [DOI] [PubMed] [Google Scholar]
- Gabor J, Cen S, Javanbakht H, Niu M, Kleiman L 2002. Effect of altering the tRNALys3 concentration in human immunodeficiency virus type 1 upon its annealing to viral RNA, GagPol incorporation, and viral infectivity. J Virol 76: 9096–9102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F, Robertson DL, Carruthers CD, Li Y, Bailes E, Kostrikis LG, Salminen MO, Bibollet-Ruche F, Peeters M, Ho DD, et al. 1998. An isolate of human immunodeficiency virus type 1 originally classified as subtype I represents a complex mosaic comprising three different group M subtypes (A, G, and I). J Virol 72: 10234–10241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F, Kasprzak W, Stupina VA, Shapiro BA, Simon AE 2012. A ribosome-binding, 3′ translational enhancer has a T-shaped structure and engages in a long-distance RNA-RNA interaction. J Virol 86: 9828–9842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron K, Ferbeyre G, Heveker N, Brakier-Gingras L 2011. The activity of the HIV-1 IRES is stimulated by oxidative stress and controlled by a negative regulatory element. Nucleic Acids Res 39: 902–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschmidt V, Paillart JC, Rigourd M, Ehresmann B, Aubertin AM, Ehresmann C, Marquet R 2004. Structural variability of the initiation complex of HIV-1 reverse transcription. J Biol Chem 279: 35923–35931 [DOI] [PubMed] [Google Scholar]
- Guo F, Cen S, Niu M, Javanbakht H, Kleiman L 2003. Specific inhibition of the synthesis of human lysyl-tRNA synthetase results in decreases in tRNALys incorporation, tRNA3Lys annealing to viral RNA, and viral infectivity in human immunodeficiency virus type 1. J Virol 77: 9817–9822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Gabor J, Cen S, Hu K, Mouland AJ, Kleiman L 2005. Inhibition of cellular HIV-1 protease activity by lysyl-tRNA synthetase. J Biol Chem 280: 26018–26023 [DOI] [PubMed] [Google Scholar]
- Guo F, Saadatmand J, Niu M, Kleiman L 2009. Roles of Gag and NCp7 in facilitating tRNALys3 annealing to viral RNA in human immunodeficiency virus type 1. J Virol 83: 8099–8107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond JA, Rambo RP, Filbin ME, Kieft JS 2009. Comparison and functional implications of the 3D architectures of viral tRNA-like structures. RNA 15: 294–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heacock D, Forsyth C, Shiba K, Musier-Forsyth K 1996. Synthesis and aminoacyl-tRNA synthetase inhibitory activity of prolyl adenylate analogs. Bioorg Chem 24: 273–289 [Google Scholar]
- Huang Y, Mak J, Cao Q, Li Z, Wainberg MA, Kleiman L 1994. Incorporation of excess wild type and mutant tRNALys3 into HIV-1. J Virol 68: 7676–7683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Shalom A, Li Z, Wang J, Mak J, Wainberg MA, Kleiman L 1996. Effects of modifying the tRNALys3 anticodon on the initiation of Human Immunodeficiency Virus Type 1 reverse transcription. J Virol 70: 4700–4706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Wang J, Shalom A, Li Z, Khorchid A, Wainberg MA, Kleiman L 1997. Primer tRNA3Lys on the viral genome exists in unextended and two-base extended forms within mature human immunodeficiency virus type 1. J Virol 71: 726–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isel C, Marquet R, Keith G, Ehresmann C, Ehresmann B 1993. Modified nucleotides of tRNA3Lys modulate primer/template loop-loop interaction in the initiation complex of HIV-1 reverse transcription. J Biol Chem 268: 25269–25272 [PubMed] [Google Scholar]
- Isel C, Ehresmann C, Keith G, Ehresmann B, Marquet R 1995. Initiation of reverse transcription of HIV-1: Secondary structure of the HIV-1 RNA/tRNA3Lys (template/primer). J Mol Biol 247: 236–250 [DOI] [PubMed] [Google Scholar]
- Iwatani Y, Rosen AE, Guo J, Musier-Forsyth K, Levin JG 2003. Efficient initiation of HIV-1 reverse transcription in vitro. Requirement for RNA sequences downstream of the primer binding site abrogated by nucleocapsid protein-dependent primer-template interactions. J Biol Chem 278: 14185–14195 [DOI] [PubMed] [Google Scholar]
- Javanbakht H, Cen S, Musier-Forsyth K, Kleiman L 2002. Correlation between tRNALys3 aminoacylation and its incorporation into HIV-1. J Biol Chem 277: 17389–17396 [DOI] [PubMed] [Google Scholar]
- Javanbakht H, Halwani R, Cen S, Saadatmand J, Musier-Forsyth K, Gottlinger H, Kleiman L 2003. The interaction between HIV-1 Gag and human lysyl-tRNA synthetase during viral assembly. J Biol Chem 278: 27644–27651 [DOI] [PubMed] [Google Scholar]
- Jiang M, Mak J, Ladha A, Cohen E, Klein M, Rovinski B, Kleiman L 1993. Identification of tRNAs incorporated into wild-type and mutant human immunodeficiency virus type 1. J Virol 67: 3246–3253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CP, Datta SA, Rein A, Rouzina I, Musier-Forsyth K 2011. Matrix domain modulates HIV-1 Gag’s nucleic acid chaperone activity via inositol phosphate binding. J Virol 85: 1594–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khorchid A, Javanbakht H, Wise S, Halwani R, Parniak MA, Wainberg MA, Kleiman L 2000. Sequences within Pr160gag-pol affecting the selective packaging of primer tRNALys3 into HIV-1. J Mol Biol 299: 17–26 [DOI] [PubMed] [Google Scholar]
- Kim S, You S, Hwang D 2011. Aminoacyl-tRNA synthetases and tumorigenesis: More than housekeeping. Nat Rev Cancer 11: 708–718 [DOI] [PubMed] [Google Scholar]
- Kleiman L, Jones CP, Musier-Forsyth K 2010. Formation of the tRNALys packaging complex in HIV-1. FEBS Lett 584: 359–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobbi L, Octobre G, Dias J, Comisso M, Mirande M 2011. Association of mitochondrial lysyl-tRNA synthetase with HIV-1 GagPol involves catalytic domain of the synthetase and transframe and integrase domains of Pol. J Mol Biol 410: 875–886 [DOI] [PubMed] [Google Scholar]
- Kovaleski BJ, Kennedy R, Khorchid A, Kleiman L, Matsuo H, Musier-Forsyth K 2007. Critical role of helix 4 of HIV-1 capsid C-terminal domain in interactions with human lysyl-tRNA synthetase. J Biol Chem 282: 32274–32279 [DOI] [PubMed] [Google Scholar]
- Li M, Kao E, Gao X, Sandig H, Limmer K, Pavon-Eternod M, Jones TE, Landry S, Pan T, Weitzman MD, et al. 2012. Codon-usage-based inhibition of HIV protein synthesis by human schlafen 11. Nature 491: 125–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride MS, Panganiban AT 1996. The human immunodeficiency virus type 1 encapsidation site is a multipartite RNA element composed of functional hairpin structures. J Virol 70: 2963–2973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miele G, Mouland A, Harrison GP, Cohen E, Lever AM 1996. The human immunodeficiency virus type 1 5′ packaging signal structure affects translation but does not function as an internal ribosome entry site structure. J Virol 70: 944–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC 1987. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res 15: 8783–8798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechushtan H, Kim S, Kay G, Razin E 2009. Chapter 1: The physiological role of lysyl tRNA synthetase in the immune system. Adv Immunol 103: 1–27 [DOI] [PubMed] [Google Scholar]
- Pagano JM, Farley BM, McCoig LM, Ryder SP 2007. Molecular basis of RNA recognition by the embryonic polarity determinant MEX-5. J Biol Chem 282: 8883–8894 [DOI] [PubMed] [Google Scholar]
- Paillart JC, Marquet R, Skripkin E, Ehresmann B, Ehresmann C 1994. Mutational analysis of the bipartite dimer linkage structure of human immunodeficiency virus type 1 genomic RNA. J Biol Chem 269: 27486–27493 [PubMed] [Google Scholar]
- Pavon-Eternod M, Wei M, Pan T, Kleiman L 2010. Profiling non-lysyl tRNAs in HIV-1. RNA 16: 267–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piekna-Przybylska D, DiChiacchio L, Mathews DH, Bambara RA 2010. A sequence similar to tRNA3Lys gene is embedded in HIV-1 U3-R and promotes minus-strand transfer. Nat Struct Mol Biol 17: 83–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puglisi EV, Puglisi JD 1998. HIV-1 A-rich RNA loop mimics the tRNA anticodon structure. Nat Struct Biol 5: 1033–1036 [DOI] [PubMed] [Google Scholar]
- Roldan A, Warren OU, Russell RS, Liang C, Wainberg MA 2005. A HIV-1 minimal gag protein is superior to nucleocapsid at in vitro annealing and exhibits multimerization-induced inhibition of reverse transcription. J Biol Chem 280: 17488–17496 [DOI] [PubMed] [Google Scholar]
- Saadatmand J, Kleiman L 2012. Aspects of HIV-1 assembly that promote primer tRNALys3 annealing to viral RNA. Virus Res 169: 340–348 [DOI] [PubMed] [Google Scholar]
- Saadatmand J, Guo F, Cen S, Niu M, Kleiman L 2008. Interactions of reverse transcriptase sequences in Pol with Gag and LysRS in the HIV-1 tRNALys3 packaging/annealing complex. Virology 380: 109–117 [DOI] [PubMed] [Google Scholar]
- Shiba K, Stello T, Motegi H, Noda T, Musier-Forsyth K, Schimmel P 1997. Human lysyl-tRNA synthetase accepts nucleotide 73 variants and rescues Escherichia coli double-defective mutant. J Biol Chem 272: 22809–22816 [DOI] [PubMed] [Google Scholar]
- Song M, Balakrishnan M, Gorelick RJ, Bambara RA 2009. A succession of mechanisms stimulate efficient reconstituted HIV-1 minus strand strong stop DNA transfer. Biochemistry 48: 1810–1819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stello T, Hong M, Musier-Forsyth K 1999. Efficient aminoacylation of tRNALys,3 by human lysyl-tRNA synthetase is dependent on covalent continuity between the acceptor stem and the anticodon domain. Nucleic Acids Res 27: 4823–4829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart-Maynard KM, Cruceanu M, Wang F, Vo MN, Gorelick RJ, Williams MC, Rouzina I, Musier-Forsyth K 2008. Retroviral nucleocapsid proteins display nonequivalent levels of nucleic acid chaperone activity. J Virol 82: 10129–10142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JR 1997. An introduction to error analysis: The study of uncertainties in physical measurements. University Science Books, Sausalito, CA [Google Scholar]
- van Weringh A, Ragonnet-Cronin M, Pranckeviciene E, Pavon-Eternod M, Kleiman L, Xia X 2011. HIV-1 modulates the tRNA pool to improve translation efficiency. Mol Biol Evol 28: 1827–1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakasugi K, Slike BM, Hood J, Otani A, Ewalt KL, Friedlander M, Cheresh DA, Schimmel P 2002. A human aminoacyl-tRNA synthetase as a regulator of angiogenesis. Proc Natl Acad Sci 99: 173–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW Jr, Swanstrom R, Burch CL, Weeks KM 2009. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 460: 711–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother 46: 1896–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson KA, Gorelick RJ, Vasa SM, Guex N, Rein A, Mathews DH, Giddings MC, Weeks KM 2008. High-throughput SHAPE analysis reveals structures in HIV-1 genomic RNA strongly conserved across distinct biological states. PLoS Biol 6: e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Korostelev A, Costantino DA, Donohue JP, Noller HF, Kieft JS 2011. Crystal structures of complexes containing domains from two viral internal ribosome entry site (IRES) RNAs bound to the 70S ribosome. Proc Natl Acad Sci 108: 1839–1844 [DOI] [PMC free article] [PubMed] [Google Scholar]