

Using a novel approach to identify exonic circular RNAs (ecircRNAs), the authors show widespread production of circular RNAs from a significant fraction of expressed genes in murine and human cells. They demonstrate that several of these noncoding transcripts are stable and abundant and can be targeted by siRNA. Bioinformatic analysis of the entire ecircRNA complement revealed gene features associated with RNA circularization.
Keywords: exon shuffling, missplicing, noncoding RNA, trans-splicing
Abstract
Circular RNAs composed of exonic sequence have been described in a small number of genes. Thought to result from splicing errors, circular RNA species possess no known function. To delineate the universe of endogenous circular RNAs, we performed high-throughput sequencing (RNA-seq) of libraries prepared from ribosome-depleted RNA with or without digestion with the RNA exonuclease, RNase R. We identified >25,000 distinct RNA species in human fibroblasts that contained non-colinear exons (a “backsplice”) and were reproducibly enriched by exonuclease degradation of linear RNA. These RNAs were validated as circular RNA (ecircRNA), rather than linear RNA, and were more stable than associated linear mRNAs in vivo. In some cases, the abundance of circular molecules exceeded that of associated linear mRNA by >10-fold. By conservative estimate, we identified ecircRNAs from 14.4% of actively transcribed genes in human fibroblasts. Application of this method to murine testis RNA identified 69 ecircRNAs in precisely orthologous locations to human circular RNAs. Of note, paralogous kinases HIPK2 and HIPK3 produce abundant ecircRNA from their second exon in both humans and mice. Though HIPK3 circular RNAs contain an AUG translation start, it and other ecircRNAs were not bound to ribosomes. Circular RNAs could be degraded by siRNAs and, therefore, may act as competing endogenous RNAs. Bioinformatic analysis revealed shared features of circularized exons, including long bordering introns that contained complementary ALU repeats. These data show that ecircRNAs are abundant, stable, conserved and nonrandom products of RNA splicing that could be involved in control of gene expression.
INTRODUCTION
Noncoding RNAs (ncRNA) form the dominant product of eukaryotic transcription, comprising over 95% of total RNA in eukaryotic cells (Warner 1999). Though the bulk of these noncoding RNAs consist of the rRNA and tRNA apparatus required for translation, it has been increasingly appreciated that noncoding products comprise a diverse set of species relevant in core biological processes and disease (Esteller 2011). These products range from short microRNAs to long intergenic noncoding RNAs (lincRNAs). Circular RNAs comprised of exonic sequence represent an understudied form of ncRNA that was discovered more than 20 years ago from a handful of transcribed genes (Nigro et al. 1991; Capel et al. 1993; Cocquerelle et al. 1993; Zaphiropoulos 1997). These species have been typically identified as RNA molecules harboring exons out of order from genomic context, a phenomenon termed “exon shuffling” or “non-colinear splicing.” Such species have generally been considered to be of low abundance and likely representing errors in splicing. No known function has been ascribed to endogenous circular RNA transcripts.
Recently, our group discovered a circular RNA species, circular ANRIL or “cANRIL,” whose expression is associated with that of products of the human INK4a/ARF locus and is correlated with the risk of human atherosclerosis (Burd et al. 2010). Production of cANRIL in humans is associated with common single nucleotide polymorphisms (SNPs) predicted to affect cANRIL splicing, suggesting the possibility that cANRIL production influences Polycomb group (PcG)-mediated repression of the INK4a/ARF locus to influence atherosclerosis risk (Burd et al. 2010). This observation led us to perform an unbiased assessment of circular RNA species in mammalian cells. Toward that end, we developed a genome-wide RNA exonuclease enrichment strategy. RNase R degrades linear RNAs through its exonuclease activity while leaving circular RNAs unaffected (Suzuki et al. 2006). This method was first optimized to enrich a known, low-copy RNA circle (cANRIL) and then coupled with long-read, high-throughput sequencing to find potential circles in human fibroblast RNA. The resulting sequencing data were mapped to the genome using bioinformatic tools to identify out-of-order exon arrangements in an unbiased manner (MapSplice) (Wang et al. 2010). These exonic circular RNAs (ecirc RNAs) were analyzed by bioinformatic and molecular tools and demonstrated to be abundant, stable, conserved, nonrandom, and potentially functional as competing endogenous RNAs.
RESULTS
Unbiased identification of RNA circles
To identify exonic circular RNAs on a genomic scale, we developed an enrichment strategy for use in mammalian cells that we have termed “CircleSeq” (Fig. 1A). While a bioinformatic approach to RNA circle identification has recently been described (Salzman et al. 2012), we reasoned that a biochemical enrichment of nonlinear RNAs might allow for the detection of more rare and diverse circular RNA forms. Toward that end, total RNA was isolated from an immortalized human fibroblast cell line (Hs68), depleted of ribosomal RNA (RiboMinus) and then treated with RNase R, an RNA exonuclease that degrades linear RNAs with short 3′ tails regardless of secondary structure but does not degrade circular species (Suzuki et al. 2006). This method was optimized to allow for >10-fold enrichment of cANRIL in cDNA prepared from RNase R-treated vs. untreated samples. Upon confirmation of enrichment of this rare circular species, we next performed high-throughput sequencing of such samples on an Illumina HiSeq yielding ∼300 million 100-bp reads per sample, which were aligned to the human genome using a de novo splice mapping algorithm, MapSplice (Wang et al. 2010). This algorithm segments reads and uses mappings of these segments to find spliced mappings as well as fusions. The algorithm gives preference to continuous mappings, then spliced mappings, and finally fusion mappings that include non-colinear splicing, long range splicing, or interchromosomal splicing (Fig. 1B).
FIGURE 1.

CircleSeq experimental approach. Experimental schema for identification of circular RNAs in cultured human fibroblasts. (A) Experimental procedure, with aliquots of ribosome-depleted RNA split into a mock treatment and RNase R treatment and run through RNAseq. (B) Resulting reads mapped with MapSplice are segmented and mapped separately with resulting possible mappings in order of preference, including spliced and backspliced reads. (C) Diagram of normalized, aggregated sequencing data producing a normalized coverage value over individual nucleotides (reads per kilobase per million mapping [RPKM]; see Materials and Methods) as well as locations of backsplice reads that were enriched in RNase R-treated samples and a normalized count of those backspliced reads (blue horizontal bracket, spliced reads per billion mapping [SRPBM]; see Materials and Methods).
Once mapped, the coverage over all genomic coordinates was calculated and normalized to the total number of reads mapping to permit comparisons between runs and displayed in the format as shown in the example in Figure 1C. Coverage plots of untreated and RNase R-treated sequencing results were normalized to allow comparisons between conditions (expressed as reads per kilobase mapped [RPKM]; see Materials and Methods). The scales (noted at right in Fig. 1C) may differ in subsequent plots to permit visualization of key features. We compiled the list of all fusion splice junctions where splice donor and acceptor occur within 2 Mb but in the non-colinear ordering. We term these junctions “backsplices.” Counts of reads mapping across an identified backsplice in untreated samples, normalized by read length and number of reads mapping (spliced reads per billion mapping [SRPBM]; see Materials and Methods), are also shown to permit quantitative comparisons between backsplices. Treatment with RNase R was expected to result in decreased coverage of linear products, enrichment of reads from exons included in circular products, and in increased reads mapping as backsplice junctions in exonuclease-treated samples, all of which are diagrammed in Figure 1C. Normalized counts of reads mapping across backsplices were compiled for all conditions and replicates and compared to identify backsplices that were enriched by exonuclease exposure.
Enrichment of circular RNAs by CircleSeq
Analysis of biological replicates revealed reproducible backsplice coverage and coverage of individual exons in the RNase R-treated and RNase R-untreated samples, respectively (Fig. 2A,B). Comparisons between treatment conditions, in contrast, showed marked enrichment of backsplices, indicating that the vast majority of these species were enriched by RNase R (Fig. 2C). Accordingly, coverage of most annotated exons showed depletion with RNase R, as would be expected of exons contained within linear RNA species (Fig. 2D). These data show that CircleSeq reproducibly identifies backsplice-containing transcripts. Such backsplices are significantly enriched by RNase R digestion, contrasting with linear transcripts, which are depleted by RNase R.
FIGURE 2.

CircleSeq enriches for backsplice junctions. Two-dimensional histograms showing normalized backspliced read count (SRPBM) or normalized exon coverage (RPKM) between two samples or replicates. (A) Coverage of backsplice reads in RNase R-treated replicates over all distinct backsplice species (R2 = 0.579). (B) Coverage of exons in mock treated replicates (R2 = 0.91). (C) Average backsplice coverage in RNase R-treated against mock treated RNA-seq showing enrichment of most backsplice species by RNase R. (D) Mean normalized exon coverage in annotated exon sequences in RNase R-treated against mock treated RNA-seq showing depletion of the majority of species by RNase R.
Using this method, we identified >100,000 unique backsplice events throughout the genome. Of these, 25,166 were present in both RNase R-treated biological replicates and were also enriched by RNase R treatment as compared with mock treatment. While backsplice species generally were significantly enriched by RNase R treatment, we noted that 31% of backsplice species observed in untreated controls were not enriched by RNase R (Fig. 2C). These species likely represent mapping artifacts or nonsequential exons harbored in linear products, resulting from either RNA trans-splicing or cleavage of ecircRNAs.
Circular RNAs contain predominantly exonic sequence
Backsplicing of known ecircRNAs such as cANRIL (Burd et al. 2010) and cETS-1 (Cocquerelle et al. 1993) was readily apparent (Fig. 3A–D). The aggregated sequencing data are shown in the format of the schematic in Figure 1C (Fig. 3A,C) along with the circular structures inferred from the data (Fig. 3B,D). Circles previously discovered in ETS-1 and ANRIL were observed, in addition to several new species of circular ANRIL products. In the case of multiexon circles, the intervening exons not directly part of the backsplice also showed enrichment by RNase R (e.g., exons 6–13 in ANRIL) as would be expected. Intervening intronic sequence, however, was almost never enriched by RNase R digestion, suggesting introns are spliced from most circular forms. We were not able to detect other known circular species (e.g., SRY and DCC), due to low expression of these genes in Hs68 cells. We also observed many single exon ecircRNAs, that is, where the donor site splices to the acceptor of the same exon (e.g., KIAA0182) (Fig. 3E,F). This method appears very sensitive; the 25,166 replicated backsplice events detected by CircleSeq included the large majority (1025 of 1319) of putative circles identified through a previously described bioinformatic approach (Salzman et al. 2012). This high degree of concordance is even more noteworthy given that the two methods examined ncRNA expression in different cell types (T cells vs. fibroblasts). These data demonstrate that backsplice-containing transcripts identified by this method are diverse, generally RNase R-resistant, and include most previously described circular RNAs.
FIGURE 3.

CircleSeq identifies previously identified species of circular RNA. Mapped read depth for RNase R-untreated (green) and -treated (orange) samples is shown at differing scales, along with bars identifying the end points of backsplice reads and their number in RNase R-untreated samples (blue). (A) Circle-Seq identification of cANRIL species in the 9p21.3 locus. (B) The imputed cANRIL circular products. (C) Circle-Seq identification of ETS-1 circular RNAs and (D) imputed circular RNA species. (E) Sequencing identifies a highly expressed circular RNA in KIAA0182, producing a single exon circular RNA (F).
Additionally, we observed backsplice events that were not enriched by RNase R; for example, the recently reported CDR1 antisense transcript (Hansen et al. 2011). Backsplices for this gene were abundant in the control samples (mean SRPBM of 198), but both the backsplice reads and nonsplicing reads within the gene were depleted by exonuclease digestion (mean SRPBM of 16). These observations are most consistent with linear trans-splicing products rather than circular RNAs or with the cleavage of this circular RNA, as has been reported (Hansen et al. 2011). Backsplices that were not enriched by RNase R digestion were considerably less common than ecircRNAs. To focus our analysis on ecircRNAs, we sought to identify exonic, circular species while excluding lariats (see below) and linear trans-splices or cleaved backspliced RNAs.
Of the 25,166 unique backsplicing events that were reproducibly enriched by RNase R digestion and appeared in both biological replicates, most of these backsplices were only found in RNase R-treated samples and were not observed in the absence of exonuclease digestion. We suspect that many of these events represent rare ecircRNAs arising from pervasive background levels of RNA circularization, which may result from an occasional error in splicing (Hsu and Hertel 2009). Others transcripts, in contrast, demonstrated coverage of backsplices and their associated circularized exons >10-fold higher than that of the canonical linear transcript from the same gene, even in control sequencing runs (KIAA0182) (Fig. 3E,F). To focus on more abundant ecircRNAs with greater potential biological relevance, we chose three stringency cutoffs (LOW, MEDIUM, HIGH), selecting backsplicing events based upon their abundance and reproducible presence in both untreated and treated samples (Table 1; Supplemental Table S1). In all cases, we required backsplices to be observed and enriched in both RNase R-treated biological replicates. We placed varying minimum coverage requirements of backsplices in the control samples, with the LOW stringency set requiring a single backsplice read in the control data and HIGH stringency requiring coverage on a par with splices in a moderately expressed gene. Bioinformatic analyses gave similar results with regard to various features (see below) regardless of which transcript list was used.
TABLE 1.
Backsplice enrichment detects thousands of circular RNA species

For each of these circular RNAs, we estimated the relative percentage of backspliced products to forward spliced products at steady state by comparing the abundance of backsplice-spanning reads to reads spanning traditionally spliced junctions. The relative rate of backsplicing over forward splicing for these sites varied enormously, from <0.1% to >3200%, with no forward splicing products observed in some cases. The relative backsplicing rates for each circular RNA are shown in Supplemental Table S1. This analysis suggests that the formation of circular RNAs is considerable; e.g., using the LOW stringency list, 14.4% of genes expressed in human fibroblasts produced circular RNA species, suggesting at least one in eight human genes produces abundant circular as well as linear transcripts.
Circular RNA validation
To verify that the RNase R-enriched backsplicing events were indicative of true circular, and not linear, trans-splicing products, we examined the physical properties of these products. Outward facing primers were designed against seven representative transcripts from the LOW stringency list (Fig. 4A). Each primer pair amplified a single, distinct product of the expected size and sequence from Hs68 cDNA, and a quantitative TaqMan-based RT-PCR assay was developed for each primer pair. Enrichment of all seven novel backsplice events as well as cANRIL was apparent following RNase R treatment, whereas the abundance of linear RNAs (i.e., TBP, GAPDH, 18S) decreased (Fig. 4B). Moreover, oligo-dT priming for reverse transcription significantly reduced the levels of backspliced products relative to poly(A)-containing transcripts (e.g., TBP and GAPDH), indicating that these species were not polyadenylated (Fig. 4C). To further exclude the possibility that these transcripts were the results of trans-spliced products, we employed a “virtual Northern” approach (Hurowitz and Brown 2003), which identifies the size of transcripts by fractionating on a denaturing agarose gel, followed by qPCR. Backsplice-containing transcripts appeared in faster migrating fractions as compared to the associated full-length linear transcripts (Fig. 4D), as would be predicted of circular RNA species composed of only a subset of exons. Trans-spliced products, in contrast, would be expected to be longer than full-length, as they contain repeated exons. Together, these data suggest that the vast majority of novel RNA transcripts comprising the LOW stringency list (1) contain backsplices, (2) are enriched by RNase R treatment, (3) are not polyadenylated, and (4) are of smaller size than linear mRNAs emanating from the same locus. These characteristics are consistent with circular RNAs, demonstrating that CircleSeq identifies ecircRNAs resulting from cis- rather than trans-splicing.
FIGURE 4.

Validation of circular RNA species and “virtual Northern.” (A) Design of Taqman assays using outward facing primers. (B) Validation of RNase R enrichment in seven novel backsplice junctions and one control circular RNA (ANRIL 14-5). Noncircular RNAs (TBP, GAPDH, and 18S) are depleted by RNase R treatment. (C) Ratio of expression of these same ecirc and control RNAs with cDNA synthesis using oligo dT primers vs. random hexamer. Note markedly decreased cDNA synthesis with oligo dT for ecircRNAs and 18S. (D) “Virtual northern” analysis of four backsplice species and their linear counterparts employing agarose gel size fractionation followed by qPCR for quantification of products in each fraction. The x-axis shows size fraction in order of decreasing size, and the y-axis indicates the calculated fraction of total species contained at that size range.
Lariat detection and branch point mapping
CircleSeq was also able to detect lariat RNA species. Lariats are created by RNA splicing and differ from RNA circles in two ways: they contain significant intronic sequence, and they involve a 2′–5′ phosphodiester linkage at a branch point. Stable lariats have been previously noted in sequencing performed on nonpolyadenylated RNA (Yang et al. 2011). In our study, lariats were apparent in sequencing as regions of RNase R enrichment within some introns. This was expected, as RNase R has been described to leave lariats intact, while degrading only the 3′ “tail” before the branch point (Suzuki et al. 2006). Lariats however, could be distinguished from ecircRNAs in that lariats exhibited RNase enrichment in the absence of observed backsplice reads.
Despite the lack of backsplice reads, lariat 2′–5′ linkages were evidenced in our data by a different kind of read, which we term branch point reads. Using the abundant, stable lariat observed in GAPDH, we analyzed reads from RNase R-treated samples for 30 nt of sequence at the 5′ end of the intron, including the splice donor. We identified 24 such reads in one replicate alone, which were mapped to the human genome using the BLAT tool in the UCSC Genome Browser. Of these 24 reads, nine included sequence consistent with reverse transcription across a branch point (Fig. 5A). These sequences included an untemplated T base previously described to occur with reverse transcription by Superscript II across branch points (Gao et al. 2008). One read of the nine did not include this T, and instead included an “AG” sequence at this location. We also observed that the base coverage of lariats markedly decreased close to the sites of 2′–5′ linkages (Fig. 5B), as would be predicted by the known inefficient traversal 2′–5′ junctions by reverse transcriptase (Lorsch et al. 1995). Lariat sequences also demonstrated an ablation of coverage 3′ from the branch point of the intron in the RNase-treated samples. This observation is the predicted result, as this section of the intron is not circularized and, therefore, is susceptible to exonuclease attack (Fig. 5B,C). These data show that the CircleSeq approach can also identify lariat branch points as evidenced by 3′ tail degradation and branch point spanning reads.
FIGURE 5.

Lariat species also identified by CircleSeq. (A) Nine read sequences including at least 30 nt from the splice donor of intron 2 of GAPDH also include sequence 5′ of the proposed branch point. (B) Intron 2 of GAPDH demonstrates enriched coverage after RNase R treatment, but no backsplice reads are detected. Plots of mapped read depth in untreated (green) and RNase R-treated (orange) samples shown at differing scales. (C) Expected effects of RNase R treatment on lariat structures.
Conservation of abundant circularized transcripts
Conservation of circular RNA production in paralogous or orthologous genes would be an indication of evolutionary preservation of circular RNA formation and a potentially important function. We observed from our results in Hs68 cells that two paralogous kinases, HIPK2 and HIPK3, both produced abundant circular RNAs. These genes were sufficiently diverged to allow unique mapping but retain a similar genomic structure, notably a large second exon that contains the start codon flanked by large introns on either side (Fig. 6A,B). Both genes expressed high levels of backspliced products, particularly HIPK3, which demonstrated a large spike of coverage in exon 2 (Fig. 6B). Of note, increased coverage of exon 2 was not seen in cDNA libraries derived from poly(A) synthesis, consistent with the predominant exon 2 species being circular (Encode Data, not shown). Based on RPKM and qRT-PCR, the circular exon 2 transcript of HIPK3 was approximately fivefold more abundant than the linear form (Fig. 6C,D). The murine orthologs of HIPK2/3 demonstrated a similar genomic structure with regard to the large second exon surrounded by large introns (Fig. 6E), and backsplice-containing transcripts from these genes could be readily detected using exon 2 outward-facing primers in cDNA prepared from murine testis. Consistent with transcripts being ecircRNAs originating from the murine Hipk2/3 genes, the amplified fragments were of the expected size and sequence and were enriched by RNase R digestion. Therefore, a predilection for RNA circularization is conserved among paralogs and orthologs in the HIPK family.
FIGURE 6.

Circularization is conserved between paralogs and in mice. CircleSeq identified high exonuclease enrichment and backsplice prevalence in paralogous kinases (A) HIPK2 and (B) HIPK3. (C) Coverage of each gene by exon in the absence of RNase R treatment, showing a marked excess of the circularized exon of HIPK3. (D) Absolute quantification by quantitative real-time PCR of circular and linear HIPK3 species. (E) Genomic structures of murine Hipk2 and Hipk3, showing conservation of long introns around a relatively long circularized second exon (arrows).
Based on this observation, we assessed circle conservation in mammals on a greater scale by applying CircleSeq to RNA isolated from murine testis. Coverage was lower for these murine tissue-derived samples (∼300 million reads total) compared to that obtained in analyses of RNA from cultured human fibroblasts (∼1 billion reads). Sequences were mapped using MapSplice and ecircRNAs were identified using low stringency criteria. As was the case for human cells, a high number (646 of 1477) of circles found through a prior bioinformatic analysis (Salzman et al. 2012) of murine brain were identified by CircleSeq of murine testis. Given the lower coverage, tissue specific effects (e.g., testis vs. fibroblast), and other technical differences between the human and murine data sets, we limited further analysis solely to a consideration of the homology of murine ecircRNAs compared with human circular species. Of 2121 human circles from the MEDIUM stringency list that could be readily mapped to the murine genome, 457 mapped to genes that produced a murine circular RNA. At 22% of potential targets, this is a significant enrichment over the expectation of 14% (P < 10−10). In particular, we identified 69 murine circular species (including Hipk3) with exactly homologous start and stop points of RNA circularization (Supplemental Table S3). These events, occurring at a rate of 15% of genes producing circular RNAs in both organisms, involved the orthologous splice donor and splice acceptor sites of identical exons to generate a conserved ecircRNA. As these genes on average involved more than 15 exons, we would instead expect a rate of <1% overlap if these events were randomly distributed through the gene. This analysis indicates a high degree of conservation of specific backsplicing events between humans and rodents.
Representative ecircRNAs are untranslated but can be targeted by siRNA
Given their high expression, conservation, and the presence of an open reading frame, we sought to explore whether some circular RNAs might be translated in vivo. Engineered circular RNAs have previously been shown to have coding potential (Chen and Sarnow 1995). To explore translation of endogenous transcripts, we isolated unbound, monosome-bound, and polysome-bound fractions by sucrose gradient centrifugation and assayed the relative quantity of both linear and circular products by RT-PCR (Fig. 7A–C). To increase the chances of detecting translated circular species, we assayed ecircRNAs that contain a translation start site. Linear products were significantly enriched in the ribosome-bound fraction for the genes assayed: HIPK3, KIAA0182, and MYO9B (Fig. 7B). Circular products, in contrast, were abundant in the unbound fractions but not detected in the bound fractions, indicating that these AUG-containing ecircRNAs are not translated (Fig. 7C).
FIGURE 7.

Circular RNAs are not associated with ribosomes and are susceptible to siRNA knockdown. Jurkat cell lysates were separated by sucrose gradient centrifugation. (A) Agarose gel to verify separation of 40S, 60S, 80S, monosome, and polysome fractions. Linear and circular RNAs were quantified by qRT-PCR and plotted by relevant quantity in each fraction. (B) Linear forms assayed were associated with monosome and polysome fractions. (C) Circular forms were predominantly unassociated with these complexes. Knockdown using three targeted siRNAs against HIPK3 or ZFY were quantified by qRT-PCR and plotted to show the effect of differentially targeted siRNA against the linear, circular, and both (lin/circ) forms of (D) HIPK3 and (E) ZFY. Quantities shown are given as ΔΔCt and are normalized to TBP and then to a nonspecific (NS) siRNA.
Although we did not find evidence of ecircRNA translation, we considered whether cytoplasmic pools of circular RNAs might regulate transcription through an effect on microRNA binding. To address this possibility, we transfected Hs68 cells with siRNA targeting two genes (HIPK3 and ZFY) that we found to produce both linear and circularized transcripts. For each gene, we designed three siRNAs: one siRNA targeting sequence only in the linear transcript, another targeting the backsplice sequence, and a third targeting sequence in a circularized exon shared by both linear and circular species. A nonspecific control siRNA sequence was also employed. As expected, siRNA directed against the linear species induced knockdown of the linear transcript only, without affecting expression of the circular species (Fig. 7D,E). Knockdown using siRNA targeted to exonic sequence shared in both the linear and circular species induced effective knockdown of both transcripts. It was even possible to design an siRNA to the backsplice junction of ZFY that specifically targeted the circular, but not linear, transcript. These results demonstrate that ecircRNAs can be targeted by RNA interference.
ecircRNAs are predominantly cytoplasmic and highly stable
We next investigated ecircRNA stability and localization in vivo. Toward that end, Hs68 cells were treated with actinomycin D, an inhibitor of transcription, and total RNA was harvested at indicated time points. While highly stable RNAs such as 18S and p16INK4a exhibited very long transcript half-lives (>48 h) as expected, the abundance of less stable transcripts c-Myc and TATA Binding Protein (TBP) decreased following actinomycin D treatment with short half-lives (<3 h) (Fig. 8A). Analysis of four ecircRNAs and their associated linear mRNAs revealed that, while the associated linear transcripts exhibited half-lives of <20 h (Fig. 8B), the circular RNA isoforms were highly stable, with transcript half-lives exceeding 48 h (Fig. 8C). Fluorescence in situ hybridization (FISH) against HIPK3 demonstrated that circular forms of HIPK3 were preferentially localized in the cytoplasm (Fig. 8D). These results are consistent with prior studies of other RNA circles (Nigro et al. 1991; Salzman et al. 2012) and suggest that ecircRNAs either undergo nuclear export or are released to the cytoplasm during mitosis, where they enjoy extraordinary stability, likely as a result of resistance to debranching enzymes and RNA exonucleases.
FIGURE 8.

Novel ecircRNAs are highly stable and cytoplasmic. RNA stability assay using actinomycin D and qRT-PCR quantification demonstrates (A) expected stability of control transcripts and (B) less stable linear gene products compared to (C) highly stable circular RNAs from the same genes. (D) RNA fluorescence in situ hybridization using a probe specific to circular HIPK3 demonstrates cytoplasmic localization (top panel). Knockdown with an siRNA (from Fig. 7D) to the linear form does not affect localization (middle panel), whereas treatment with an siRNA targeting both the linear and circular forms extinguishes detection of cytoplasmic species (bottom panel).
Bioinformatic analyses of ecircRNAs
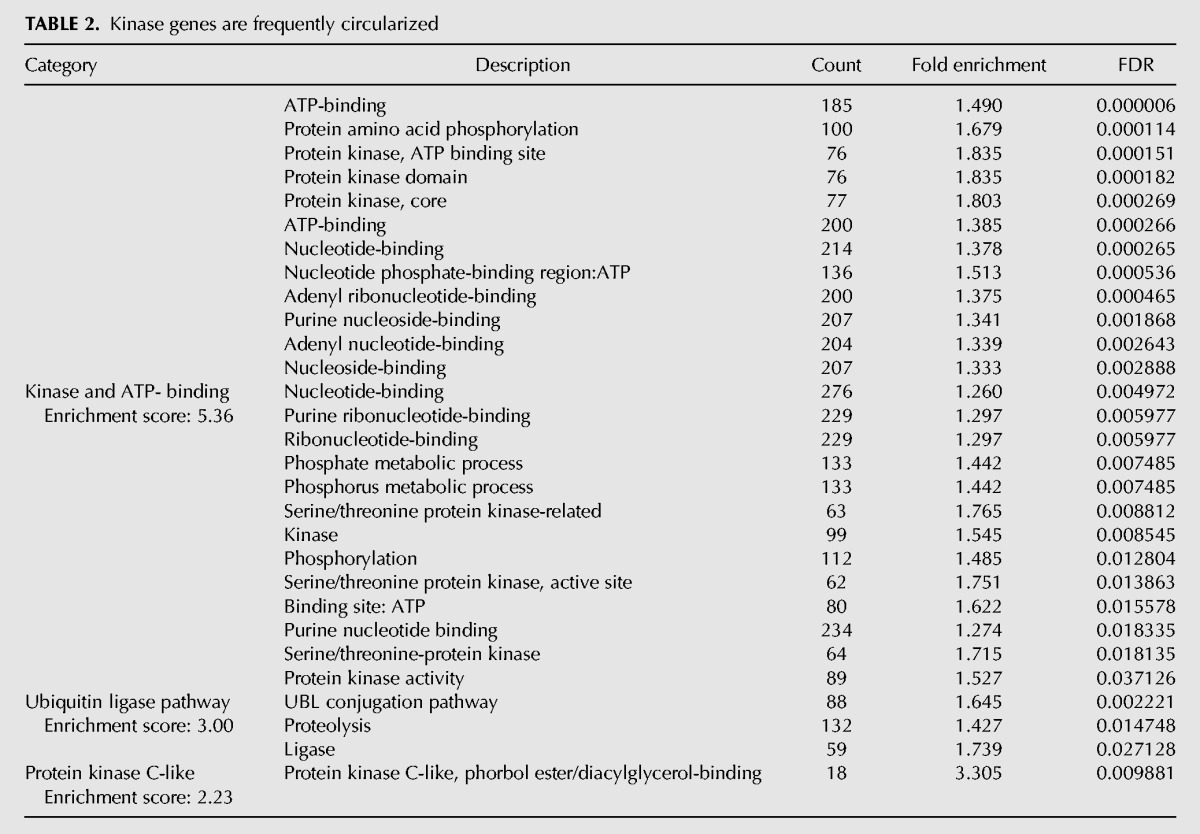
Having explored properties of ecircRNAs in vivo, we attempted to bioinformatically identify shared genomic features that might further elucidate function or mechanism of formation. DAVID analysis (Huang da et al. 2009) revealed an enrichment of protein kinases and related proteins among the set of genes producing ecircRNAs (Table 2), as compared to the set of expressed genes in the sample (see Materials and Methods). Although protein kinase transcripts as a whole were more likely to exhibit abundant circularization, no specific subfamily of kinase was particularly associated with ecircRNA production. Therefore, RNA circles are produced throughout the genome but are not randomly distributed among genes and may be disproportionately associated with kinase expression.
TABLE 2.
Kinase genes are frequently circularized
To explore the mechanisms of formation, we sought to identify cis-sequence elements proximal to backsplice events. Sequences in the 200 bp preceding (upstream of) or following (downstream from) backsplice sites were analyzed for enriched motifs compared to similar windows flanking noncircularized, expressed exons. This analysis revealed several enriched motifs (Supplemental Table S4), but the highest information-bearing motif was shared by both the upstream and downstream introns and was identified as the canonical ALU repeat (Fig. 9A). Regardless of whether the LOW, MEDIUM, or HIGH stringency transcript list was analyzed, the intronic flanks adjacent to circularized exons were approximately twofold more likely to contain an ALU repeat than noncircularized exons (P < 10−11) (Fig. 9B,C). This preference to contain flanking ALU repeats was noted for both single exon as well as multiple exon ecircRNAs (data not shown). Furthermore, when ALU polarity was considered, we found that pairs of ALU elements taken from introns flanking circularized exons were significantly more likely to be complementary (in inverted orientation) than noncomplementary. This held for all choices of transcript list or size of the flanking intronic window (Fig. 9D). For example, using a 500-bp window, 20% of introns flanking ecircRNAs contained complementary ALU pairs vs. 8% with noncomplementary pairs (P < 10−10). Circularized exons were sixfold more likely to contain complementary ALUs than control, noncircularized exons. While a role for inverted repeats on RNA circularization had been suggested in vitro (Dubin et al. 1995), these data confirm and extend this relationship at the genome-wide scale.
FIGURE 9.

Backsplices are flanked by paired ALU elements and long introns. (A) The highest information-bearing motif discovered within 200 bp upstream of and downstream from backsplice locations shows high homology to ALU elements. Frequency of RepeatMasker-annotated ALU elements in flanking sequences 50, 100, 200, and 500 bp (B) upstream of or (C) downstream from these expression categories of backsplice events, as compared to control splice sites of expressed genes. (D) The frequency of complementing and noncomplementing ALU pairs located on opposite sides of a backsplice within a flank. (E) Annotated length of introns flanking backsplices as compared to introns flanking control exons generally. (*) P < 10−5, (**) P < 10−10, (***) P < 10−20.
In addition to the presence of ALU repeats in flanking introns, we also noted distinct characteristics of exon and intron length related to ecircRNA production. Consistent with findings at HIPK2/3 (Fig. 6A,B,E) and KIAA0182 (Fig. 3E), the upstream and downstream introns flanking circularized exons tended to be large—on average more than approximately threefold longer than introns flanking control exons (P < 10−15) (Fig. 9E), regardless of choice of backsplice list for analysis. Consistent with prior in vitro studies (Pasman et al. 1996), circularized exons also were larger than expected. When restricted to an analysis of single exon ecircRNAs, we noted that circularized exons were approximately threefold longer than expressed exons overall, at an average length of 690 nt (P < 10−15) (data not shown). In aggregate, these results suggest that the genomic structure of long exons flanked by long introns harboring inverted repeat elements facilitates RNA circularization.
Circular RNAs have been suggested to result from exon-skipping events. These events produce an exon-containing lariat, which could then itself be internally spliced to an exon circle (see Fig. 10, Model 1; Zaphiropoulos 1997). To test this notion, we determined if exon-skipped linear transcripts (e.g., the exon 1–4 transcript in Model 1, Fig. 10) could be identified in the setting of ecircRNAs (containing exon 2–3 in Model 1, Fig. 10). For 45% of ecircRNAs (regardless of stringency list used), we identified the corresponding, predicted colinear splicing reads characteristic of an exon-skipping event. This suggests that circularity may sometimes result from exon-skipping. Given the greater stability of ecircRNAs over associated linear transcripts, the linear transcript resulting from an exon-skipping event might be less abundant than the resulting circular molecules and, therefore, undetectable in our analysis. Therefore exon-skipping may be more common than estimated by this analysis. The set of ecircRNAs for which an associated skipped transcript could be identified was slightly more likely to harbor complementary ALU repeats (24% vs. 21%, P < 0.005), suggesting that intronic pairing may contribute to circularization with or without exon-skipping.
FIGURE 10.

Backsplicing and ecircRNA formation mechanisms. Models of backsplice formation. In Model 1, exon-skipping leads to a lariat whose restricted structure promotes circularization. In Model 2, exon-skipping is not required, with ALU complementarity or other RNA secondary structures bringing nonsequential donor-acceptor pairs into apposition, allowing for circularization. See Discussion for further explanation.
DISCUSSION
Here, we have shown that nonlariat circular RNAs emanate from >14% of transcribed genes in fibroblasts, comprising a highly abundant but unappreciated class of (presumably) noncoding RNA. We find that nearly all circular RNAs are comprised of exonic sequences, containing one or more exons, and always featuring a single non-colinear fusion of exons otherwise joined by canonical splicing. Salient features of our method allowing for unbiased identification of circular species are the use of ribosome depletion (RiboMinus), RNA exonuclease enrichment, cDNA synthesis using random primers rather than oligo dT, long (100-bp) reads with high coverage (>300 million reads per sample), use of MapSplice for de novo junction detection, and methodological optimization on a known circle (cANRIL). To consider a species circular, we required that it contain a backsplice and exhibit RNase R enrichment. These criteria discriminated circles from linear, trans-spliced species (which would possess a backsplice but not enrichment) as well as lariats (enrichment in the absence of a backsplice sequence but with branch junction reads). Use of this approach to identify circular transcripts in other cell types and species is an area of active ongoing investigation, as is applying exonuclease sequencing to the problem of branch point mapping.
Other groups have recently identified sets of non-colinear RNA species. A recent high-throughput sequencing analysis of non-colinear splicing in mammalian cells has recently been described (Al-Balool et al. 2011), but this analysis did not discriminate trans-splicing from circular events, since libraries were made from poly(A)-selected RNA, and RNA exonuclease resistance was not established. High-throughput sequencing of nonpolyadenylated RNA identified enrichment of lone exons within some genes (Yang et al. 2011), a finding that is likely explained by abundant circular RNA containing these exons. This work also made note of the apparent stability of some lariat sequences, as we also have observed. A recent high-throughput sequencing analysis in Archaea employed RNase enrichment to identify circular RNA forms (Danan et al. 2011), establishing that the production of RNA circles is evolutionarily ancient. Most recently, Brown and colleagues reported the identification of >1000 transcription products favoring circularization (Salzman et al. 2012). In this report, the authors relied on paired-end reads of high-throughput sequencing data to find probable circles and showed that some of these species exhibited RNase R resistance. Our study expands on this result by assessing RNase R sensitivity and, therefore, circularity on the genome-wide scale. We identify >100,000 circular RNA species within RNase R-treated samples that are beyond the limit of detection in untreated samples. These studies, in concert with the present work, establish that RNA circles are highly abundant, evolutionarily old, and can be tractably identified using high-throughput sequencing strategies.
While our data suggest mechanisms of ecircRNA production in vivo, these results are consistent with at least two distinct paths to the generation of RNA circles. In theory, any exon-skipping event has potential to produce an ecircRNA (Fig. 10, Model 1). In this model, the spliced lariat containing skipped exons undergoes internal splicing more rapidly than debranching to form an ecircRNA. Alternatively, ecircRNA could also be formed by alternative, 5′ to 3′ splicing of nascent transcripts (Fig. 10, Model 2). In this model, alternative splicing to a circular molecule is favored presumably because of intronic motifs bordering the circularized exon(s), which may interact to bring the two exons (or the ends of the same exon) close together. Key differences between these models are (1) the order of splicing events (i.e., which splice donor [SD] attacks first, of the circularized or noncircularized exon), (2) the need for a splice acceptor (SA) site 3′ from the circularized exons, and (3) the obligate production of an associated linear transcript containing the predicted exon-skipped fusion (e.g., exon 1–4 in Model 1). We believe the identification of long flanking introns and increased complementary ALU repeats about circularized exons is consistent with either model. Both features have been shown to influence exon-skipping (Ganguly et al. 2003; Fox-Walsh et al. 2005), but hybridization of complementary ALUs in long flanking introns would also favor the production of secondary structures in pre-mRNAs needed to drive the initiating 5′ to 3′ splicing event characteristic of Model 2.
The findings that ecircRNAs can be considerably more abundant (>10-fold) than associated linear transcripts and are evolutionarily conserved suggests selection for persistence of these RNAs and, hence, possible function. For example, circularization could regulate gene expression. Alternative splicing has been associated with diverse biologic processes (Faustino and Cooper 2003; Evsyukova et al. 2010; Ebert and Bernard 2011; Kalsotra and Cooper 2011), and direct circularization (Model 2) appears to represent a novel form of “alternative” splicing. Because our results show that ecircRNAs are significantly more stable than the associated linear mRNAs (Fig. 8B,C), we were not able to rely on transcript abundance to estimate the ratio of circular to linear transcripts produced from these genes per transcriptional event. Nevertheless, our observations suggest that a significant fraction of pre-mRNA transcripts created by traversal of RNA polymerase through exon 2 of HIPK2/3 or exon 3 of KIAA0182 (Fig. 3E) undergo “alternative” splicing to circularize rather than canonical splicing to produce the canonical linear transcript. As circularization of HIPK2/3 appears to preclude production of the usual protein-encoding transcript (as it would lack the ATG and significant N-terminal sequence) and is conserved between humans and rodents, this form of “alternative splicing” could regulate expression of genes like HIPK2/3. Observation of high homology between 69 murine circular RNAs and their human counterparts suggests that this may be a conserved mechanism of regulation in these genes. Application of exonuclease enrichment to a more diverse spectrum of organisms could further clarify the conservation and function of processes leading to circular RNAs.
Additionally, data from plants and mammals suggest that expression of some ncRNAs can perturb miRNA function by “target mimicry,” where ncRNAs such as those produced by pseudogenes sequester miRNAs to prevent translational repression of target coding RNA species (Ebert et al. 2007; Franco-Zorrilla et al. 2007; Poliseno et al. 2010). Results of siRNA knockdown show that ecircRNAs can be knocked down by RNAi (Fig. 7D,E), consistent with Ago2's RNA endonuclease “slicer” activity (Liu et al. 2004; Meister et al. 2004). We also show that siRNAs which target sequence in both the linear and circular transcripts of a given gene are able to induce knockdown of both species. As ecircRNAs are, in some cases, abundant, highly stable, and harbor miRNA binding sites, we expect these species could attenuate endogenous miRNA-mediated transcriptional repression through target mimicry. Additionally, ecircRNAs could sequester RNA-binding proteins, as we have suggested may be a function for cANRIL with respect to PcG proteins (Burd et al. 2010). We believe this is a particularly tantalizing prospect given the prior association of RNA splicing with PcG-mediated silencing (Isono et al. 2005).
Lastly, although it appears many circular forms are not protein encoding (Fig. 7A–C), it is not clear that all RNA circles are noncoding. Protein encoding RNA circles are well-established as viroids (e.g., the Hepatitis Δ agent) and have been synthesized experimentally to undergo translation in human cells (Chen and Sarnow 1995). It is also worth remarking in this regard that many circular RNAs contain a start codon and sometimes even the canonical AUG of the associated linear transcript (e.g., HIPK2/3). Whether endogenous circular RNAs are translated is an area of active ongoing study.
In summary, this genome-wide analysis based on RNase R resistance establishes circular RNA species as a common and abundant form of noncoding RNA. Using a conservative estimate, ecircRNAs appear to originate from >14% of transcribed genes in human fibroblasts and, in many cases, are more highly expressed than the associated, canonical mRNA. Circularization of transcribed exons is conserved between human and mice, is correlated with the presence of inverted repeats within long flanking introns, and may result from exon-skipping via a lariat structure or direct circularization driven by intronic pairing. In aggregate, these findings suggest that ecircRNAs are not the product of mere accidents of splicing but instead may regulate gene expression by affecting translation, RNAi, or through sequestration of RNA binding proteins.
MATERIALS AND METHODS
Cell culture
Telomerized Hs68 human fibroblasts cells were obtained from G. Peters and grown as previously described (Brookes et al. 2002). Human T lymphocytes (Jurkat E6-1) were cultured in RPMI 1640 containing 10% fetal bovine serum. Cells were grown at 37°C and 3% CO2 and subcultured at 3-d intervals. Jurkat cells were harvested at a density of 5 × 105 cell/mL.
RNA isolation and RNase R enrichment
RNA was isolated with RNAeasy system (Qiagen), including on-column DNAse digestion (Qiagen). For our first biological replicate, total RNA (60 µg) was depleted for ribosomal RNAs using the RiboMinus kit in six separate 10-μg reactions per the standard protocol (Invitrogen) and repooled. In the second biological replicate, total RNA (20 µg) was depleted for ribosomal RNAs using the RiboMinus kit. For both replicates, we prepared six 14.3-μL RNase R reactions. Diluted samples alone were briefly heated to 70°C to denature, then cooled to 40°C on a thermocycler. We then added 10× RNase R buffer (1.7 µL). For one of the six reactions, we added 1 µL water and added 1 µL RNase R to the remaining five reactions. The reaction was allowed to proceed at 40°C for 1 h.
Library preparation and sequencing
After RNase R digestion, libraries were prepared using the TruSeq library preparation protocol (Illumina) using a modified protocol. Elute Fragment Prime buffer was added immediately to the exonuclease treatment reaction, which then proceeded with reverse transcription and second-strand synthesis per the TruSeq protocol. Before end-repair, however, we pooled fragmented, reverse transcribed cDNA from the same replicate and treatment condition. These were then prepared by the TruSeq protocol. Sequencing was performed on an Illumina HiSeq instrument with 100-bp paired end reads. Sequencing data will be publicly available pending completed submission to the Short Read Archive.
Sequence mapping
Sequencing reads from each replicate and treatment were mapped independently using the MapSplice spliced alignment algorithm (version 2.0 beta) (Wang et al. 2010) against the GRChr37/hg19 human reference genome or NCBI37/mm9 mouse reference genome using the –fusion –noncanonical –bam options. To improve discovery rate with the mouse genome only, we also used the –gene-gtf option with the RefSeq annotation of mouse transcriptome. MapSplice first aligns reads to exons and regular splice junctions. It then detects “fusion junctions” using the reads that fail to map in the first step. Fusion junctions allow backsplice junctions, inversions, and long-range and inter-chromosome junctions. The strandedness of a junction is determined using the immediately flanking sequences donor-acceptor motifs (e.g., GT-AG sequence), which are required by MapSplice to call a junction.
Data analysis
Reads from the “fusion” output category in MapSplice were culled for splice junctions on the same strand and within 2 Mb, but in non-colinear ordering (backsplices). We calculated a spliced reads per billion mapping metric, computed as (Spliced reads/Total mapped reads) × 109, for each backsplice junction. Junctions enriched by this metric in both replicate pools were considered circular RNAs. Those that were observed in at least one mock treated replicate were assigned to one of three categories based on that expression, with “LOW” expression ecircRNAs observed in one mock replicate, “MEDIUM” expression observed in both replicates, and “HIGH” expression observed in both with an SRPBM of 10 in each. “Local RPKM” statistic was computed by [Read coverage at base × 106/(Total mapped reads × Read length)].
Bioinformatic analysis
Data from spliced alignment were parsed using custom PERL scripts and visualized using the Integrative Genomics Viewer (Robinson et al. 2011). Identification of annotated exon junctions among the “medium” expression stringency sites was performed using the KnownGene database downloaded from the UCSC Genome Browser's table viewer December 2011. The same database was used to compute coverage of annotated exons using the BedTools suite (Quinlan and Hall 2010), followed by normalization using RPKM. Exons with RPKM ≥ 0.1 were considered “expressed.” Expressed exon annotations and medium expression stringency backsplice annotations were compared using the DAVID tool for enrichment of GO terms and other annotations. Genes with the protein kinase annotation from the highest expression stringency category were plotted using Reaction Biology's Kinome Mapper tool according to backsplice expression level (http://www.reactionbiology.com/webapps/main/pages/LaunchKir.aspx). Percentage of expressed genes showing circularization was computed by first converting KnownGene annotations for expressed exons and low stringency backsplices to gene symbol. The percent circularized was then taken as percentage of gene symbols in both sets relative to all those in expressed genes.
Backsplices were compared to findings reported in the supplementary tables of Salzman et al. (2012), which were translated from RefSeq to hg19 coordinates. Backsplice sites were compared using BEDtools.
To calculate the relative abundance of backspliced vs. traditionally spliced products at steady state, we made use of the spliced mapping produced by the MapSplice analysis. We identified the total number of reads mapping to forward spliced junctions upstream of and downstream from the sites involved in the spliced junction. These were used to compute the ratio of backsplices as a percentage of forward splices for a given species for each replicate. These were averaged and are included in Supplemental Table S1.
Identification of enriched cis-elements
Sequences 200 bp upstream of and 200 bp downstream from backsplice events were pulled using BEDtools. Similarly flanking sequences from 20% of expressed exons (hereafter, “control exons”) were also extracted as a background set. These were compared for enriched motifs flanking backsplices using the CisFinder (Sharov and Ko 2009) algorithm. Motifs were then clustered to form longer elements.
Analysis of intronic ALU repeats near backsplice events
Annotated repeat sites were taken from the UCSC Genome Browser's RepeatMasker track using the table viewer December 2011. Sequences flanking backsplices and control exons that overlapped an ALU element were identified using BEDtools, and significance was tested using a χ2 test. For ALU pairing analysis, both flanks on either side of a backsplice were analyzed for RepeatMasker ALU elements using BEDtools in conjunction with custom perl scripts. Complementary ALU pairs were identified as at least one plus and one minus stranded ALU family element on opposite sides of the backsplice. Backsplices with such features were tallied, as were backsplices with noncomplementary pairs. Significant differences in the rates of these events were compared to an equal rate null hypothesis using a χ2 test. This analysis was similarly performed on control exons.
Analyisis of flanking intron length
Introns were identified using KnownGene annotations for colinear splice sites sharing a junction with a backsplice. In cases where multiple annotations were possible (multiple isoforms of the source gene), the shortest intron length was used. Significant differences in flanking intron length between backsplice sets and control exons were computed using a t-test.
Analysis of exon-skipping
To identify exon-skipping events, the set of all identified colinear splices was taken from MapSplice output and compared to the set of observed backsplices using BEDtools. To be included as a possible product of exon-skipping, a backsplice had to be completely traversed by a colinear splice on the same strand. Strandedness was identified by GT-AG donor-acceptor motifs. Overlap with the set of backsplices with complementary ALU pairs was identified using custom perl scripts. Significant enrichment of complementary ALU pairs in skipped backsplices vs. all backsplices was computed by a χ2 test.
Quantitative real-time PCR
RNA was isolated from Hs68 cells as above and treated with ImPromII reverse transcriptase using either random hexamer (Invitrogen) and oligo dT (Invitrogen) or oligo dT alone as indicated. Primers used in qRT-PCR were designed to span at least one intron, and primers assaying for circular products were designed to cross the backsplice junction (Supplemental Table S2). Real-time PCR was carried out in triplicate on an ABI 7900HT thermocyler. The resulting PCR products were cloned into the TOPO-TA cloning kit (Invitrogen) and sequenced for validation. Cloned fragments for HIPK3 circular and HIPK3 linear qPCR were linearized with HindIII (NEB) and used to create quantitative standards ranging in concentration from 100 molecules per μL to 200,000,000 molecules per μL. These were used to generate a standard curve for estimating absolute quantities of circular vs. linear molecules in Hs68 cells. Standard curve R2 values were >0.99.
Virtual northern RT-PCR
RNA was isolated into 17 fractions by “virtual Northern” analysis as previously described (Hurowitz et al. 2007). The size-fractioned gel was then cut into 17 gel slices of roughly equivalent size. Each slice was dissolved in buffer RLT of the RNAeasy system (Qiagen) and purified on column per protocol. Equal volume quantities of eluted sample were reverse transcribed with ImPromII reverse transcriptase and analyzed for circular and linear forms by TaqMan as described above. To identify the size of each product, the relative quantity of product in each fraction was approximated by the equation: Expressionfraction = 2(40–Mean Ct). To determine the percentage of the total sample in each size fraction, we used the equation: Percentagefraction = Expressionfraction/∑fractions Expressionfraction.
RNA stability
For assay of RNA stability, 5 × 105 cells were plated on 3.5-cm cell culture plates and treated with 10 µg/mL actinomycin D. Cells were then harvested at time 0, 6, 12, 24, and 48 h time points, and RNA was isolated and subjected to qRT-PCR as described above, with equal quantities RNA from the five time points provided to each RT reaction. Each RNA was normalized against the 0-h time point to calculate log2(fold enrichment).
Cell localization
Cytoplasmic and nuclear RNA was isolated from two biological replicates of 2.5 × 106 Hs68 cells using the Norgen Biotek cytoplasmic and nuclear RNA purification kit. Equal quantities of RNA were provided to RT as described above, and targets were quantified by qRT-PCR.
Conservation of an abundant ecRNA
C57Bl/6 strain mouse testes were isolated and homogenized using the gentleMACs dissociator system (Militenyi), and RNA was isolated and reverse transcribed as above. For analysis of murine HIPK2 and HIPK3 homologs, outward-facing primers were designed against the first coding exon of Hipk2 and Hipk3 in the mm9 reference genome (Fig. 6E; Supplemental Table S2). For sequencing analysis, RNA was treated using the RiboMinus kit (Invitrogen) and split into RNase R and mock treatment aliquots, from which TruSeq (Illumina) sequencing libraries were prepared and sequenced together in a single lane on a HiSeq system high-throughput sequencer (Illumina) as described above. We focused our analysis on circular RNAs observed in RNase R-treated samples that were enriched by RNase R, without restricting to those also observed in untreated controls, due to the more limited sequence from those samples. We identified 1275 circular RNAs from murine testis and cross referenced these against 2121 human circular RNA products in the MEDIUM expression category that could be successfully converted to mouse coordinates using the UCSC Genome Browser's LiftOver utility. Significance estimates of overlap were performed using a χ2 test. Estimates of the frequency of exact overlap in backsplice sites assumed a random permutation over splice donors and acceptors with an average number of exons per gene greater than 11 (consistent with UCSC KnownGene annotations of genes with overlap).
Cell fractionation
Jurkat cells were centrifuged at 4°C for 10 min (500g) then washed 1× in PBS, followed by resuspension in Homogenization Buffer (HB) containing 400 mM KOAc (pH 7.5), 25 mM K-HEPES, 15 mM Mg(OAc)2, 1 mM DTT, 200 µM cycloheximide, 1% NP-40, 0.5% deoxycholate, 1 mM PMSF, and 50 units/mL RNasin (Promega). Cells were incubated on ice for 10 min then centrifuged at 4°C for 10 min (12,000g) to remove nuclei.
Polysome analysis
Jurkat cells were grown to a density of 5 × 105 cells/mL and treated with 200 µM cycloheximide to stabilize polysome complexes. Cytoplasmic supernatant equal to 30 × 106 cells was loaded onto 10-mL continuous 15%–50% sucrose gradients containing 400 mM KOAc (pH 7.5), 25 mM K-HEPES, 15 mM Mg(OAc)2, 200 µM cycloheximide and 50 units/mL RNasin (Promega). Gradients were centrifuged at 4°C for 3 h at 100,000g in an SW41 rotor (Beckman). Gradient fractions were collected using a Brandel Fractionation System and an Isco UA-6 ultraviolet detector used to produce polysome profiles for gradients (data not shown). Total RNA was extracted from fractions essentially as described in Chomczynski and Sacchi (1987). Briefly, fractions were supplemented with an equal volume (total volume of 750 µL) of Extraction Buffer (EB) containing 4M guanidinium thiocyanate, 25 mM sodium-citrate (pH 7), 0.5% N-Lauryl-sarcosine, 5 mM EDTA, and 0.1M β-mercaptoethanol. Two hundred and fifty µL of phenol (pH 4.5) was added to the homogenized fractions and rotated 15 min at room temperature. Two hundred µL of chloroform was added, followed by centrifugation at 10,000g for 10 min at 4°C. The aqueous layer was transferred to new centrifuge tubes and precipitated at −20°C for 1 h with a 1/10th-volume of sodium acetate (pH 5.2) and an equal volume of isopropanol. Samples were centrifuged at 12,000g, aspirated, and washed 1× with 70% ethanol. Samples were resuspended in DEPC-H2O. The polysome profile produced by the Isco UA-6 ultraviolet detector and the identity of the individual fractions were confirmed by loading 45 µL of sucrose fractions on an agarose gel and staining with ethidium bromide to visualize the ribosomal RNA (Fig. 7A). Isolated RNA fractions were reverse transcribed with equal volumes of input RNA by ImPromII reverse transcriptase. The quantity of circular and linear forms was determined by TaqMan as described above. Abundance of the fractions was normalized against the sum of the total signal as with the virtual Northern (above).
Knockdown of linear and circular species by siRNA
Three siRNAs were employed for targeted knockdown of HIPK3 and ZFY. Constructs to knockdown circular RNA were directed specifically against the backsplice. Knockdown of linear RNA used downstream exons of the gene not included in the predicted circular product. Joint knockdown used exonic sequences predicted to be included in the circular form. Experiments were performed in HS86 cells using Ambion siRNA and RNA Max kit (Invitrogen, Cat. # 13778-075). A quantity of 1.25 nM of each siRNA (see Supplemental Table S2) with 0.2 µL of RNA Max lipafectamine were added to plates in antibiotic-free Optimem media and incubated for 20 min. HS68 cells were then plated with antibiotic-free DMEM with 10% FBS and incubated overnight. After 24 h, media were changed. Cells were harvested 48 h after treatment. RNA was isolated and quantified by qPCR as described above. Ct values were first normalized against TBP to correct for reverse transcription efficiency and then normalized to values for nonspecific knockdown.
RNA in situ hybridization
A probe antisense to the backsplice junction of HIPK3 was labeled by in vitro transcription of the construct also used as the qPCR standard for the HIPK3 circular RNA. Labeling was performed with Digoxinogen RNA Labeling Mix (Roche).
Hs68 cells were grown on cover slips and treated with siRNA knockdown against HIPK3 linear, both HIPK3 circular and linear, or nonspecific (NS) targets as above, and fixed 48 h after media change. Cells were in freshly prepared 4% PFA in 0.1M PBS for 10 min and permeabilized in 0.01M HCl and 0.1% pepsin at 37°C for 2 min. After rinsing three times in PBS with 0.05% Triton X-100 for 5 min, then once in 2× SSC for 5 min, cells were prehybridized in the hybridization buffer at room temperature for 1 h. Digoxigenin-labeled probes and cells were denatured at 75°C for 3 min at the same time, and the probes were applied onto the cells and hybridized for 16 h at 37°C in the humidified chamber. After hybridization, cells were washed three times in 2× SSC for 10 min at 37, twice in 0.1% SSC for 5 min at 60°C, and twice in TNT (0.1M Tris-HCl, 0.15M NaCl, and 0.1% Tween 20, pH 7.5) for 10 min at room temperature. To detect the hybridized probes, cells were then incubated with a 1:1000 dilution of alkaline phosphatase conjugated sheep anti-digoxigenin (Roche Diagnostics) in blocking buffer at room temperature for 3 h and developed in HNPP/Fast Red TR (Roche) for 5 h. Cells were counterstained with DAPI and mounted in aqueous mounting medium (Faramount aqueous mounting medium, DAKO). Slides were imaged by confocal microscopy, and stacks were flattened by maximum intensity Z-projection and composited to form final images.
DATA DEPOSITION
Data have been uploaded to the Short Read Archive with accession number SRA050270 and are pending validation and release.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Drs. Derek Chiang, Gordon Peters, Jan Prins, and Zefeng Wang for advice and expertise. This work was supported by grants from the NIA (AG024379 and AG041567) and the Paul Glenn Foundation.
REFERENCES
- Al-Balool HH, Weber D, Liu Y, Wade M, Guleria K, Nam PL, Clayton J, Rowe W, Coxhead J, Irving J, et al. 2011. Post-transcriptional exon shuffling events in humans can be evolutionarily conserved and abundant. Genome Res 21: 1788–1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes S, Rowe J, Ruas M, Llanos S, Clark PA, Lomax M, James MC, Vatcheva R, Bates S, Vousden KH, et al. 2002. INK4a-deficient human diploid fibroblasts are resistant to RAS-induced senescence. EMBO J 21: 2936–2945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd CE, Jeck WR, Liu Y, Sanoff HK, Wang Z, Sharpless NE 2010. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet 6: e1001233 10.1371/journal.pgen.1001233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capel B, Swain A, Nicolis S, Hacker A, Walter M, Koopman P, Goodfellow P, Lovell-Badge R 1993. Circular transcripts of the testis-determining gene Sry in adult mouse testis. Cell 73: 1019–1030 [DOI] [PubMed] [Google Scholar]
- Chen CY, Sarnow P 1995. Initiation of protein synthesis by the eukaryotic translational apparatus on circular RNAs. Science 268: 415–417 [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N 1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162: 156–159 [DOI] [PubMed] [Google Scholar]
- Cocquerelle C, Mascrez B, Hetuin D, Bailleul B 1993. Mis-splicing yields circular RNA molecules. FASEB J 7: 155–160 [DOI] [PubMed] [Google Scholar]
- Danan M, Schwartz S, Edelheit S, Sorek R 2011. Transcriptome-wide discovery of circular RNAs in Archaea. Nucleic Acids Res 40: 3131–3142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin RA, Kazmi MA, Ostrer H 1995. Inverted repeats are necessary for circularization of the mouse testis Sry transcript. Gene 167: 245–248 [DOI] [PubMed] [Google Scholar]
- Ebert B, Bernard OA 2011. Mutations in RNA splicing machinery in human cancers. N Engl J Med 365: 2534–2535 [DOI] [PubMed] [Google Scholar]
- Ebert MS, Neilson JR, Sharp PA 2007. MicroRNA sponges: Competitive inhibitors of small RNAs in mammalian cells. Nat Methods 4: 721–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M 2011. Non-coding RNAs in human disease. Nat Rev Genet 12: 861–874 [DOI] [PubMed] [Google Scholar]
- Evsyukova I, Somarelli JA, Gregory SG, Garcia-Blanco MA 2010. Alternative splicing in multiple sclerosis and other autoimmune diseases. RNA Biol 7: 462–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faustino NA, Cooper TA 2003. Pre-mRNA splicing and human disease. Genes Dev 17: 419–437 [DOI] [PubMed] [Google Scholar]
- Fox-Walsh KL, Dou Y, Lam BJ, Hung SP, Baldi PF, Hertel KJ 2005. The architecture of pre-mRNAs affects mechanisms of splice-site pairing. Proc Natl Acad Sci 102: 16176–16181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco-Zorrilla JM, Valli A, Todesco M, Mateos I, Puga MI, Rubio-Somoza I, Leyva A, Weigel D, Garcia JA, Paz-Ares J 2007. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat Genet 39: 1033–1037 [DOI] [PubMed] [Google Scholar]
- Ganguly A, Dunbar T, Chen P, Godmilow L, Ganguly T 2003. Exon skipping caused by an intronic insertion of a young Alu Yb9 element leads to severe hemophilia A. Hum Genet 113: 348–352 [DOI] [PubMed] [Google Scholar]
- Gao K, Masuda A, Matsuura T, Ohno K 2008. Human branch point consensus sequence is yUnAy. Nucleic Acids Res 36: 2257–2267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen TB, Wiklund ED, Bramsen JB, Villadsen SB, Statham AL, Clark SJ, Kjems J 2011. miRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. EMBO J 30: 4414–4422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu SN, Hertel KJ 2009. Spliceosomes walk the line: Splicing errors and their impact on cellular function. RNA Biol 6: 526–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- Hurowitz EH, Brown PO 2003. Genome-wide analysis of mRNA lengths in Saccharomyces cerevisiae. Genome Biol 5: R2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurowitz EH, Drori I, Stodden VC, Donoho DL, Brown PO 2007. Virtual Northern analysis of the human genome. PLoS One 2: e460 10.1371/journal.pone.0000460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isono K, Mizutani-Koseki Y, Komori T, Schmidt-Zachmann MS, Koseki H 2005. Mammalian polycomb-mediated repression of Hox genes requires the essential spliceosomal protein Sf3b1. Genes Dev 19: 536–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsotra A, Cooper TA 2011. Functional consequences of developmentally regulated alternative splicing. Nat Rev Genet 12: 715–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, Song JJ, Hammond SM, Joshua-Tor L, Hannon GJ 2004. Argonaute2 is the catalytic engine of mammalian RNAi. Science 305: 1437–1441 [DOI] [PubMed] [Google Scholar]
- Lorsch JR, Bartel DP, Szostak JW 1995. Reverse transcriptase reads through a 2′-5′ linkage and a 2′-thiophosphate in a template. Nucleic Acids Res 23: 2811–2814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister G, Landthaler M, Patkaniowska A, Dorsett Y, Teng G, Tuschl T 2004. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol Cell 15: 185–197 [DOI] [PubMed] [Google Scholar]
- Nigro JM, Cho KR, Fearon ER, Kern SE, Ruppert JM, Oliner JD, Kinzler KW, Vogelstein B 1991. Scrambled exons. Cell 64: 607–613 [DOI] [PubMed] [Google Scholar]
- Pasman Z, Been MD, Garcia-Blanco MA 1996. Exon circularization in mammalian nuclear extracts. RNA 2: 603–610 [PMC free article] [PubMed] [Google Scholar]
- Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP 2010. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 465: 1033–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM 2010. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO 2012. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One 7: e30733 10.1371/journal.pone.0030733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharov AA, Ko MS 2009. Exhaustive search for over-represented DNA sequence motifs with CisFinder. DNA Res 16: 261–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Zuo Y, Wang J, Zhang MQ, Malhotra A, Mayeda A 2006. Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res 34: e63 10.1093/nar/gkl151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, He X, Mieczkowski P, Grimm SA, Perou CM, et al. 2010. MapSplice: Accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res 38: e178 10.1093/nar/gkq622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner JR 1999. The economics of ribosome biosynthesis in yeast. Trends Biochem Sci 24: 437–440 [DOI] [PubMed] [Google Scholar]
- Yang L, Duff MO, Graveley BR, Carmichael GG, Chen LL 2011. Genomewide characterization of non-polyadenylated RNAs. Genome Biol 12: R16 10.1186/gb-2011-12-2-r16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaphiropoulos PG 1997. Exon skipping and circular RNA formation in transcripts of the human cytochrome P-450 2C18 gene in epidermis and of the rat androgen binding protein gene in testis. Mol Cell Biol 17: 2985–2993 [DOI] [PMC free article] [PubMed] [Google Scholar]