

Abstract
Toxin-antitoxin (TA) modules consist of a pair of genes that encode two components: a protein toxin and an antitoxin, which may be in the form of either a labile protein or an antisense small RNA. Here we describe, to the best of our knowledge, the first functional chromosomal type I TA system in streptococci. Our model organism is the oral pathogen Streptococcus mutans. Our results showed that the genome of S. mutans UA159 reference strain harbors a previously unannotated Fst-like toxin (Fst-Sm) and its cis-encoded small RNA antitoxin (srSm) converging towards the end of the toxin gene in IGR176, a small intergenic region of 318 nt. Fst-Sm is a small hydrophobic peptide of 32 amino acid residues with homology to the Fst toxin family. Transcripts of ∼200 nt and ∼70 nt specific to fst-Sm mRNA and srSm RNA, respectively, were detected by Northern blot analysis throughout S. mutans growth. The toxin mRNA was considerably more stable than its cognate antitoxin. The half-life of srSm RNA was determined to be ∼30 min, while fst-Sm mRNA had a half-life of ∼90 min. Both fst-Sm and srSm RNAs were transcribed across direct tandem repeats providing a region of complementarity for inhibition of toxin translation. Overproduction of Fst-Sm had a toxic effect on E. coli and S. mutans cells which can be neutralized by coexpression of srSm RNA. Deletion of fst-Sm/srSm locus or overexpression of Fst-Sm/srSm had no effect on S. mutans cell growth in liquid medium and no differences in the total biofilm biomass were noted. In contrast, mild-overproduction of Fst-Sm/srSm type I TA system decreases the levels of persister cells tolerant to bacterial cell wall synthesis inhibitors.
Introduction
Prokaryotic chromosomes contain small genetic elements encoding two components: a stable toxin and its less stable cognate antitoxin. These modules are called toxin-antitoxin (TA) systems [1]. Typically, the toxin inhibits an essential microbial cellular function. TA pairs form a stabilized complex in the cell preventing toxicity under normal growth conditions. However, when the balance between the toxin and antitoxin is perturbed, usually following cellular damage or stressful conditions, the toxin is released from the TA complex leading to cell growth arrest and/or cell death. The physiological significance and importance of TA modules in microbial physiology is reflected by the fact that they are ubiquitously found on the chromosome of bacteria [2]. Five different types of bacterial TA systems have been described thus far, depending of the nature and mode of action of the antitoxin component. While toxins are always proteins, antitoxins are either RNAs (type I, type III) or proteins (type II, type IV, type V). RNA antitoxins suppress toxin expression (type I) [3]–[5] or interfere with the toxin activity instead of preventing its expression (type III) [6], [7]. Protein antitoxins neutralize the toxicity of the cognate toxin by forming a stable complex (type II) [8]–[10], by functioning as antagonists for the toxin activity (type IV) [11], [12], or by inhibiting the toxin by cleaving specifically its mRNA (type V) [13]. Current hypotheses propose that TAs are stress-response elements that help bacteria cope with environmental stress either by promoting altruistic death of a subpopulation or by inducing formation of dormant persister cells [14]–[16]. The dormant state allows bacteria to survive even high doses of the antibiotic. It has been suggested that persister cells may play a significant role in the recalcitrance of chronic biofilm infections to antimicrobials [17]. In E. coli, multiple type II TA systems have been linked to the formation of persister cells [18], [19].
The oral cavity environment is inarguably a dynamic and complex ecosystem with a wide range of environmental challenges. Our model organism is the oral pathogen Streptococcus mutans. S. mutans depends on a biofilm lifestyle for its survival and persistence in its natural habitat [20]. In presence of fermentable dietary carbohydrates, this acid-producing bacterium can cause damage (cavities) to the tooth’s hard tissues [21], [22]. In fact, S. mutans is a major human pathogen that infects more than half of the world’s human population. We recently characterized a locus encoding a functional type II TA system, the S. mutans chromosomal MazEF module. We demonstrated that MazF protein was a toxic nuclease arresting cell growth through the mechanism of RNA cleavage, and that MazE antitoxin protein inhibited the ribonuclease activity of MazF by forming a protein complex [23]. Our most recent data showed that ectopic mild overexpression of chromosomal MazEF and RelBE type II TA systems induced formation of multidrug tolerant persister cells in S. mutans [24].
In this study, we are pursuing our investigation of the chromosomal TA systems in S. mutans. We were interested in discovering type I TA systems in the chromosome of S. mutans. In type I TA systems, antitoxins are small untranslated RNAs acting as antisense RNAs. RNA antitoxins can be encoded directly opposite the coding sequence of the toxin, opposite the 5′ UTR, or opposite the 3′ UTR of the toxin mRNA, or even divergent to the toxin gene but with long stretches of complementarity to the toxin mRNA [3]. The RNA antitoxins are acting as antisense RNAs that anneal with the corresponding toxin mRNA, thereby inhibiting its translation or promoting its degradation. All type I toxins are small, hydrophobic peptides of 19–38 amino acid residues. Overexpression of these hydrophobic toxins has been shown to lead to membrane depolarization or membrane disruption resulting in a loss of cell viability [3]. Type I TA modules were first discovered on plasmids where they were found to stabilize various plasmids in Gram-negative bacteria (post-segregational killing activity) [25]. The RNAI-RNAII encoded by the par locus of the Enterococcus faecalis plasmid pAD1 was the first type I TA system identified in Gram-positive bacteria. RNAI encodes a 33-aa peptide toxin (Fst), while RNAII codes for the ∼70-nt regulatory antisense RNA [26], [27]. The par addiction module kills plasmid-free cells by affecting membrane permeability and cell division [28], [29]. Recently, Weaver et al. (2009) [30] identified pAD1-like TA systems on the chromosomes and plasmids of Enterococcus, Lactobacillus, and Staphylococcus species suggesting that Fst-like toxin may be prevalent in Gram-positive bacteria.
Our results showed that the genome of S. mutans UA159 reference strain harbors an unannotated Fst-like toxin based on the presence of a conserved open reading frame. Examination of the genetic context revealed conserved features similar to the Fst type I TA system. The aim of this study was to investigate the functionality of this putative S. mutans type I TA system; we proposed the name Fst-Sm/srSm for Fst S . mutans and small RNA S. mutans. The possible link between Fst-Sm/srSm TA system and the development of dormant persister cells was also investigated. The Fst-Sm/srSm module is to our knowledge the first type I TA system characterized in Streptococcus.
Materials and Methods
Bacterial Growth Conditions
S. mutans strains were grown in Todd-Hewitt broth supplemented with 0.3% yeast extract (THYE) and incubated statically at 37°C in a 5% CO2 atmosphere. E. coli strains were cultivated aerobically in Luria-Bertani (LB) medium at 37°C. When needed, antibiotics were used as follows: chloramphenicol (20 µg/ml) or kanamycin (50 µg/ml) for E. coli, and erythromycin (10 µg/ml), or chloramphenicol (10 µg/ml) for S. mutans. Cell growth was monitored through optical density at 600 nm (OD600). Cell viability was assessed by counting CFU on replica agar plates.
Plasmid and Strain Construction
A summary of bacterial strains and plasmids is provided in Table 1. Primers used for the generation of PCR products indicated below are listed in Table 2. A nonpolar insertion-deletion IGR176 mutant (ΔIGR176) was constructed in S. mutans UA159 wild-type (WT) strain by PCR ligation mutagenesis using the primer pairs CMT-576/CMT-577 and CMT-578/CMT-579. All plasmids were constructed in E. coli strain DH10B or TOP10. Plasmids were introduced into E. coli by transformation using electroporation or chemical transformation. Plasmids were transferred to S. mutans by natural transformation as described previously [31].
Table 1. Bacterial strains and plasmids used in this study.
| Strain or plasmid | Relevant characteristic(s)a | Source or reference |
| Strains | ||
| S. mutans | ||
| UA159 | Wild-type S. mutans reference strain | Lab stock |
| ΔIGR176 mutant | In-frame IGR176 deletion mutant derived from S. mutans UA159; Emr | This study |
| UA159(pIB166) | UA159 harboring pIB166; Cmr | [23] |
| ΔIGR176(pIB166) | ΔIGR176 harboring pIB166; Emr, Cmr | This study |
| ΔIGR176(pSK10) | ΔIGR176 harboring pSK10; Emr, Cmr | This study |
| E. coli | ||
| DH10B | Host for cloning and plasmid production | Lab stock |
| TOP10 | Host for cloning and plasmid production | Invitrogen |
| LMG194 | Host strain for pBAD expression | Invitrogen |
| LMG194(pSK1) | LMG194 harboring pSK1; Kmr | This study |
| LMG194(pSK2) | LMG194 harboring pSK2; Kmr | This study |
| LMG194(pSK8) | LMG194 harboring pSK8; Kmr | This study |
| DH10B(pSK3) | DH10B harboring pSK3; Cmr | This study |
| DH10B(pSK7) | DH10B harboring pSK7; Kmr | This study |
| DH10B(pHSG299)(pSK3) | DH10B harboring pHSG299, pSK3; Kmr Cmr | This study |
| DH10B(pHSG299)(pSK4) | DH10B harboring pHSG299, pSK4; Kmr Cmr | This study |
| DH10B(pHSG299)(pSK5) | DH10B harboring pHSG299, pSK5; Kmr Cmr | This study |
| DH10B(pSK3)(pSK6) | DH10B harboring pSK3, pSK6; Cmr Kmr | This study |
| DH10B(pSK4)(pSK6) | DH10B harboring pSK4, pSK6; Cmr Kmr | This study |
| DH10B(pSK5)(pSK6) | DH10B harboring pSK5, pSK6; Cmr Kmr | This study |
| Plasmids | ||
| pBAD202/D-TOPO | Expression vector linearized and topoisomerase-activated; Kmr | Invitrogen |
| pHGS299 | High-copy-number cloning vector; Kmr | Takara Bio USA |
| pPROBE-NT’ | Promoterless GFP vector; Kmr | [32] |
| pIB166 | Streptococcus-E. coli shuttle plasmid; Cmr | [33] |
| pSK1 | Fst-Sm cloned under the control of araBAD promoter into pBAD202/D-TOPO; Kmr | This study |
| pSK2 | Fst-Sm/srSm cloned under the control of araBAD promoter into pBAD202/D-TOPO; Kmr | This study |
| pSK3 | GFP cassette cloned into pIB166; Cmr | This study |
| pSK4 | Pfst+DRI-II transcriptionally fused to gfp into pSK3; Cmr | This study |
| pSK5 | Pfst transcriptionally fused to gfp into pSK3; Cmr | This study |
| pSK6 | srSm cloned into pHSG299; Kmr | This study |
| pSK7 | Fst-Sm G16A mutation into pSK1; Kmr | This study |
| pSK8 | Fst-Sm A11G/G16A mutation into pSK1; Kmr | This study |
| pSK9 | fst-Sm cloned under the control of its own promoter into pIB166; Cmr | This study |
| pSK10 | fst-Sm and srSm cloned under the control of their own promoter into pIB166; Cmr | This study |
Emr, erythromycin resistance; Cmr, chloramphenicol resistance; Kmr, kanamycin resistance.
Table 2. Primers used in this study.
| Primer | Gene | Sequence (5′→3′)a,b | Purpose |
| CMT-576 | fst-Sm/srSm | GCACAATGGGAATCTGGAAG | Gene deletion |
| CMT-577 | fst-Sm/srSm | GGCGCGCC AGGCATGATTTCTTTATTCGCA | Gene deletion |
| CMT-578 | fst-Sm/srSm | GGCCGGCC TATCATATTCTCCACAAACGATA | Gene deletion |
| CMT-579 | fst-Sm/srSm | GACTTATGGTCATTTGGTTGC | Gene deletion |
| CMT-19 | erm | GGCGCGCC CCGGGCCCAAAATTTGTTTGAT | Gene deletion |
| CMT-20 | erm | GGCCGGCC AGTCGGCAGCGACTCATAGAAT | Gene deletion |
| CMT-499 | fst-Sm | CACCATGTGGCATAATTTCTTTATATA | pBAD cloning |
| CMT-500 | fst-Sm | ATCGTCGTCTTTCTTATCCA | pBAD cloning |
| CMT-581 | fst-Sm/srSm | CACCAGTTGCCAAATGTGGCATAAT | pBAD cloning |
| CMT-582 | fst-Sm/srSm | TTGACAAAACCAAAATAAAAAGA | pBAD cloning |
| CMT-621 | fst-Sm | CGCACCAATTTTTGTTGCGATAGTGCTTGCGCTAT | Mutagenesis |
| CMT-622 | fst-Sm | ATAGCGCAAGCACTATCGCAACAAAAATTGGTGCG | Mutagenesis |
| CMT-627 | fst-Sm | CATAATTTCTTTATATATATCGTCGGACCAATTTTTGTTGCGATAGTG | Mutagenesis |
| CMT-628 | fst-Sm | CACTATCGCAACAAAAATTGGTCCGACGATATATATAAAGAAATTATG | Mutagenesis |
| CMT-449 | gfp | CTCGAG GTCACGACGTTGTAAAACGAC | GFP reporter |
| CMT-450 | gfp | CCGCGG CAGCTATGACCATGATTACGC | GFP reporter |
| CMT-596 | fst-Sm | GAATTC GACTGAAGATGAATTTGAAAAC | GFP reporter; fst-Sm, fst-Sm/srSm expression |
| CMT-597 | fst-Sm | GGTACC ATATATAAAGAAATTATGCCACA | GFP reporter |
| CMT-598 | fst-Sm | GGTACC CACCCCCTCTCTAAGGCG | GFP reporter |
| CMT-599 | srSm | GGATCC TTGACAAAACCAAAATAAAAAGA | GFP reporter; srSm expression |
| CMT-600 | srSm | GAATTC TAGCTCGGACGCAGTATTC | GFP reporter; srSm expression |
| CMT-595 | fst-Sm | AAGCTT ACTTAATCGTCGTCTTTCTTAT | Fst-Sm expression |
| CMT-594 | fst-Sm/srSm | AAGCTT TTGACAAAACCAAAATAAAAAGA | fst-Sm/srSm expression |
| CMT-497 | fst-Sm | ATGTGGCATAATTTCTTTATATA | RT-PCR, Northern |
| CMT-498 | fst-Sm | TTAATCGTCGTCTTTCTTATCC | RT-PCR, Northern |
| CMT-583 | fst-Sm | TTAATCGTCGTCTTTCTTATC | 5′RACE-PCR |
| CMT-585 | srSm RNA | AGCTAGGGCTTTTCCGTTG | 5′RACE-PCR |
| CMT-180 | Poly-G tail | GAATTCGAATTC CCCCCCCCCCC | 5′ RACE-PCR |
| CMT-181 | Poly-T tail | GAATTCGAATTC AAAAAAAAAAAA | 5′ RACE-PCR |
| CMT-584 | fst-Sm | GAATTC AGCCATTCAGAAAATAGCGC | 5′ RACE-PCR |
| CMT-586 | srSm | GAATTC GGGCTTTTCCGTTGCCAAA | 5′ RACE-PCR |
| CMT-497 | fst-Sm | ATGTGGCATAATTTCTTTATATA | Northern blot |
| CMT-498 | fst-Sm | TTAATCGTCGTCTTTCTTATCC | Northern blot |
| CMT-572 | srfSm | /5BiosG/AGCTAGGGCTTTTCCGTTGCCA | Northern blot |
| CMT-558 | srfSm | /5BiosG/ATATTAAGGCATGATTTCTTTAT | Northern blot |
| CMT-672 | 5S rRNA | CCTAGGGGAGACACCTGT | Northern blot |
| CMT-673 | 5S rRNA | AGCTAAACTCCCTCTTGCT | Northern blot |
Restriction sites are underlined.
Modified residues are shown in bold and underlined.
(i) Construction of expression vectors for induction in E. coli
To generate inducible expression constructs for induction of fst-Sm and fst-Sm/srSm in E. coli, fragments containing the open reading frame of fst-Sm and fst-Sm/srSm locus were first PCR amplified using UA159 genomic DNA (gDNA) as a template and the primer pairs CMT-499/CMT-500 for fst-Sm gene and CMT-581/CMT-582 for fst-Sm/srSm locus. The PCR products were purified, cloned in-frame upstream from the His6 sequence under the control of araBAD promoter into pBAD202/D-TOPO vector (Invitrogen), and transferred into E. coli TOP10. The recombinant plasmids were sequenced on both strands for confirmation. The plasmids designated pSK1 (fst-Sm in pBAD) and pSK2 (fst-Sm/srSm in pBAD) were then transferred into electrocompetent E. coli LMG194 cells for induction by arabinose.
(ii) Construction of a non-toxic Fst peptide
A non-toxic Fst peptide was generated by site-directed mutagenesis using the QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies) following the manufacturer’s recommendations. Briefly, plasmid pSK1 carrying the wild-type copy of fst-Sm gene was used as a template for the replacement of G16A using the two mutagenic PCR primers CMT-621 and CMT-622. The clone used to produce Fst-Sm G16A mutant was designated pSK7 and was confirmed by sequencing. The clone pSK7 was next used as a template with the two mutagenic PCR primers CMT-627 and CMT-628 for the replacement of A11G. The clone used for production of Fst-Sm G16A A11G mutant was designated pSK8 and was confirmed by sequencing. The clone pSK8 was then electrotransferred into LMG194 cells for induction by arabinose to produce the recombinant non-toxic Fst peptide (rNT-Fst).
(iii) Construction of GFP reporter constructs
First, a promoterless GFP vector was constructed by PCR amplifying the promoterless gfp cassette flanked by its upstream and downstream terminator sequences using pPROBE-NT’ [32] as template and the primer pair CMT-449/CMT-450. The PCR product was purified, double digested with XhoI/SacII, and then cloned into the shuttle vector pIB166 [33] pre-cut by the same enzymes. The recombinant plasmid pSK3 was confirmed by restriction digestion. To construct the fst-Sm promoter reporter fusions (Pfst+DRI/II and Pfst), the promoter region of fst-Sm with and without the direct repeats (DRI+DRII) was PCR amplified using UA159 gDNA as template and the primer pairs CMT-596/CMT-597 and CMT-596/CMT-598, respectively. The PCR products were double digested with EcoRI/KpnI and cloned into pSK3 precut with the same enzymes to generate the recombinant plasmids pSK4 (Pfst+DRI/II) and pSK5 (Pfst). To construct the srSm RNA expression vector, the srSm locus was PCR amplified using UA159 gDNA and the primer pair CMT-599/CMT-600. The PCR product was double digested with BamHI/EcoRI, purified, and cloned into pHGS299 precut by the same enzymes. The recombinant plasmid pSK6 was confirmed by sequencing.
(iv) Construction of vectors for expression in S. mutans
The full-length coding region and promoter region of fst-Sm and the fst-Sm/srSm locus were PCR amplified using UA159 gDNA as a template and the primer pairs CMT-596/CMT-595 and CMT-596/CMT-594, respectively. The PCR products were purified, double digested with EcoRI/HindIII, and cloned into pIB166 precut with the same enzymes. The recombinant plasmids pSK9 (fst-Sm in pIB166) and pSK10 (fst-Sm/srSm in pIB166) were sequenced on both strands for verification.
RT-PCR
To confirm transcription of the putative fst-Sm toxin, reverse transcription-PCR (RT-PCR) was performed using the primers CMT-497 and CMT-498 (Table 2). Total RNA was isolated from UA159 WT strain cultures at early log (OD600 ∼ 0.1), mid-log (OD600 ∼ 0.5), and early stationary (OD600 ∼ 1.5) phases using TRIzol reagent (Invitrogen), DNase treated with RQ1 DNase (Promega), and converted to cDNA by using a RevertAid H Minus First Strand cDNA Synthesis Kit (Fermentas). Negative controls without reverse transcriptase enzyme were included in all experiments. As a positive control, the same RT-PCR primers were used to directly PCR amplify the UA159 gDNA. PCR products of 99 bp were resolved on a 2% (wt/vol) agarose gel.
5′RACE-PCR
A 5′ rapid amplification of cDNA ends (5′RACE)-PCR technique was used to define the transcriptional start site of fst-Sm mRNA and srSm RNA. Primers used for the 5′RACE-PCR assays are listed in Table 2. The assays were carried out with total RNA isolated from mid-log phase (OD600 ∼ 0.5) UA159 WT cultures by using TRIzol reagent (Invitrogen). DNA-free RNA (5 µg) was reverse transcribed by using the RACE outer primers CMT-583 and CMT-585 for fst-Sm gene and srSm RNA, respectively, and RevertAid H Minus First Strand cDNA Synthesis Kit (Fermentas) according to supplier’s instructions. RNase H and RNase T1 (Ambion) were then added, followed by incubation at 37°C for 30 min. The cDNAs were purified by using a GeneJET PCR Purification Kit (Fermentas) according to the manufacturer’s instructions. Tailing of purified cDNA was performed using the terminal deoxynucleotidyl transferase (Invitrogen) and dGTP/dTTP according to the manufacturer’s instructions. Tailed cDNAs were then PCR amplified by using the RACE universal primers CMT-180 (Poly-G tail) and CMT-181 (Poly-T tail), and the RACE inner primers CMT-584 (fst-Sm gene) and CMT-586 (srSm RNA). The amplicons were analyzed by agarose gel electrophoresis, and sequenced using the RACE inner primers CMT-584 and CMT-586.
Northern Blot Analysis
Total RNA was isolated from UA159 WT strain cultures at early log (OD600 ∼ 0.1), mid-log (OD600 ∼ 0.5), early stationary (OD600 ∼ 1.5), and late stationary (OD600 ∼ 1.1) phases using TRIzol reagent (Invitrogen). Ten micrograms of total RNA was loaded by lane and resolved on a 12% (wt/vol) polyacrylamide denaturing gel containing 8 M urea. Size-fractionated RNA was transferred to a positively charged nylon membrane (Roche) using a Bio-Rad Mini Trans-Blot Cell and subjected to UV cross-linking. Membranes were pre-hybridized for 30 min at 42°C and followed by hybridization with biotin-labeled DNA probes (1 ng/ml for PCR probe and 10 ng/ml for oligoprobes) in ULTRAhyb hybridization buffer (Ambion) at 42°C overnight. The fst-Sm probe (99 bp DNA fragment) was PCR amplified from UA159 gDNA using CMT-497 and CMT-498 primers (Table 2) and labeled with Psoralen-biotin using a BrightStar Psoralen-Biotin Nonisotopic Labeling Kit (Ambion) according to supplier’s instructions. The biotin-labeled DNA oligoprobes CMT-558 and CMT-572 (Table 2) directed against the 5′ and 3′ UTR of srSm RNA, respectively, were purchased from Integrated DNA Technologies (Coralville, IA). The BrightStar BioDetect Kit (Ambion) was used for the detection of fst-Sm mRNA and srSm RNA following the manufacturer’s instructions.
RNA Half-life Determination
An overnight culture of S. mutans UA159 WT strain was diluted (1∶20) into fresh THYE broth and incubated at 37°C until mid-log phase (OD600 ∼ 0.5) was reached. Rifampicin was added to a final concentration of 300 µg/ml and total RNA was isolated from 20-ml aliquots of culture taken at 0, 2, 5, 10, 15, 20, 40, 60, 90, 120, and 150 min after the addition of rifampicin. Northern blotting was performed as described above. For the detection of fst-Sm mRNA and 5S rRNA, probes were PCR amplified from UA159 gDNA using the primer pairs CMT-497/CMT-498 and CMT-672/CMT-673, respectively, and labeled with Psoralen-biotin using a BrightStar Psoralen-Biotin Nonisotopic Labeling Kit (Ambion) according to supplier’s instructions. For the detection of srSm RNA, the biotin-labeled DNA oligoprobe CMT-558 purchased from Integrated DNA Technologies (Coralville, IA) was used. Quantification of the signal was performed using the ChemiDoc XRS System and Quantity One software provided by Bio-Rad.
Toxicity Assays in E. coli
Overnight cultures of LMG194(pSK1), LMG194(pSK2), and LMG194(pSK8) were diluted (1∶100) into fresh LB medium supplemented with kanamycin at 50 µg/ml and grown aerobically at 37°C. At an OD600 of ∼0.5, 0.2% (wt/vol) glucose (uninduced control) or 0.002% (wt/vol) arabinose was added to induce the expression of recombinant fusion proteins. Protein expression was verified by SDS-PAGE using the buffer system of Laemmli. The protein bands were visualized by staining with Coomassie brilliant blue. Aliquots of cultures were removed at 0, 15 min, 30 min, 45 min, 1 h, 2 h, and 3 h post-induction, serially diluted and plated on LB agar plates. Colonies were counted after 16 h of incubation at 37°C. All experiments were performed in triplicate from three independent experiments.
Toxicity Assays in S. mutans
Overnight cultures of S. mutans were diluted (1∶20) into fresh THYE broth and grown at 37°C until an OD600 of ∼0.1 was reached. Cultures were then divided into 0.5-ml aliquots: (i) pIB166 (control), (ii) pSK9 (fts-Sm in pIB166), (iii) pSK10 (fts-Sm/srSm in pIB166). Portions (1 µg) of plasmids were added to the cultures, which were grown for a further 2.5 h at 37°C in the presence of synthetic competence-stimulating peptide at a final concentration of 0.5 µg/ml. Cultures were serially diluted and plated on THYE agar plates. The transformation efficiency was calculated as the percentage of chloramphenicol-resistant transformants divided by the total number of recipient cells, which was determined by the number of CFU on antibiotic-free THYE agar plates. All assays were performed in triplicate from three independent experiments.
GFP-based Assay
Overnight cultures of E. coli strains harboring the GFP reporter constructs were diluted (1∶100) into fresh LB broth supplemented with chloramphenicol and kanamycin. Cells were grown aerobically at 37°C until an OD600 of ∼0.4 was reached (∼ 6×107 CFU/ml). Cells (2-ml aliquots) were harvested by centrifugation, washed once with sterile phosphate-buffered saline (PBS), and resuspended in 1 ml of sterile PBS. Green protein fluorescence was detected in terms of relative fluorescence units (RFU) with a fluorescence spectrophotometer. The excitation and emission filters were 488 nm and 511 nm. All experiments were performed in triplicate from two independent experiments.
Persistence Assay
Overnight cultures of S. mutans (WT vs. ΔIGR176; WT(pIB166) vs. ΔIGR176(pSK10)) were diluted (1∶20) into fresh THYE broth containing oxacillin (2 µg/ml), cefotaxime (2 µg/ml), or vancomycin (20 µg/ml) and incubated for 24 h at 37°C. Aliquots were removed at the indicated times, serially diluted, and plated on THYE agar plates. The colonies were counted after 48 h of incubation. All assays were performed in triplicate from three independent experiments.
Results
Detection of RNAs from the Intergenic Region IGR176
A study by Weaver et al. (2009) [30] previously identified pAD1-like TA systems on the chromosomes and plasmids of Enterococcus, Lactobacillus, and Staphylococcus species suggesting that Fst-like toxins may be prevalent in Gram-positive bacteria. Recently, an exhaustive PSI-BLAST and TBLAST searches across 774 bacterial genomes identified several homologs of type I toxins [4], . Analysis of the genome of S. mutans UA159 reference strain predicted a putative type I toxin belonging to the Fst family in the intergenic region IGR176. This small intergenic region (318 nt) is located between the genes SMU.219 and SMU.220 (Fig. 1A) encoding a Zn-dependent protease of the COG2856 family [8] and a hypothetical protein of unknown function, respectively. SMU.219 is mostly co-transcribed with an upstream gene, SMU.218, encoding a HTH domain-containing protein of the Xre family. The SMU.218/SMU.219 gene pair is predicted to function as bona fide type II TA system (TADB database: http://bioinfo-mml.sjtu.edu.cn/TADB/) [34].
Figure 1. Analysis of S. mutans IGR176 region.

(A) Schematic representation of the location of the fst-Sm/srSm locus on the S. mutans chromosome. Arrows indicate the direction of transcription. The fst-Sm and srSm promoter sequences are indicated by Pfst-Sm and PsrSm, respectively. A predicted stem-loop bidirectional terminator is indicated between srSm and fst-Sm. Shown at the bottom are the boundaries of the intergenic region IGR176. (B) Nucleotide and amino acid sequences of the S. mutans fst-Sm/srSm type I TA locus located in the intergenic region IGR176 (from 211452 to 211769) of UA159 genome. The conserved APUU(A/V)GUU motif present in Fst-Sm peptide is boxed. Putative promoter sites of fst-Sm toxin (–35, –10) and srSm antitoxin (–10), ribosome binding site (RBS) of Fst-Sm toxin, and a factor-independent bidirectional terminator (double underlined) are indicated. The transcriptional start site (+1) of fst-Sm and srSm identified by 5′ RACE-PCR are indicated below the sequence. The regions encoding the DRI and DRII repeats are boxed. The primers CMT-497 and CMT-498 used in the RT-PCR experiments are underlined. (C) Proposed RNA:RNA interactions (in shaded regions) between fst-Sm mRNA and srSm RNA.
Bioinformatic analysis revealed that the intergenic region IGR176 contains an unannotated open reading frame of 99 bp, which we named ORF176 (Fig. 1B). ORF176 encodes a putative peptide of 32 amino acid residues with a predicted MW of 3613.3 Da. The putative peptide showed homology to the Fst toxin family [4], [5]. The APUU(A/V)GUU motif (where U equals Ile, Leu, Val, or Phe) present in the hydrophobic region of the Fst family of RNA-regulated peptides [30] was also identified in the putative translated ORF176 peptide. We first determined whether or not an mRNA was transcribed by RT-PCR using gene-specific primers and RNA isolated from S. mutans WT cells during early-log, mid-log, and early stationary phases. A single PCR product of 99-bp corresponding to ORF176 was detected in all growth phases; the identity of the amplicon was confirmed by DNA sequencing (data not shown). To map the 5′ end of ORF176, we performed 5′RACE-PCR. The apparent start site (+1) was located 7 nucleotides 3′ proximal to a canonical –10 sequence (TATAAT). An obvious –35 sequence (TTGTTT) is also present spaced by 21 nucleotides from the –10 box (Fig. 1B). Taken together, these results provide genetic evidence for the presence of a previously unannotated fst-like toxin gene in the intergenic region IGR176 of S. mutans. We proposed the name fst-Sm to designate the gene encoding a putative Fst-like toxin in S. mutans, a predicted type I toxin.
We next hypothesized that the putative Fst-Sm toxin could be regulated post-transcriptionally by small RNA base pairing. Therefore, we performed a visual inspection of the DNA sequence immediately surrounding the fst-Sm toxin gene and searched for a sequence that would generate an RNA that could potentially bind the fst-Sm mRNA. A putative small RNA, which we named srSm (small RNA S. mutans), was found directly opposite the coding sequence of fst-Sm (Fig. 1B). The sequence was predicted to encode a small untranslated RNA as it contains potential –35 and –10 promoter elements but no open reading frame (ATG start codon) or ribosome binding site (RBS). To test further whether srSm is an untranslated RNA, we searched for open reading frames preceded with an alternative translational start codon (GTG or TTG). The fact that the longest open reading frame was only 10 codons in length (using TTG as putative initiation codon) and no putative RBS could be identified, suggest that srSm RNA is not translated. Using 5′RACE-PCR, we next determined the transcriptional start site of srSm. The srSm RNA starts at the G position, 108 bp upstream the fst-Sm stop codon (Fig. 1B) confirming transcription of srSm RNA. Moreover, the 5′ end mapping results indicate that both fst-Sm mRNA and srSm RNA are transcribed across direct tandem repeats, DRI and DRII (Fig. 1B), at which interactions mostly occur. The direct repeats overlap the start codon of fst-Sm mRNA leading most likely to suppression of translation.
Northern blotting was next performed to investigate expression of fst-Sm gene and srSm RNA during growth, and to determine the sizes of the transcripts produced from the fst-Sm/srSm locus. As shown in Fig. 2A, transcripts specific to fst-Sm and srSm RNAs were detected throughout S. mutans growth under nutrient-rich conditions. Interestingly, a smaller transcript was also observed for fst-Sm gene during late stationary growth phase suggesting that specific processing may also occur. Transcripts of ∼200 nt and ∼70 nt specific to fst-Sm mRNA and srSm RNA, respectively, were detected by Northern blot analysis (Fig. 2B). The signals obtained are in agreement with the predicted size of the RNAs inferred from the experimentally mapped +1 transcriptional start site of fst-Sm and srSm RNAs. Altogether, these results confirm that the intergenic region IGR176 expresses the fst-Sm gene, a previously unannotated open reading frame, and a cis-encoded small RNA. Consequently, the S. mutans fst-Sm/srSm locus encodes a predicted type I TA system.
Figure 2. Detection of RNAs in IGR176 by Northern blot analysis using biotin-labeled DNA probes.
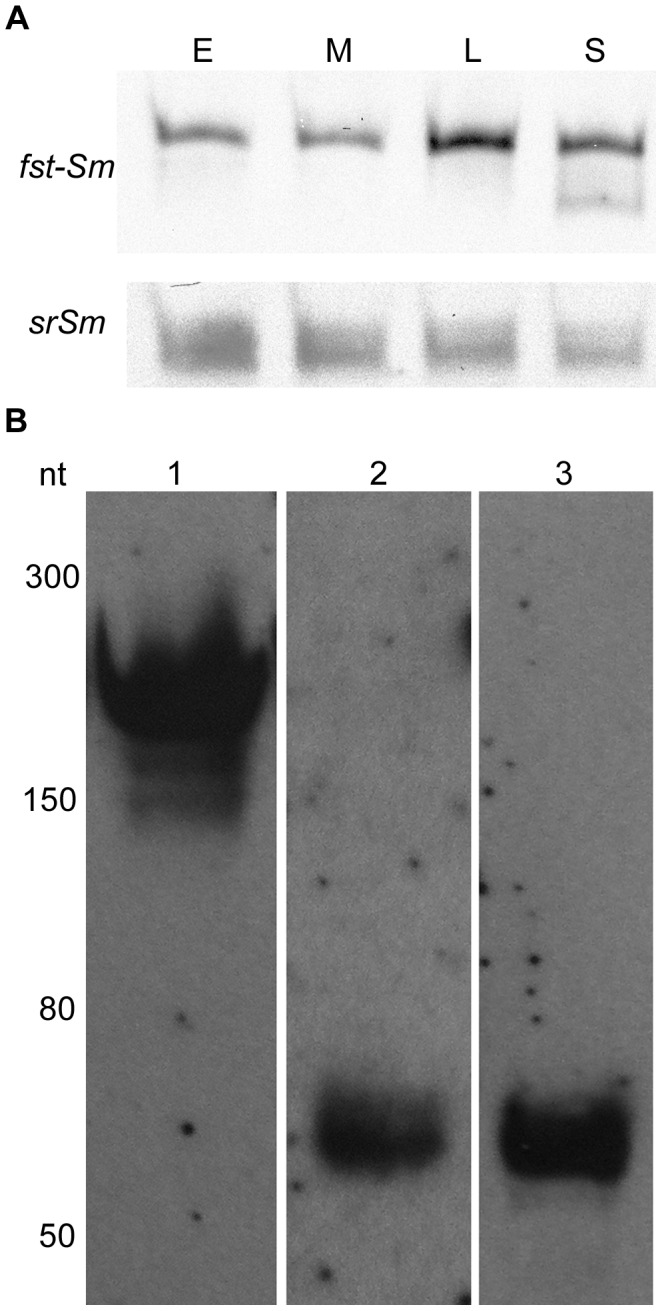
Total RNA from S. mutans WT strain was resolved on a 12% polyacrylamide denaturing gel containing 8 M urea. (A) Total RNA was extracted from UA159 WT cells during the early log (E), mid-log (M), late log (L), and stationary (S) phase of growth. The blot of fst-Sm was probed with a PCR-amplified double-stranded DNA that corresponds to the full-length coding region, while the blot of srSm was probed with CMT-558 DNA oligoprobe. The blots shown are representative of three independent experiments. (B) Total RNA isolated from UA159 WT cells grown to mid-log phase. fst-Sm mRNA was detected with a PCR-amplified double-stranded DNA that corresponds to the full-length coding region (lane 1). Oligoprobes CMT-558 (lane 2) and CMT-572 (lane 3) corresponding to the 5′ and 3′ UTR of srSm, respectively, were used to detect the srSm RNA. Low Range ssRNA Ladder (New England Biolabs) are indicated in nucleotides on the left.
Stability of the fst-Sm and srSm RNAs
The relative stabilities of both the toxin mRNA and antitoxin small RNA are an important aspect of TA regulation. In TA systems, the antitoxin is less stable than the toxin in the cell, and consequently it has to be constantly produced to inhibit the toxin [3], [35]. In order to test whether srSm RNA is less stable and might therefore represent an antisense RNA antitoxin, we determined the half-life of fst-Sm mRNA and srSm RNA by Northern blotting. WT strain was cultivated in THYE medium until mid-log growth phase was reached. Rifampicin was then added to prevent the initiation of RNA synthesis, time samples were taken, and RNA was extracted and subjected to Northern blot analysis. The half-life of srSm RNA was determined to be ∼ 30 min, while fst-Sm mRNA had a half-life of ∼ 90 min under the conditions tested (Fig. 3). Antisense RNAs of bacterial plasmids generally exhibit very short half-lives (<2 min), whereas long half-lives (20–60 min) were reported for regulatory sRNAs encoded by bacterial chromosomes [36], [37]. Northern blot analyses of fst-Sm transcript also indicated the presence of another RNA size in addition to the full-length transcript (Fig. 3A) suggesting that fst-Sm mRNA undergoes specific processing, most likely affecting its RNA stability. These data indicate that the fst-Sm toxin mRNA is much more stable than its srSm antitoxin RNA. These results are consistent with the findings of previous studies on other type I TA systems [3], [35].
Figure 3. RNA half-life determination.
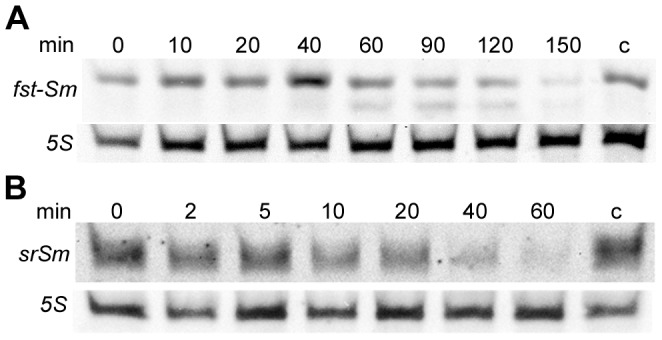
Stability of fst-Sm mRNA (A) and srSm RNA (B) by Northern blot analysis. Total RNA was extracted from WT mid-log cells at the indicated times after addition of 300 µg/ml rifampicin. Time points of sampling are indicated above each lane. Biotin-labeled DNA probes were used for RNA detection. The probing for 5S RNA confirmed equal loading. Control RNA extraction represents total RNA extracted from cells cultivated without rifampicin at time-point 150-min (fst-Sm mRNA detection) and 60-min (srSm RNA detection). Blots shown represent results from three experiments.
srSm RNA Expressed in Trans Represses GFP Expression
Our results suggest that fst-Sm/srSm locus encodes a type I TA system, in which srSm RNA antitoxin functions through an antisense base-pairing mechanism that results in inhibition of toxin translation. As both fst-Sm and srSm RNAs are transcribed across the direct tandem repeats DRI and DRII (5′ end mapping results, Fig. 1B), we hypothesized that srSm RNA (antisense) could bind to the DRI/DRII region in fst-Sm mRNA (sense) thereby controlling its expression (Fig. 1C). To test this hypothesis, we designed a two-plasmid gene reporter system to investigate whether the srSm RNA acts on the fst-Sm mRNA. The promoter of fst-Sm and its 5′ mRNA coding region with the DRI and DRII repeats were transcriptionally fused to gfp gene into the promoterless-GFP plasmid pSK3. The resulting recombinant plasmid, pSK4, was then introduced into an E. coli strain carrying the pHGS299 vector expressing srSm RNA (pSK6). Cells were grown until mid-log phase and fluorescence was measured. We reasoned that, if srSm repressed GFP expression in this E. coli strain, it would indicate that srSm RNA acts directly on the fst-Sm mRNA. Indeed, when srSm was introduced in E. coli(pSK4), approximately two-fold repression of GFP occurred (Fig. 4). We next cloned the promoter of fst-Sm and its 5′ mRNA coding region without the DRI/II repeats in frame with gfp. The recombinant plasmid, pSK5, was then introduced into E. coli(pSK6) and fluorescence was measured. No repression was observed compared with an E. coli strain containing the empty vector (Fig. 4). Based on the above data, we can predict that srSm directly binds to the DRI/II repeats to inhibit Fst-Sm expression.
Figure 4. The srSm RNA represses GFP expression.

Fluorescence from plasmid-encoded DRI/II-gfp (pSK4) or ΔDRI/II-gfp (pSK5) transcriptional fusions was measured in E. coli DH10B carrying pHSG299 vector expressing srSm (pSK6) or the empty vector. The promoterless GFP vector (pSK3) was used as negative control. All experiments were performed in triplicate from two independent experiments. The means ± SDs are shown.
Induced fst-Sm Expression Confers Toxicity in E. coli
The S. mutans fst-Sm/srSm locus was next investigated to determine whether it constitutes a functional type I TA system. The fst-Sm gene was cloned into the pBAD expression system for induction of gene expression using araBAD promoter dose-dependent regulation. The recombinant plasmid designated pSK1 was used to transform E. coli LMG194 strain. Overexpression of Fst-Sm resulted in inhibition of colony formation (data not shown). In cell viability assays, growth of E. coli was markedly affected when Fst-Sm was overexpressed from the araBAD promoter (Fig. 5). Overexpression of Fst-Sm resulted in a reduction of cell viability of ∼ 90% and ∼ 99% by 30 min and 45 min post-induction, respectively. By contrast, LMG194(pSK1) cultivated in presence of glucose (uninduced control) did not show any growth defects (Fig. 5). This indicated that Fst-Sm is a toxin. Furthermore, we constructed a plasmid for arabinose-inducible overexpression of a mutated Fst-Sm toxin. Single-site mutations of amino acid residues were introduced by two rounds of PCR in the conserved hydrophobic sequence of Fst toxin since this region of Fst has been shown to be important for Fst toxicity [30]. First, the conserved glycine at position 16 was changed to alanine. The alanine mutant (G16A) was then used to mutate alanine to glycine at position 11. The resulting pSK8 construction (G16A, A11G) was finally transferred to LMG194 strain and tested using our cell viability assay. As shown in Fig. 5, LMG194(pSK8) cultivated in liquid LB medium supplemented with arabinose did not present any growth defects and behaved similarly to the control curve (uninduced glucose control). These results highlight the importance of this conserved hydrophobic region in Fst-Sm toxicity.
Figure 5. Characterization of the Fst-Sm/srSm TA system in E. coli.

Cells of LMG194 containing pSK1 (Fst-Sm), pSK2 (Fst-Sm/srSm), and pSK8 (NT-Fst) were grown to mid-log phase, at which time arabinose (induced) and glucose (uninduced control) were added. After induction, appropriate dilutions were plated on LB agar for determination of the number of CFU per ml. The curves presented are the averages and standard deviations of results from three independent cultures.
We next measured the toxicity of Fst-Sm peptide when overexpressed in pBAD plasmid with its cis-encoded srSm RNA under the control of its own promoter. The effect of overexpressing Fst-Sm/srSm on growth was significantly less (∼ 25% reduced cell viability by 30 min post-induction), but Fst-Sm still affected growth (∼ 90% reduced cell viability by 3 h post-induction) most probably due to insufficient levels of srSm RNA transcribed from its own promoter (Fig. 5). Based on these results, we can conclude that Fst-Sm confers toxicity to E. coli and expression of its cis-encoded srSm is able to curb Fst toxicity.
Overexpression of fst-Sm is Lethal to S. mutans Cells
We next measured the toxicity of Fst-Sm toxin in S. mutans. We designed our toxicity assay based on the natural transformation ability of S. mutans [23]. The individual fst-Sm gene and the fst-Sm/srSm locus were placed under the control of the constitutive P23 promoter into the plasmid pIB166. Overnight cultures of S. mutans WT strain and its ΔIGR176 mutant were diluted in fresh medium and grown to early log phase prior to addition of pIB166 (empty plasmid), pSK9 (fst-Sm in pIB166), or pSK10 (fst-Sm/srSm in pIB166). Cells were grown for a further 2.5 h before differential plating. Our results showed that WT and ΔIGR176 mutant cells were readily transformed with pIB166 and pIB166 bearing fst-Sm/srSm locus. Indeed, the transformation efficiencies of WT and ΔIGR176 mutant for the empty vector were similar to the efficiencies obtained with the pSK10 construct (Table 3). This was not the case when the pSK9 construct was transferred to WT strain and ΔIGR176 mutant. The transformation efficiency of the WT strain was reduced by more than 1,000-fold for pSK9 bearing the fst-Sm toxin gene only, corresponding to less than 0.07% of the pIB166 control value. For ΔIGR176 mutant, less than 5 colonies were obtained when undiluted cells were plated (Table 3). The fact that the transformation efficiency was much higher for WT strain compared with the ΔIGR176 mutant for pSK9 (almost no colonies could be detected for ΔIGR176 after 48 h of incubation) suggested that the chromosomal copy of the srSm RNA (trans-encoded) in WT strain might confer some protective effects. These results clearly showed a growth-inhibitory effect of Fst-Sm on S. mutans cells and that the toxic effect elicited by Fst-Sm can be neutralized by coexpression of its srSm RNA. Hence, fst-Sm/srSm encodes a functional type I TA system in S. mutans.
Table 3. S. mutans toxicity assay based on natural competence.
| Construct | Description | Transformation Efficiency ± SDa | |
| WT | ΔIGR176 | ||
| pIB166 | Empty vector | (1.1±0.6) ×100 | (2.2±0.6) ×10−2 |
| pSK9 | fst-Sm gene | (8.5±1.5) ×10−4 * | <5 colonies ¶ |
| pSK10 | fst-Sm/srSm locus | (7.0±0.4) ×10−1 | (4.0±0.7) ×10−3 |
The transformation efficiency was expressed as the percentage of chloramphenicol-resistant transformants divided by the total number of recipient cells. All experiments were performed in triplicate from three independent experiments. Statistical significance: * WT(pSK9) vs. WT(pIB166); ¶ΔIGR176(pSK9) vs. ΔIGR176(pIB166).
Deletion and Mild-overexpression of IGR176 Region has no Effect on S. mutans Cell Growth
To study the cellular function of Fst-Sm/srSm system, we first investigated microbial growth kinetics of WT, ΔIGR176 mutant, and IGR176 complemented strains. The absence of Fst-Sm/srSm TA system or its mild-overexpression did not affect cell growth in liquid medium under the conditions tested (data not shown). No effect on cell growth was observed when S. mutans cells were exposed to extracellular recombinant Fst-Sm peptide at concentration up to 160 µg/ml. We next conducted a long-term survival assay using monocultures of WT and its ΔIGR176 mutant cultivated in a nutrient-rich medium (THYE) or a chemically defined medium (CDM). After 14 days, there was no difference in survival between WT and mutant strains under both nutrient growth conditions (data not shown). Biofilm formation was investigated using static biofilms developed in polystyrene microtiter plates. Assessment of the biofilm-forming capacity of ΔIGR176 mutant and IGR176 complemented strain relative to WT showed that both mutants formed stable and reproducible biofilms typical of the parent strain in ¼-THYE-sucrose and ¼-THYE-glucose. No significant differences in the total biomass of the mutant biofilms compared with the WT biofilms were noted for 24-h-, 48-h-, and 72-h-old biofilms (data not shown).
Ectopic Expression of Fst-Sm/srSm Type I TA Module Decreases Persister Cell Formation in S. mutans
Persister cells are phenotypic variants that are extremely tolerant to high concentrations of antibiotics. They make up a small part of the bacterial population [38]. In S. mutans, type II TA systems have been linked to persister formation. Indeed, mild-overexpression of the MazEF and RelBE type II TA modules resulted in an increase in multidrug-tolerant persister levels [24]. To examine whether the Fst-Sm/srSm type I TA system was also involved in persister cell formation, we first tested an IGR176 knockout mutant. As expected, deletion of Fst-Sm/srSm system did not affect the persister levels after challenge with a high dose of the cell wall synthesis inhibitors oxacillin, vancomycin, and cefotaxime (data not shown) due most likely to redundant pathways for bacterial persister formation. Similar results were obtained using the ΔmazEF and ΔrelBE deletion mutants [24], reinforcing the hypothesis that more than one single mechanism is responsible for persister formation. In contrast, the ectopic expression of Fst-Sm/srSm module caused a dramatic decrease in the number of persister cells. Our data showed that overexpression of Fst-Sm/srSm system decreased the number of oxacillin-tolerant (∼145-fold decrease at 24 h), cefotaxime-tolerant (∼240-fold decrease at 24 h), and vancomycin-tolerant (∼50-fold decrease at 24 h) persisters (Fig. 6). The fact that the MIC values of oxacillin, cefotaxime, and vancomycin against ΔIGR176(pSK10) mutant were not different from those for the WT strain confirmed that the complemented strain was a true persister mutant.
Figure 6. Effects of mild-overexpression of Fst-Sm/srSm type I TA system on S. mutans persister formation.
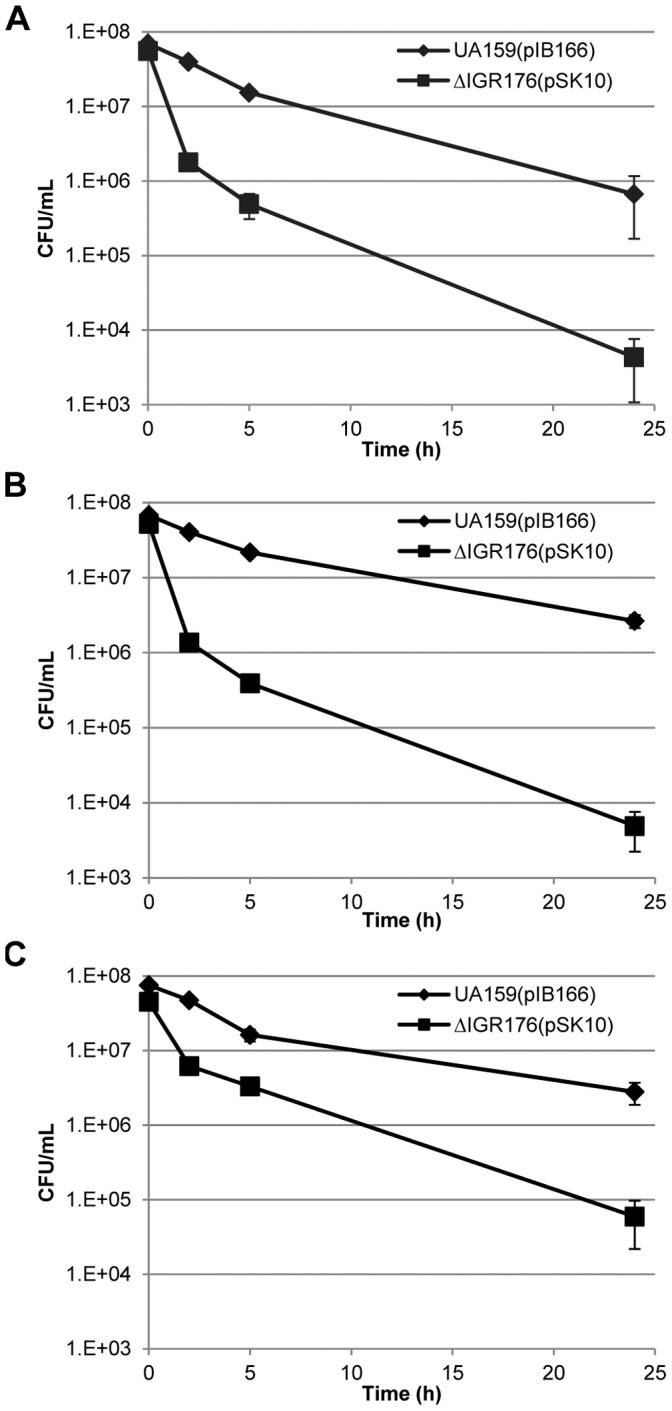
Oxacillin-treated (A), cefotaxime-treated (B), and vancomycin-treated (C) cells were removed at the indicated time points, serially diluted, spot plated onto THYE agar plates, and the number of CFU per ml was determined from plate counts. The curves presented are the averages and standard deviations of results from three independent cultures.
Discussion
TA systems are small genetic modules that are widespread in the prokaryotic kingdom [4], [8], [9]. There are of five different types depending of the nature and mode of action of the antitoxin component. The type III, IV, and V were discovered very recently and are represented so far by unique examples [6], [7], [11]–[13]. While type II TAs are highly represented in bacterial chromosomes, little is known about the distribution of type I TA loci. One possible reason could be that all the toxins of the type I TAs consist of very small hydrophobic peptides [3] and gene annotation software frequently fails to identify short protein-coding genes in microbial genomes [39]. Moreover, the development of computational approaches to identify novel type I TA systems is challenging owing to the short, hydrophobic character of the toxins, and the difficulty in predicting the antitoxin small RNAs [4]. In the present study, we reported to our knowledge the first functional type I TA system in streptococci. TAs are commonly described as constituents of the prokaryotic mobilome [4]. The chromosomal fst-Sm/srSm locus is located in a transposon-related genomic island of S. mutans UA159 strain suggesting that this type I TA module may have moved through horizontal gene transfer. IGR176 could not be found in other sequenced S. mutans genomes by searching public genomic sequence. This result was not surprising since transposon element movement is one of the major causes for intraspecies sequence variations in the intergenic regions. Collectively our results support that Fst-Sm/srSm forms a functional type I TA pair in the oral pathogen S. mutans. These results are: i) fst-Sm encodes a small hydrophobic peptide belonging to the Fst toxin family; ii) a cis-encoded small RNA was found converging towards the end of the toxin gene; iii) srSm antisense RNA is less stable than fst-Sm mRNA; iv) Fst-Sm has a toxic effect on S. mutans which can be neutralized by expression of srSm antitoxin; v) both fts-Sm mRNA and srSm RNA are transcribed across direct tandem repeats providing a region of complementarity between the srSm antisense RNA and the translation initiation region of fst-Sm mRNA; vi) srSm RNA most likely binds to the direct tandem repeats in the fst-Sm mRNA occluding Fst-Sm start codon. Although our results using the GFP constructs suggest that pairing of srSm RNA with the 5′ end of fst-Sm mRNA suppresses toxin translation, additional experiments will be required to determine if this pairing leads to translational silencing or mRNA degradation. It is also possible that degradation is secondary to translation inhibition [40], [41]. Indeed, the srSm:fst-Sm duplex is a good target for cleavage by RNase III, an endoribonuclease specific for double stranded RNA. Attempts to construct a S. mutans RNase III mutant were unsuccessful, suggesting that this may be an essential gene in S. mutans.
The toxic phenotype of many chromosomal type I toxins have been reported upon overexpression from a multicopy plasmid. So far all type I toxins characterized from E. coli are predicted to function through the same mechanism as phage holins, forming a pore to destroy the membrane potential and to inhibit ATP synthesis [42]–[45]. In E. faecalis, the intracellular overproduction of the plasmid-encoded Fst toxin compromised the integrity of the cell membrane and specific defects in chromosome partitioning and cell division were also observed [28], [29]. At present, the mechanism of action of Fst-Sm toxicity is not known. Our data suggest that Fst-Sm functions intracellularly since extracellular addition of recombinant Fst-Sm toxin to cells in cultures had no effect on growth of S. mutans and E. coli. Preliminary examination of Fst-induced E. coli cells by scanning electron microscopy showed no evidence of ghost cell formation or any other abnormal cellular morphology compared with the non-toxic Fst-induced cells (data not shown). Further experiments will be required to identify the primary target of Fts-Sm toxin in its native host. For instance, construction of a tightly regulated inducible expression system for S. mutans will be necessary to study the effects of Fst-Sm induction on S. mutans cellular morphology. It would also be interesting to investigate whether Fst-Sm toxin could affect cell division by inhibiting the polymerization of S. mutans cytoskeleton proteins (e.g., FtsQ, FtsA, FtsZ) in future studies.
The physiological function of chromosomal TA systems remains unclear. In fact, the functions of chromosomal TAs are diverse and may depend on the type of TA, its genomic location, and host species. Several roles have been discovered for type II TA systems, including stress survival, growth control, programmed cell death, persister formation, stabilization of the genome, biofilm formation, phage abortive infection, and anti-addiction system [1], [2], [15], [46]. We previously demonstrated that ectopic mild-overexpression of type II TA systems generated greater number of persister cells in S. mutans. The picture that emerged from our work and from several other studies is clearly pointing out to a multi-gene function and suggests that S. mutans did not evolve a dedicated mechanism allowing it to adopt a persistence phenotype [24]. Not surprisingly, the deletion of fst-sm/srSm locus did not affect the persister levels (data not shown). To date, the E. coli MqsR/MqsA type II TA is the only system that affects (decreases) persister formation upon deletion [47]. Our prediction was that mild-overexpression of Fst-Sm/srSm system would increase persister levels, promoting antibiotic tolerance. Quite to the contrary, when we overexpressed Fst-Sm/srSm system, we observed a decrease in persister levels surviving cell wall-acting antibiotic treatments. In a typical persister experiment, a biphasic killing curve is obtained consisting of an initial drop in viable cell counts (the antimicrobial susceptibility of the bulk of the population) followed by a surviving subpopulation of persisters that decreased slowly over time. The decrease in persister levels obtained when the Fst-Sm/srSm system is overexpressed could be related to persister cells awakening from dormancy. These persister cells that start to grow in the presence of the antibiotic are then killed more rapidly. Indeed, the second phase revealing the resuscitation rate of persisters produced by the overexpressing mutant has a steeper slope in comparison to the WT control curve. Assuming that persister cells could provide a reservoir of viable bacteria that can acquire resistance by random mutation or horizontal gene transfer [17], the results from this study reinforce the idea that TA systems represent an attractive target for designing new drugs that could kill persister cells that have woken up. The clinical implications of TA systems and persister cell formation in the context of chronic infections are thus highly significant.
Acknowledgments
We thank Delphine Dufour and Vincent Leung for careful reading of the manuscript. CML is a recipient of a Canada Research Chair.
Funding Statement
This work was supported by a Canadian Institutes of Health Research grant MOP-93555 to CML and a Natural Sciences and Engineering Research Council of Canada grant RGPIN 355968 to CML. CML is a recipient of a Canada Research Chair. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Yamaguchi Y, Park JH, Inouye M (2011) Toxin-antitoxin systems in bacteria and archae. Annu Rev Genet 45: 61–79. [DOI] [PubMed] [Google Scholar]
- 2. Van Melderen L (2010) Toxin-antitoxin systems: why so many, what for? Curr Opin Microbiol 13: 781–785. [DOI] [PubMed] [Google Scholar]
- 3. Fozo EM, Hemm MR, Storz G (2008) Small toxic proteins and the antisense RNAs that repress them. Microbiol Mol Biol Rev 72: 579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fozo EM, Makarova KS, Shabalina SA, Yutin N, Koonin EV, et al. (2010) Abundance of type I toxin-antitoxin systems in bacteria: searches for new candidates and discovery of novel families. Nucleic Acids Res 38: 3743–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kwong SM, Jensen SO, Firth N (2010) Prevalence of Fst-like toxin-antitoxin systems. Microbiology 156: 975–977. [DOI] [PubMed] [Google Scholar]
- 6. Fineran PC, Blower TR, Foulds IJ, Humphreys DP, Lilley KS, et al. (2009) The phage abortive infection system, ToxIN, functions as a protein-RNA toxin-antitoxin pair. Proc Natl Acad Sci U S A 106: 894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blower TR, Short FL, Rao F, Mizuguchi K, Pei XY, et al. (2012) Identification and classification of bacterial type III toxin-antitoxin systems encoded in chromosomal and plasmid genomes. Nucleic Acids Res 40: 6158–6173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Makarova KS, Wolf YI, Koonin EV (2009) Comprehensive comparative-genomic analysis of type 2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes. Biol Direct 4: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leplae R, Geeraerts D, Hallez R, Guglielmini J, Drèze P, et al. (2011) Diversity of bacterial type II toxin-antitoxin systems: a comprehensive search and functional analysis of novel families. Nucleic Acids Res 39: 5513–5525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Syed MA, Lévesque CM (2012) Chromosomal bacterial type II toxin-antitoxin systems. Can J Microbiol 58: 553–562. [DOI] [PubMed] [Google Scholar]
- 11. Tan Q, Awano N, Inouye M (2011) YeeV is an Escherichia coli toxin that inhibits cell division by targeting the cytoskeleton proteins, FtsZ and MreB. Mol Microbiol 79: 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Masuda H, Tan Q, Awano N, Wu KP, Inouye M (2012) YeeU enhances the bundling of cytoskeletal polymers of MreB and FtsZ, antagonizing the CbtA (YeeV) toxicity in Escherichia coli . Mol Microbiol 84: 979–989. [DOI] [PubMed] [Google Scholar]
- 13.Wang X, Lord DM, Cheng HY, Osbourne DO, Hong SH, et al.. (2012) A new type V toxin-antitoxin system where mRNA for toxin GhoT is cleaved by antitoxin GhoS. Nat Chem Biol doi: 10.1038/nchembio.1062. [DOI] [PMC free article] [PubMed]
- 14. Gerdes K, Christensen SK, Lobner-Olesen A (2005) Prokaryotic toxin-antitoxin stress response loci. Nat Rev Microbiol 3: 371–382. [DOI] [PubMed] [Google Scholar]
- 15.Hu MX, Zhang X, Li EL, Feng YJ (2010) Recent advancements in toxin and antitoxin systems involved in bacterial programmed cell death. Int J Microbiol doi:10.1155/2010/781430. [DOI] [PMC free article] [PubMed]
- 16. Van Melderen L, Saavedra De Bast M (2009) Bacterial toxin-antitoxin systems: more than selfish entities? PLoS Genet 5: e1000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fauvart M, De Groote VN, Michiels J (2011) Role of persister cells in chronic infections: clinical relevance and perspectives on anti-persister therapies. J Med Microbiol 60: 699–709. [DOI] [PubMed] [Google Scholar]
- 18. Shah D, Zhang Z, Khodursky A, Kaldalu N, Kurg K, et al. (2006) Persisters: a distinct physiological state of E. coli. . BMC Microbiology 6: 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gerdes K, Maisonneuve E (2012) Bacterial persistence and toxin-antitoxin loci. Annu Rev Microbiol 66: 103–123. [DOI] [PubMed] [Google Scholar]
- 20. Smith EG, Spatafora GA (2012) Gene regulation in S. mutans: complex control in a complex environment. J Dent Res 91: 133–141. [DOI] [PubMed] [Google Scholar]
- 21. Fitzgerald RJ, Keyes PH (1960) Demonstration of the etiologic role of streptococci in experimental caries in the hamster. J Am Dent Assoc 61: 9–19. [DOI] [PubMed] [Google Scholar]
- 22. Takahashi N, Nyvad B (2008) Caries ecology revisited: microbial dynamics and the caries process. Caries Res 42: 409–418. [DOI] [PubMed] [Google Scholar]
- 23. Syed MA, Koyanagi S, Sharma E, Jobin MC, Yakunin AF, et al. (2011) The chromosomal mazEF locus of Streptococcus mutans encodes a functional type II toxin-antitoxin addiction system. J Bacteriol 193: 1122–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Leung V, Lévesque CM (2012) A stress-inducible quorum-sensing peptide mediates the formation of persister cells with noninherited multidrug tolerance. J Bacteriol 194: 2265–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gerdes K, Larsen JE, Molin S (1985) Stable inheritance of plasmid R1 requires two different loci. J Bacteriol 161: 292–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Weaver KE, Jensen KD, Colwell A, Sriram SI (1996) Functional analysis of the Enterococcus faecalis plasmid pAD1-encoded stability determinant par . Mol Microbiol 20: 53–63. [DOI] [PubMed] [Google Scholar]
- 27. Greenfield TJ, Ehli E, Kirshenmann T, Franch T, Gerdes K, et al. (2000) The antisense RNA of the par locus of pAD1 regulates the expression of a 33-amino acid toxic peptide by an unusual mechanism. Mol Microbiol 37: 652–660. [DOI] [PubMed] [Google Scholar]
- 28. Weaver KE, Weaver DM, Wells CL, Waters CM, Gardner ME, et al. (2003) Enterococcus faecalis plasmid pAD1-encoded Fst toxin affects membrane permeability and alters cellular responses to lantibiotics. J Bacteriol 185: 2169–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Patel S, Weaver KE (2006) Addiction toxin Fst has unique effects on chromosome segregation and cell division in Enterococcus faecalis and Bacillus subtilis . J Bacteriol 188: 5374–5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Weaver KE, Reddy SG, Brinkman CL, Patel S, Bayles KW, et al. (2009) Identification and characterization of a family of toxin-antitoxin systems related to the Enterococcus faecalis pAD1 par addiction module. Microbiology 155: 2930–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dufour D, Cordova M, Cvitkovitch DG, Lévesque CM (2011) Regulation of the competence pathway as a novel role associated with a streptococcal bacteriocin. J Bacteriol 193: 6552–6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miller WG, Leveau JH, Lindow SE (2000) Improved gfp and inaZ broad-host-range promoter-probe vectors. Mol Plant Microbe Interact 13: 1243–1250. [DOI] [PubMed] [Google Scholar]
- 33. Biswas I, Jha JK, Fromm N (2008) Shuttle expression plasmids for genetic studies in Streptococcus mutans . Microbiology 154: 2275–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shao Y, Harrison EM, Bi D, Tai C, He X, et al. (2011) TADB: a web-based resource for Type 2 toxin-antitoxin loci in bacteria and archaea. Nucleic Acids Res 39: D606–D611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brantl S (2002) Antisense RNAs in plasmids: control of replication and maintenance. Plasmid 48: 165–173. [DOI] [PubMed] [Google Scholar]
- 36. Vogel J, Bartels V, Tang TH, Churakov G, Slagter-Jäger JG, et al. (2003) RNomics in Escherichia coli detects new sRNA species and indicates parallel transcriptional output in bacteria. Nucleic Acids Res 31: 6435–6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wagner EGH, Flärdh K (2002) Antisense RNAs everywhere? Trends Genet 18: 223–226. [DOI] [PubMed] [Google Scholar]
- 38. Lewis K (2010) Persister cells. Annu Rev Microbiol 64: 357–372. [DOI] [PubMed] [Google Scholar]
- 39. Overbeek R, Bartels D, Vonstein V, Meyer F (2007) Annotation of bacterial and archaeal genomes: improving accuracy and consistency. Chem Rev 107: 3431–3447. [DOI] [PubMed] [Google Scholar]
- 40. Evguenieva-Hackenberg E, Klug G (2011) New aspects of RNA processing in prokaryotes. Curr Opin Microbiol 14: 587–592. [DOI] [PubMed] [Google Scholar]
- 41. Gottesman S (2005) Micros for microbes: non-coding regulatory RNAs in bacteria. Trends Genet 21: 399–404. [DOI] [PubMed] [Google Scholar]
- 42. Fozo EM, Kawano M, Fontaine F, Kaya Y, Mendieta KS, et al. (2008) Repression of small toxic protein synthesis by the Sib and OhsC small RNAs. Mol Microbiol 70: 1076–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kawano M, Aravind L, Storz G (2007) An antisense RNA controls synthesis of an SOS-induced toxin evolved from an antitoxin. Mol Microbiol 64: 738–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kawano M, Oshima T, Kasai H, Mori H (2002) Molecular characterization of long direct repeat (LDR) sequences expressing a stable mRNA encoding for a 35-amino-acid cell-killing peptide and a cis-encoded small antisense RNA in Escherichia coli . Mol Microbiol 45: 333–349. [DOI] [PubMed] [Google Scholar]
- 45. Unoson C, Wagner EGH (2008) A small SOS-induced toxin is targeted against the inner membrane in Escherichia coli . Mol Microbiol 70: 258–270. [DOI] [PubMed] [Google Scholar]
- 46. Wang X, Wood TK (2011) Toxin-antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl Environ Microbiol 77: 5577–5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kim Y, Wood TK (2010) Toxins Hha and CspD and small RNA regulator Hfq are involved in persister cell formation through MqsR in Escherichia coli . Biochem Biophys Res Commun 391: 209–213. [DOI] [PMC free article] [PubMed] [Google Scholar]