

SUMMARY
The genetic diversity of Streptococcus mutans has been extensively studied using a variety of genotyping methods. Repetitive extragenic palindromic-polymerase chain reaction (rep-PCR) is a genotyping approach used for screening large numbers of bacterial isolates. This 2-part study used multilocus sequence typing (MLST) analysis to evaluate genotypes previously identified as unique using rep-PCR. In part one an isolate was selected from each of the 22 S. mutans rep-PCR genotype groups representing 8,000 clinical isolates. For part two, four additional isolates were selected from the 6 most commonly occurring genotype groups (GG) for further analysis. Real-time PCR was performed using eight housekeeping S. mutans gene loci and the amplicons sequenced. Sequence data analysis was performed using CLC DNA Workbench and alleles were compared with the PubMLST database for Oral Streptococcus using the Nakano scheme. Concatenated sequences were evaluated with MEGA using minimum evolution method with bootstrap.
All 22 rep-PCR genotypes were unique by MLST analysis. Within rep-PCR GGs, MLST matched rep-PCR in 3 groups demonstrating clonality; 3 groups exhibited more diversity with MLST. The discovery of 3 clonal groups is unique to this study and suggests that S. mutans genotypes are shared between unrelated subjects. Furthermore, MLST defined 19 new alleles and 26 new STs that have been confirmed and registered with PubMLST. Methods for processing were streamlined and a process for using MLST with rep-PCR is suggested. In conclusion, MLST verified that rep-PCR is a reliable and cost effective method for screening large numbers of S. mutans strains for epidemiological study.
Keywords: molecular oral microbiology, dental caries, Streptococcus mutans, genotypes, DiversiLab, phylogenetics
INTRODUCTION
Dental caries is a global infectious disease. The multi-factorial risk elements of dental caries make prevention efforts difficult, particularly in lower socio-economic groups where dietary factors, oral healthcare education and access to regular dental care are inadequate (Beltran-Aguilar et al. 2005). The mutans streptococci (MS), i.e., Streptococcus mutans and Streptococcus sobrinus are the main etiological agents associated with dental caries with S. mutans most commonly implicated (Loesche 1986). The genetic diversity of S. mutans has been widely studied, but a longitudinal view is necessary to develop effective caries risk assessment tools and treatments.
Many methods for genotyping have been utilized to determine genetic diversity of S. mutans including arbitrarily primed-polymerase chain reaction (AP-PCR)(Li and Caufield 1995; Mitchell et al. 2009), randomly amplified polymorphic DNA (RAPD)(Redmo Emanuelsson et al. 2003), and restriction endonuclease analysis (Kulkarni et al. 1989). Pulsed-field gel electrophoresis (PFGE), a method that has served as the “gold standard” for genetic fingerprinting with improved reproducibility due to “in gel” DNA extraction and reaction with restriction enzymes, was used to evaluate mother-to-child transmission of S. mutans (Goering 2000; Mitchell et al. 2009). PFGE, like other gel-based genotyping procedures, is expensive and labor intensive making successful application impractical for high-throughput studies. Importantly, these gel-based methods have less longitudinal discriminatory power for population and evolutionary studies than a typing scheme that compares isolates at the sequence level (Maiden 2006).
Repetitive extragenic palindromic-polymerase chain reaction (rep-PCR) is an extragenic typing method commonly used to profile bacterial isolates. Standardized rep-PCR is more rapid and economical than other forms of gel electrophoresis, yet it remains subject to the same concerns with discrimination in genetic profiling (Maiden 2006). A standardized commercial method for performing rep-PCR has been developed by DiversiLab including a web-based interface and analysis software (Healy et al. 2005). This system has been used to evaluate several pathogenic organisms including Salmonella enterica, Escherichia coli, and methicillin-resistant Staphylococcus aureus. (Ross et al. 2005; Doleans-Jordheim et al. 2009; Te Witt et al. 2009; Tenover et al. 2009; Kilic et al. 2010). Recently, S. mutans genotypes have also been evaluated with this system (Moser et al. 2010; Cheon et al. 2011), demonstrating its usefulness and consistency in identifying unique genotypes.
Multilocus sequence typing (MLST) is a molecular typing method that compares the nucleotide sequences of 6–10 housekeeping genes (intragenic) for phylogenetic analysis. MLST has the potential for greater discriminatory power since data is generated directly from nucleotide sequences of conserved genes responsible for basic metabolic functions. MLST provides an effective means of data collection, analysis and sharing through Internet databases (Kilian et al. 2006; Maiden 2006). It is easily reproducible; however, it is labor and cost intensive.
A MLST scheme for the analysis of S. mutans was developed in an evolutionary study of S. mutans serotypes using 8 housekeeping genes loci selected from the genomic sequence of S. mutans UA159 (Nakano et al. 2007). These loci were later used to evaluate mother-to-child transmission of S. mutans in a small collection of clinical isolates (Lapirattanakul et al. 2008). However, no studies have compared rep-PCR with MLST using S. mutans clinical isolates. Such a comparison is useful since all genotyping method should be confirmed by an alternate method (Foley et al. 2006; Van Bambeke 2006).
For this study, MLST analysis was used to evaluate commonality among and consistency between S. mutans isolates previously identified as unique “genotypes” using rep-PCR. In addition, a recommendation is made for the collaborative use of rep-PCR and MLST for the determination of S. mutans genotypes in a large-scale epidemiological study.
MATERIALS & METHODS
Sample Selection and Processing
S. mutans strains were collected from 83 school children (5–6 year old) and 200 household family members in Uniontown, Alabama with informed consent provided in accordance with the regulations established by the University of Alabama at Birmingham (UAB) Institutional Review Board. Uniontown was selected as a high caries risk population because it is a rural isolated African-American community with limited access to dental health care. Plaque, saliva and tongue samples were obtained from children; plaque and tongue samples from household family members.
Samples were transported to the UAB School of Dentistry for processing within 24 hours. Sample processing included selection of individual presumed S. mutans colonies based on morphology following incubation on Gold’s Media (Gold et al. 1973). Individual colonies were inoculated into Todd Hewitt Broth (THB, Beckton Dickinson, Sparks, Maryland) then incubated anaerobically (10% H2, 10% CO2, 80% N2) at 37°C for 24–48 hours. Isolates were stored at −80°C until processed.
DNA was extracted from isolates, confirmed as S. mutans by SYBR Green real-time PCR and analyzed with rep-PCR analysis to determine a library of possible genotypes as previously described by Moser et al. Briefly, the DiversiLab system was used for rep-PCR typing using the Streptococcus DNA fingerprinting kit (bioMerieux, Durham, NC, USA). Data analysis was performed by DiversiLab Web-based software. Distinct genotypes were determined based on a >2 minor (<100 fluorescence units) band difference or 1 major band (>100 fluorescence units) difference (Moser et al. 2010).
For MLST analysis, a two-part study was designed. Twenty-two unique rep-PCR library genotypes have been established from over 8,000 S. mutans isolates analyzed in an ongoing longitudinal study. For part one, a representative isolate or “library strain” was selected for each library (L) genotype (n=22, from 21 individual subjects) for MLST analysis to evaluate uniqueness of genotypes (Figure 1, Table S1).
Fig. 1.

Rep-PCR Percent Similarity Index Dendrogram with Virtual Gel Images. Rep-PCR genotypes (GT) and MLST Strain Types (ST) noted. Genotype groups partitioned with unique Library Isolates between. ID designations: L = Library Isolates, G = additional isolates.
In part two, 6 subgroups of genotypes from the library were evaluated to determine similarity of MLST results within genotype groups (GG). Four additional isolates from each GG were selected for MLST analysis(total n= 24) (Figure 1, Table S2). These GG were the 6 most commonly occurring genotypes (G1, G6, G11, G13, G18 and G22) representing 71% (975 isolates from 55 children) of all S. mutans isolates obtained from dental plaque in children at baseline and 6 months. Isolates chosen were from 16 individual subjects (including the 6 library isolates). G1, G6, G13 and G18 isolates were from 3 subjects; G11 and G22 were from 2 subjects (selected isolates matched the library isolates). Each group consisted of the library isolate and 2 isolates from two subjects in order to evaluate rep-PCR genotypes within the same individual and between unrelated subjects.
PCR for MLST and Sequence Analysis
Extracted DNA was diluted to 20ng/μl for MLST analysis. The primer sets used for the eight housekeeping S. mutans UA159 gene loci were: tkt (transketolase), glnA (glutamine synthetase subunit 1a), gltA (glutamate synthase), glk (glucose kinase), gyrA (DNA gyrase subunit A), aroE (shikimate 5-dehydrogenase), murI (glutamate racemase), and lepC (signal peptidase 1) (Nakano et al. 2007). A 25μl reaction was prepared for PCR amplification using 2X Maxima™ SYBR Green qPCR master mix (Thermo Scientific, Lafayette, CO) for each housekeeping gene primer set. Amplification was performed as follows: 10 min at 95°C; 30 cycles of 30 sec at 95°C, 30 sec at 55°C, 45 sec at 72°C, followed by 7 min at 72°C. Melting curve analysis was performed at 55°C. Positive control S. mutans UA159 and negative control 6715 (S. sobrinus) were used. Threshold cycle (Cq) values between 8–18 with a melting temperature (Tm) matching positive controls were considered acceptable.
PCR products were purified using Ultra Clean PCR Clean-up Kit (MoBio Laboratories, Inc., Carlsbad, CA) according to the manufacturer’s protocol. Final PCR products were eluted using 50μl molecular grade ddH2O and stored until ready for sequencing at −20°C.
Sequencing was performed by the UAB Heflin Genetics Core Lab using the Big Dye Terminator v3.1 Ready Reaction kit (Applied Biosystems, Foster City, CA). The same primers used for MLST PCR were used for sequencing.
DNA Sequence Data Analysis
Sequence data analysis was performed using CLC DNA Workbench software version 5.7.1 with MLST module (CLC bio USA, Cambridge, MA). Raw sequence data for each primer set were assembled into isolates, trimmed and checked for sequencing errors by alignment with a reference strain of UA159 obtained from Kyoto Encyclopedia of Genes and Genomes (KEGG). Sequence data for each allele and Sequence Type (STs) were checked in the PubMLST database for Oral Streptococcus using the Nakano scheme (www.pubmlst.org). For existing alleles and STs, corresponding numeric designation were assigned. New alleles were repeated from PCR through sequencing then confirmed and registered with PubMLST. STs with up to 2 allelic differences were considered to be in the same clonal complex (Nakano et al. 2007). Allelic profiles were exported to the Sequence Type Analysis and Recombinational Tests Version 2 (START2) software for Un-weighted Pair Group Method using Arithmetic averages (UPGMA) alignments (Jolley et al. 2001).
The 8 gene loci were concatenated (3366 bp) and exported to Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0 for phylogenetic analysis by Minimum Evolution (ME) algorithm with bootstrap test (1000 replicates) (Tamura et al. 2007). Concatenated sequence data was also analyzed on the DiveIn website to generate informative site (conflict sites occurring in 2 or more strains) and private site (conflict sites occurring in only 1 strains) data (Deng et al. 2010).
RESULTS
A total of 47 S. mutans isolates were analyzed with both rep-PCR and MLST. MLST data was compared with sequence data available on PubMLST resulting in the discovery of 19 new alleles and 26 new STs (Table 1). Twelve of the new STs were the result of new combinations of previously published alleles. The remaining 14 STs contained new alleles.
Table 1.
New Sequence Types (STs) and Alleles Registered with PubMLST.
| PubMLST ST | Isolate ID | tkt | glnA | gltA | glk | aroE | murI | lepC | gyrA |
|---|---|---|---|---|---|---|---|---|---|
| 148 | L-2 | 1 | 1 | 1 | 1 | 11 | 3 | 3 | 6 |
| 149 | L-6 | 1 | 1 | 5 | 8 | 26 | 3 | 1 | 1 |
| 150 | G6-1 | 1 | 1 | 1 | 1 | 2 | 3 | 1 | 1 |
| 151 | G6-3 | 19 | 2 | 10 | 8 | 8 | 3 | 5 | 1 |
| 152 | L-7 | 2 | 3 | 25 | 8 | 11 | 2 | 5 | 4 |
| 153 | L-8 | 1 | 1 | 31 | 1 | 27 | 2 | 11 | 1 |
| 154 | L-9 | 16 | 2 | 15 | 8 | 1 | 2 | 11 | 3 |
| 155 | L-10 | 1 | 2 | 15 | 8 | 1 | 11 | 11 | 1 |
| 156 | L-11 | 3 | 8 | 15 | 8 | 2 | 5 | 21 | 1 |
| 157 | L-12 | 2 | 2 | 4 | 5 | 4 | 5 | 1 | 1 |
| 158 | L-13 | 20 | 2 | 24 | 8 | 4 | 3 | 1 | 1 |
| 159 | G13-1 | 18 | 23 | 1 | 23 | 14 | 21 | 11 | 21 |
| 160 | G13-3 | 1 | 1 | 5 | 8 | 4 | 3 | 31 | 1 |
| 161 | G13-4 | 1 | 1 | 5 | 24 | 4 | 3 | 31 | 1 |
| 162 | L-14 | 1 | 3 | 15 | 8 | 4 | 2 | 19 | 1 |
| 163 | L-15 | 1 | 1 | 32 | 8 | 4 | 19 | 5 | 4 |
| 164 | L-16 | 1 | 8 | 33 | 8 | 4 | 3 | 11 | 1 |
| 165 | L-17 | 18 | 3 | 1 | 1 | 2 | 3 | 32 | 1 |
| 166 | L-18 | 3 | 24 | 5 | 25 | 1 | 1 | 30 | 15 |
| 167 | L-19 | 1 | 16 | 23 | 1 | 11 | 3 | 1 | 15 |
| 168 | L-20 | 3 | 2 | 15 | 1 | 28 | 3 | 11 | 1 |
| 169 | L-21 | 1 | 1 | 1 | 1 | 2 | 3 | 11 | 1 |
| 170 | G22-1 | 3 | 10 | 15 | 24 | 21 | 2 | 1 | 1 |
| 171 | G22-2 | 3 | 10 | 15 | 8 | 21 | 2 | 1 | 1 |
| 172 | L-23 | 21 | 3 | 20 | 4 | 27 | 5 | 1 | 1 |
| 173 | L-24 | 3 | 21 | 1 | 21 | 2 | 4 | 11 | 19 |
A total of 26 new STs were confirmed and registered with PubMLST. The 19 new alleles are highlighted. Other new STs are the result of new combinations of previously registered alleles. Isolates identified “L” are Library strains; “G” are genotype group strains.
Figure 1 shows the rep-PCR dendrogram based on percent similarity with virtual gel images for both library and GGs isolates. The rep-PCR genotype (GT) and the MLST ST (ST) are listed. For the 22 rep-PCR library genotypes, a total of 22 unique MLST STs were found (Figure 1, Table S1). Library strain L5 and S. mutans control UA159 were both ST 1. Overall, sequences in gyrA and glk loci had the least variation; glt and lepC loci the greatest variation for genotypes analyzed by MLST.
MLST analysis of the 30 isolates from the 6 most common GGs (L1, L6, L11, L13, L18, and L22) resulted in the addition of 7 new STs (Figure 1, Table S2). For groups L1, L11, and L18 the 4 additional isolates had identical STs to the library isolate. However, additional STs were found for groups L6, L13, and L22 with 3–8 allelic differences each. STs were consistent for isolates obtained from the same subject.
Concatenated sequences (3,366 bp fragment) were aligned with S. mutans reference strain UA159. A total of 72 base pair (bp) differences were observed and ranged between 6 (gyrA) to 14 (glt) conflict sites per loci. Conflict sites represented 1.38% (gyrA) to 3.60% (glt) of the total base-pair content per gene. Fifty-five informative (conflict) sites occurred in 2 or more sequences that varied from UA159. Seventeen were private sites.
Figure 2 represents the 22 Library strains based on final concatenated sequences analyzed with MEGA software. The percentage of replicate trees in which the taxa clustered together is indicated on the branches. Bootstrap values varied from 0–61 indicating no statistical significance of the clusters between isolates except for L-5 and UA159. Figure 3 represents MEGA analysis for GG, which demonstrated significant clonal clusters for groups L1, L11, and L18. Groups L6, L13, and L22 were further diversified. Isolates from the same individual clustered together. These findings were consistent with those observed with START2 analysis based on UPGMA evaluation of allelic profiles (not shown).
Fig. 2.
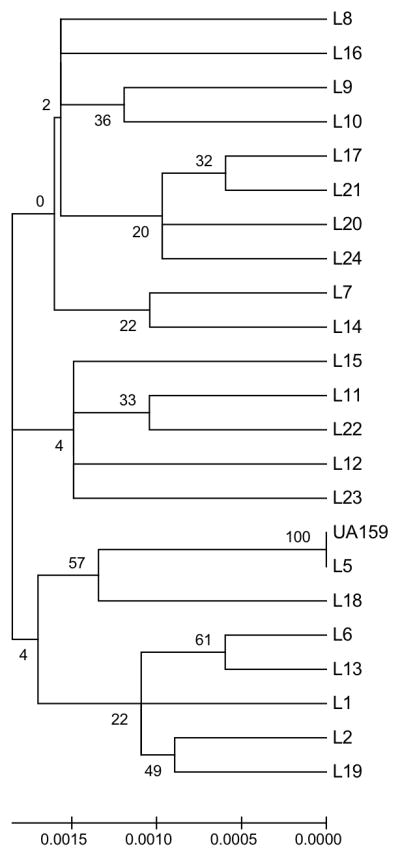
Library Strains (n=23) MEGA Minimum Evolution Bootstrap (1000 replicates) Consensus Tree. Bootstrap values are noted on branches. Only L5 and control strain UA159 clustered significantly.
Fig. 3.
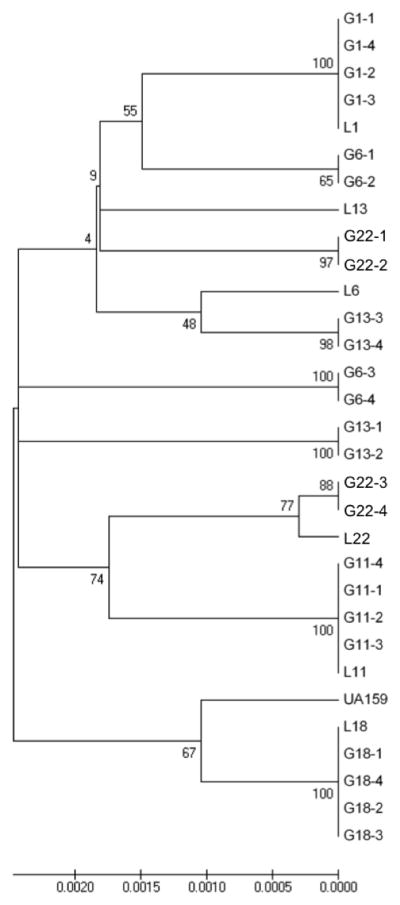
Genotype Group Strains (n=31) MEGA Minimum Evolution Bootstrap (1000 replicates) Consensus Tree. Bootstrap values are noted on internal branches of tree. Clonal clusters are demonstrated for L-1, L-11, and L-18. Clusters for GG L-6, L-13, and L-22 are not statistically supported.
DISCUSSION
DiversiLab rep-PCR has been shown to be a rapid, reproducible method for large-scale epidemiological studies. Several studies have used MLST to compare and validate rep-PCR using the DiversiLab system. It has been suggested that rep-PCR be used as a screening method for larger-scale studies to minimize the number of genotypes for further analysis. (Goldberg et al. 2006; Te Witt et al. 2009; Ben-Darif et al. 2010; Lau et al. 2010).
In this study, 47 S. mutans strains were analyzed with MLST resulting in 29 STs as compared with 22 rep-PCR genotypes (Figure 1). Nineteen previously unreported allelic profiles were reported, confirmed, and registered with the PubMLST Database (Table 1). Twenty-six new STs resulted from the new alleles or new combinations of previously reported alleles. Three STs (1, 92, & 97) observed were previously reported by Nakano (2007) and Lapirattanakul (2010)(Figure 1, Tables S1, S2).
MLST versus rep-PCR
In part one of this study, evaluation of the 22 rep-PCR library isolates with MLST analysis demonstrated 100% concordance with rep-PCR in identifying library isolates as unique (Figure 2), although tree branching order varied by method used.
For part two, MLST analysis of the GG (library strain plus 4 additional isolates) grouped all 5 isolates for groups L1, L11 and L18 together regardless of the analysis method used demonstrated clonality of the S. mutans population within individuals and identical clones in un-related subjects (Figure 3). Genotype groups L1 and L18 isolates were collected from 3 subjects; L11 from 2 subjects. While the sample size of this study is small (n=30 for genotype groups) the fact that 3 of the 6 rep-PCR genotype groups were clonal is noteworthy, as this result has not been demonstrated in the other larger studies of S. mutans, especially comparing S. mutans isolates from unrelated subjects. Lapirattanakul demonstrated vertical transmission between 14 mother-child pairs by presence of same STs (Lapirattanakul et al. 2008). Nakano and Do each found ST clusters containing 2–3 isolates, but neither discussed whether there was any relationship between subjects (Nakano et al. 2007; Do et al. 2010).
For groups L6, L13, and L22, 7 additional STs were found with MLST resulting in an overall concordance of 79% for all isolates tested (67% for genotypes only). This overall concordance between MLST and rep-PCR is lower than the 92% reported by Ben-Darif and 87.5% reported by Goldberg for other pathogens (Goldberg et al. 2006; Ben-Darif et al. 2010). The additional STs may be explained by the differences in rep-PCR and MLST methodologies. Rep-PCR evaluates extragenic sequences with genotypes being similar but not identical (Figure 1). In contrast, MLST compares stable intragenic sequences (housekeeping genes) where a single nucleotide difference results in a new ST.
Using rep-PCR with MLST
MLST appears to be more discriminatory for the evaluation of rep-PCR GG, which is congruent with Goldberg’s findings with E. coli (Goldberg et al. 2006). Although the current study is small, it is significant to note that the rep-PCR dendrogram clustered isolates similarly to MLST when only percent similarity is considered (Figure 1). However, percent similarity alone cannot be used to determine unique genotypes for rep-PCR isolates in large-scale studies since this value shifts when additional isolates are added to a GG. So the additional criteria used for rep-PCR genotypes (>2 minor, 1 major band)(Moser et al. 2010) leads to clustering of rep-PCR genotype groups (50%) which may be further distinguished with MLST analysis. The ability of the rep-PCR dendrogram to group genotypes like MLST indicates that rep-PCR may be used to screen large collections of isolates in epidemiological studies of S. mutans to rapidly determine strains for subsequent verification using MLST. Using the percent similarity dendrogram from rep-PCR, two isolates may be selected from extremes of each GG for MLST confirmation. If the two isolates are the same by MLST then it may be reasonable to assume that all isolates within the GG are the same (e.g. G1, G11, and G18). If the two isolates are different by MLST (e.g. G6, G13, and G22), then the percent similarity dendrogram may be used to determine subgroups within the rep-PCR GG. Then two new isolates may be selected from the extremes of each genotype subgroup. By this method, the number of isolates requiring subsequent MLST confirmation may be drastically reduced. However, further study with more isolates is warranted to validate this method.
Another application of MLST may be to confirm newly discovered rep-PCR genotypes are unique over the course of the study. MLST may also be used when a new isolate is difficult to assign with rep-PCR. MLST may be used to compare the ambiguous isolate with the closest possible genotype to determine if it belongs or if it is a new genotype.
This study demonstrates that rep-PCR offers a cost-effective way to perform large-scale studies. While MLST analysis is a commonly utilized genotyping method, it is not cost beneficial for large-scale epidemiological studies. Cost benefit analysis demonstrates that performing rep-PCR instead of MLST resulted in a savings of supply, labor, and equipment capital costs at 4x, 12x and 4x respectively. For example, processing with rep-PCR has a supply cost of $18 (US dollars)/sample versus $85/sample with MLST. Additionally, initial screening with rep-PCR can limit which isolates require further testing. Thus, MLST processing costs may be reduced considerably.
Advances and Limitations
In this report, the MLST PCR method previously used by Nakano (Nakano et al. 2007) was streamlined by using the same gene specific primers for both real-time PCR and sequencing, eliminating the need for gel electrophoresis, extraction and plasmid cloning. Sequence data included forward and reverse primer sequences to standardize fragment lengths, although only partial extension of the primer region was observed for most sequences.
A limitation to consider is that rep-PCR virtual gel images are visually analyzed and therefore subject to technical error. For both rep-PCR and MLST, variability between branching patterns and significance depend on the method used for evaluation.
Another MLST scheme for S. mutans was published after initiation of this study using 6 different housekeeping genes with 2 virulence genes (Do et al. 2010). However, MLST is typically defined as using 6–10 housekeeping genes (Maiden 2006). A future study is planned to compare the two MLST schemes to determine which scheme will be best for continued study (Nakano et al. 2007; Do et al. 2010). Both schemes are currently available through the PubMLST website (www.pubmlst.org).
Conclusion
In summary, library rep-PCR genotypes were also unique by MLST analysis. In analysis of GG, the discovery of 3 clonal groups in this sample population is unique to this study and suggests that S. mutans genotypes are shared between unrelated patients. Isolates obtained from the same subject had identical or similar STs. Furthermore, 19 new alleles and 26 new STs were discovered and registered with PubMLST. In addition, MLST processing time is shortened and an efficient method of using rep-PCR with MLST is proposed. This study supports the use of the DiversiLab rep-PCR system in large-scale epidemiological studies of S. mutans for economically rapid and reproducible results to select for the number of samples required for further study with MLST analysis.
Supplementary Material
Acknowledgments
This investigation was supported by Research Grant DE016684 from the National Institute of Dental and Craniofacial Research, National Institutes of Health, Bethesda, MD 20892. We especially appreciate all the clinical and laboratory participants of this study: Dr. Susan Hollingshead, Ms. Stephanie McLean, Dr. Bibi Rahima, Dr. Mohammed Salam, Ms. Tonya Wiley, Dr. Robert Osgood, Mr. Colm Atkins, Dr. Steve Mitchell, Dr. Sonia Makhija, Dr. Rosalyn Bassett, Ms. Mary Slater, Ms. Frances Jackson, and the pediatric dental residents of the UAB School of Dentistry. Special thanks to Dr. Jinthana Lapirattanakul for directing the confirmation and registration of isolates with PubMLST.
Footnotes
The authors declare no conflicts of interest.
Additional Supporting Information may be found in the online version of this article:
Table S1: MLST ST Allele Profile Assignments for 22 S. mutans Library Strains
Table S2: MLST ST Allele Profile Assignments for 6 S. mutans Genotype Groups (GG)
References
- Beltran-Aguilar ED, Barker LK, Canto MT, Dye BA, Gooch BF, Griffin SO, Hyman J, Jaramillo F, Kingman A, Nowjack-Raymer R, Selwitz RH, Wu T. Surveillance for dental caries, dental sealants, tooth retention, edentulism, and enamel fluorosis--United States, 1988–1994 and 1999–2002. MMWR Surveill Summ. 2005;54:1–43. [PubMed] [Google Scholar]
- Ben-Darif E, De Pinna E, Threlfall EJ, Bolton FJ, Upton M, Fox AJ. Comparison of a semi-automated rep-PCR system and multilocus sequence typing for differentiation of Salmonella enterica isolates. J Microbiol Methods. 2010;81:11–16. doi: 10.1016/j.mimet.2010.01.013. [DOI] [PubMed] [Google Scholar]
- Cheon K, Moser SA, Whiddon J, Osgood RC, Momeni S, Ruby JD, Cutter GR, Allison DB, Childers NK. Genetic Diversity of Plaque Mutans Streptococci with rep-PCR. J Dent Res. 2011;90:331–335. doi: 10.1177/0022034510386375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Maust BS, Nickle DC, Learn GH, Liu Y, Heath L, Kosakovsky Pond SL, Mullins JI. DIVEIN: a web server to analyze phylogenies, sequence divergence, diversity, and informative sites. BioTechniques. 2010;48:405–408. doi: 10.2144/000113370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do T, Gilbert SC, Clark D, Ali F, Fatturi Parolo CC, Maltz M, Russell RR, Holbrook P, Wade WG, Beighton D. Generation of diversity in Streptococcus mutans genes demonstrated by MLST. PLoS One. 2010;5:e9073. doi: 10.1371/journal.pone.0009073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doleans-Jordheim A, Cournoyer B, Bergeron E, Croize J, Salord H, Andre J, Mazoyer MA, Renaud FN, Freney J. Reliability of Pseudomonas aeruginosa semi-automated rep-PCR genotyping in various epidemiological situations. Eur J Clin Microbiol Infect Dis. 2009;28:1105–1111. doi: 10.1007/s10096-009-0755-z. [DOI] [PubMed] [Google Scholar]
- Foley SL, White DG, McDermott PF, Walker RD, Rhodes B, Fedorka-Cray PJ, Simjee S, Zhao S. Comparison of subtyping methods for differentiating Salmonella enterica serovar Typhimurium isolates obtained from food animal sources. J Clin Microbiol. 2006;44:3569–3577. doi: 10.1128/JCM.00745-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goering RV. The molecular epidemiology of nosocomial infection: past, present and future. Reviews in Medical Microbiology. 2000;11:145–152. [Google Scholar]
- Gold OG, Jordan HV, Van Houte J. A selective medium for Streptococcus mutans. Arch Oral Biol. 1973;18:1357–1364. doi: 10.1016/0003-9969(73)90109-x. [DOI] [PubMed] [Google Scholar]
- Goldberg TL, Gillespie TR, Singer RS. Optimization of analytical parameters for inferring relationships among Escherichia coli isolates from repetitive-element PCR by maximizing correspondence with multilocus sequence typing data. Appl Environ Microbiol. 2006;72:6049–6052. doi: 10.1128/AEM.00355-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy M, Huong J, Bittner T, Lising M, Frye S, Raza S, Schrock R, Manry J, Renwick A, Nieto R, Woods C, Versalovic J, Lupski JR. Microbial DNA typing by automated repetitive-sequence-based PCR. J Clin Microbiol. 2005;43:199–207. doi: 10.1128/JCM.43.1.199-207.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolley KA, Feil EJ, Chan MS, Maiden MC. Sequence type analysis and recombinational tests (START) Bioinformatics. 2001;17:1230–1231. doi: 10.1093/bioinformatics/17.12.1230. [DOI] [PubMed] [Google Scholar]
- Kilian M, Frandsen EV, Haubek D, Poulsen K. The etiology of periodontal disease revisited by population genetic analysis. Periodontol 2000. 2006;42:158–179. doi: 10.1111/j.1600-0757.2006.00159.x. [DOI] [PubMed] [Google Scholar]
- Kilic A, Bedir O, Kocak N, Levent B, Eyigun CP, Tekbas OF, Gorenek L, Baylan O, Basustaoglu AC. Analysis of an outbreak of Salmonella enteritidis by repetitive-sequence-based PCR and pulsed-field gel electrophoresis. Intern Med. 2010;49:31–36. doi: 10.2169/internalmedicine.49.2743. [DOI] [PubMed] [Google Scholar]
- Kulkarni GV, Chan KH, Sandham HJ. An investigation into the use of restriction endonuclease analysis for the study of transmission of mutans streptococci. J Dent Res. 1989;68:1155–1161. doi: 10.1177/00220345890680070401. [DOI] [PubMed] [Google Scholar]
- Lapirattanakul J, Nakano K, Nomura R, Hamada S, Nakagawa I, Ooshima T. Demonstration of mother-to-child transmission of Streptococcus mutans using multilocus sequence typing. Caries Res. 2008;42:466–474. doi: 10.1159/000170588. [DOI] [PubMed] [Google Scholar]
- Lau SH, Cheesborough J, Kaufmann ME, Woodford N, Dodgson AR, Dodgson KJ, Bolton EJ, Fox AJ, Upton M. Rapid identification of uropathogenic Escherichia coli of the O25:H4-ST131 clonal lineage using the DiversiLab repetitive sequence-based PCR system. Clin Microbiol Infect. 2010;16:232–237. doi: 10.1111/j.1469-0691.2009.02733.x. [DOI] [PubMed] [Google Scholar]
- Li Y, Caufield PW. The fidelity of initial acqusition of mutans streptococci by infants from their mothers. J Dent Res. 1995;74:681–685. doi: 10.1177/00220345950740020901. [DOI] [PubMed] [Google Scholar]
- Loesche W. Role of Streptococcus mutans in human dental decay. Microbiol Rev. 1986;50:353–380. doi: 10.1128/mr.50.4.353-380.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiden MC. Multilocus sequence typing of bacteria. Annu Rev Microbiol. 2006;60:561–588. doi: 10.1146/annurev.micro.59.030804.121325. [DOI] [PubMed] [Google Scholar]
- Mitchell SC, Ruby JD, Moser S, Momeni S, Smith A, Osgood R, Litaker M, Childers N. Maternal transmission of mutans Streptococci in severe-early childhood caries. Pediatr Dent. 2009;31:193–201. [PMC free article] [PubMed] [Google Scholar]
- Moser SA, Mitchell SC, Ruby JD, Momeni S, Osgood RC, Whiddon J, Childers NK. Repetitive extragenic palindromic PCR for study of Streptococcus mutans diversity and transmission in human populations. J Clin Microbiol. 2010;48:599–602. doi: 10.1128/JCM.01828-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano K, Lapirattanakul J, Nomura R, Nemoto H, Alaluusua S, Gronroos L, Vaara M, Hamada S, Ooshima T, Nakagawa I. Streptococcus mutans clonal variation revealed by multilocus sequence typing. J Clin Microbiol. 2007;45:2616–2625. doi: 10.1128/JCM.02343-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmo Emanuelsson IM, Carlsson P, Hamberg K, Bratthall D. Tracing genotypes of mutans streptococci on tooth sites by random amplified polymorphic DNA (RAPD) analysis. Oral Microbiol Immunol. 2003;18:24–29. doi: 10.1034/j.1399-302x.2002.180104.x. [DOI] [PubMed] [Google Scholar]
- Ross TL, Merz WG, Farkosh M, Carroll KC. Comparison of an automated repetitive sequence-based PCR microbial typing system to pulsed-field gel electrophoresis for analysis of outbreaks of methicillin-resistant Staphylococcus aureus. J Clin Microbiol. 2005;43:5642–5647. doi: 10.1128/JCM.43.11.5642-5647.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Te Witt R, Kanhai V, van Leeuwen WB. Comparison of the DiversiLab system, Pulsed-Field Gel Electrophoresis and Multi-Locus Sequence Typing for the characterization of epidemic reference MRSA strains. J Microbiol Methods. 2009;77:130–133. doi: 10.1016/j.mimet.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Tenover FC, Gay EA, Frye S, Eells SJ, Healy M, McGowan JE., Jr Comparison of typing results obtained for methicillin-resistant Staphylococcus aureus isolates with the DiversiLab system and pulsed-field gel electrophoresis. J Clin Microbiol. 2009;47:2452–2457. doi: 10.1128/JCM.00476-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bambeke F. Glycopeptides and glycodepsipeptides in clinical development: a comparative review of their antibacterial spectrum, pharmacokinetics and clinical efficacy. Curr Opin Investig Drugs. 2006;7:740–749. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.