

SUMMARY
It was postulated that the highly virulent JP2 genotype of Aggregatibacter actinomycetemcomitans may possess a constellation of distinct virulence determinants not found in non-JP2 genotypes. This study compared the genome content and the transcriptome of serotype b JP2 genotype and the closely related serotype b non-JP2 genotype of A. actinomycetemcomitans. A custom-designed pan-genomic microarray of A. actinomycetemcomitans was constructed and validated against a panel of 11 sequenced reference strains. The microarray was subsequently used for comparative genomic hybridization of serotype b strains of JP2 (6 strains) and non-JP2 (6 strains) genotypes, and for transcriptome analysis of strains of JP2 (3 strains) and non-JP2 (2 strains). Two JP2-specific and two non-JP2-specific genomic islands were identified. In one instance, distinct genomic islands were found to be inserted into the same locus among strains of different genotypes. Transcriptome analysis identified 5 operons, including the leukotoxin operon, to have at least two genes with an expression ratio of 2 or greater between genotypes. Two of the differentially expressed operons were members of the membrane-bound nitrate reductase system (nap operon) and the Tol-Pal system of Gram-negative bacterial species. This study is the first to demonstrate the differences in the full genome content and gene expression between A. actinomycetemcomitans strains of JP2 and non-JP2 genotypes. The information is essential for designing hypothesis-driven experiments to examine the pathogenic mechanisms of A. actinomycetemcomitans.
Keywords: aggressive periodontitis, horizontal gene transfer, genomic islands, gene expression
INTRODUCTION
The Gram-negative facultative species Aggregatibacter actinomycetemcomitans is a major etiologic agent of localized aggressive periodontitis (LAP) and other forms of periodontitis, but can also be found in the oral cavity of healthy individuals (Slots, 1999, Chen et al., 2010b). The species comprises discrete clonal lineages represented by different serotypes (Kaplan et al., 2002, Rylev & Kilian, 2008). Substantial variation in genomic content and arrangement has been found among strains (Kittichotirat et al., 2011, Kittichotirat et al., 2010). For example, the gene content of a given strain may differ as much as 19.5% from another strain (Kittichotirat et al., 2011). The patterns of genomic variation among strains correlate with the major clonal lineages of A. actinomycetemcomitans, suggesting that much of the variation was fixed in the populations long ago.
A. actinomycetemcomitans strains can be distinguished based on a variety of genotyping methods such as AP-PCR and RFLP (DiRienzo et al., 1994a, Asikainen et al., 1995, Paju et al., 2000). These typing methods were commonly used to examine the association of specific genotypes of A. actinomycetemcomitans with distinct periodontal disease conditions, leading to substantial evidence for variable virulence potentials among strains. For example, serotype b strains with a 530-bp deletion in the promoter region of the leukotoxin operon (designated as JP2 clone) were found to be associated with aggressive periodontitis in patients of African descent (Haubek et al., 1997). In prospective studies, infection by strains of the JP2 clone posed a greater risk for aggressive periodontitis or conversion from periodontal health to disease than non-JP2 clones (Haubek et al., 2008, DiRienzo et al., 1994b). While it is tempting to attribute the high virulence of the JP2 clone to its high leukotoxicity, there is no clear evidence that this is the only determinant. It is possible that other virulence determinants also play a role in the pathogenesis of periodontal infection by the JP2 clone.
In the literature, the term “non-JP2” has been loosely applied to all strains that do not have the characteristic 530-bp deletion in the promoter of leukotoxin operon, irrespective of their serotypes. However, it is clear that strains of different serotypes represent genetically distinct groups (Kittichotirat et al., 2011). To focus on JP2-specific genomic features, we here use the terms “JP2 genotype” and “non-JP2 genotype” to refer to serotype b strains that are distinguished by the structure of the promoter of leukotoxin operon.
This study tested the hypothesis that JP2 and non-JP2 genotypes differ in their genome content and gene expression patterns. A customized pan-genome microarray designed based on genome sequences of 18 strains of A. actinomycetemcomitans was used to compare the genome content and the transcriptomes of strains with JP2 and non-JP2 genotypes. The results have helped narrow the field of interest to a few, but potentially important, differences between the genomes and expression patterns of JP2 and non-JP2 strains. The information we have obtained is essential for hypothesis-driven experimental design to dissect the virulence mechanisms of A. actinomycetemcomitans.
METHODS
Bacterial strains and genomic DNA isolation
A. actinomycetemcomitans strains were grown on tryptic soy agar plates with 0.6% yeast extract for two days at 37 °C in an atmosphere supplemented with 5% CO2, and were harvested by washing the bacteria off the plates with PBS buffer. Genomic DNA was isolated using the Qiagen DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA 91355 USA) according to the manufacturer’s manual. Serotypes were determined by PCR as previously described (Kaplan et al., 2002). JP2 and non-JP2 classification was determined by PCR analysis of the ltx promoter using the primers 5′-TCCATATTAAATCTCCTTGT-3′ and 5′-AACCTGATAACAGTATT-3′ (Brogan et al., 1994) to detect the characteristic 530-bp deletion. Serotype b strains with the characteristic 530-bp deletion were designated JP2 genotype, in contrast to strains of non-JP2 genotype that had the long ltx promoter. Of the 14 previously sequenced strains 11 were used for comparative genomic hybridization (CGH) by microarray as part of the validation process. Subsequently, CGH was performed for 8 additional serotype b strains to examine their genome content (see Table 1 for a summary of strains).
Table 1.
A. actinomycetemcomitans strains used for CGH and transcriptome analysis with the pan-genome microarray
| Strain | Genome | Race/Ethnicity | Age | Periodontal Diagnosisa | Serotype/Genotypeb | Genbank accession number |
|---|---|---|---|---|---|---|
| I23C | Draft | Caucasian | 48 | CP | b/non-JP2 | AEJQ00000000 |
| SCC1398 | Draft | Caucasian | 25 | LAP | b/non-JP2 | AEJP00000000 |
| ANH9381 | Complete | Caucasian | NA | H | b/non-JP2 | CP003099 |
| ATCC29524 | N/A | NA | NA | NA | b/non-JP2 | N/A |
| 194 | N/A | NA | NA | AP | b/non-JP2 | N/A |
| G104-2 | N/A | NA | 28 | CP | b/non-JP2 | N/A |
| S067 | N/A | NA | NA | AP | b/JP2 | N/A |
| A26 | N/A | African | 19 | AP | b/JP2 | N/A |
| G111-1 | N/A | Caucasian | 21 | AP | b/JP2 | N/A |
| G121-2 | N/A | African | 38 | AP | b/JP2 | N/A |
| D28S-1 | N/A | African-American | 21 | LAP | b/JP2 | N/A |
| D41S-1 | N/A | Hispanic | 9 | AP | b/JP2 | N/A |
| HK1651 | Complete | African | 18 | LAP | b/JP2 | N/A |
| D7S-1 | Complete | African-American | 29 | GAP | a | CP003496 |
| D11S-1 | Complete | African-American | 16 | GAP | c | CP001733 |
| I63B | Draft | Caucasian | 50 | H | d | AEJL00000000 |
| SCC393 | Draft | Caucasian | 40 | CP | e | AEJN00000000 |
| D18P-1 | Draft | Asian-American | 20 | GAP | f | AEJO00000000 |
| SCC2302 | Draft | Caucasian | 33 | G | c | AEJR00000000 |
| D17P-2 | Draft | Asian-American | 24 | LAP | c | ADOB00000000 |
CP: chronic periodontitis. LAP: localized aggressive periodontitis. AP: aggressive periodontitis. GAP: generalized aggressive periodontitis. H: healthy. G: gingivitis.
Genotype was based on the analysis of the promoter structure of leukotoxin operon by PCR analysis. The promoter structure in strains other than serotype b was confirmed to be identical to that found in strains of non-JP2 genotype.
Pan-genome microarray of A. actinomycetemcomitans
Information from 14 previously examined genomes of A. actinomycetemcomitans (Kittichotirat et al., 2011) and 4 strains that were sequenced de novo (see supplemental Table S1 for sequencing results) was used to design a pan-genome microarray. The de novo sequenced strains S23A(Genbank accession number AJMH00000000), SCC4092 (AJMF00000000), AAS4A(AJMG00000000), and A160 (AJME00000000) were recovered from the same individuals as the previously studied strains I23C, SCC1398, SCC2302, and SCC393, respectively. Genomic variation of strains within individuals is a subject of another study and will not be described here. The 18 genomes were searched for genes and the genes were grouped based on sequence homology to identify unique gene clusters as described previously (Kittichotirat et al., 2011). A total of 42,668 predicted genes were grouped into 3,426 homologous gene clusters and the result can be found at http://expression.washington.edu/genetable/script/gene_table_viewer?organism=aa_ha&build=10_07_28 (please deselect column NJ8700 and ATCC33389 as these represent two different strains of Aggregatibacter aphrophilus). This set of 3,426 homologous gene clusters represents the pan-genome of A. actinomycetemcomitans, which is much larger than the total number of genes in any single genome of this species. In the text below, we refer to each gene in the A. actinomycetemcomitans pan-genome by using the p-cluster ID as shown in our homologous gene cluster result.
The longest sequence in each gene cluster was selected as a representative sequence and used for microarray probe design. A total of 13,960 probes (60 bp in length) for 3,121 homologous gene clusters were initially generated using eArray (https://earray.chem.agilent.com/earray/). The tool ArrayOligoSelector (Bozdech et al., 2003) was then used to design 20 probes for 5 additional clusters. The remaining 300 gene clusters, for which no probes were generated by either software, were often (71%) small genes (<300bp) with no known function (annotated as hypothetical protein). All designed probes were then synthesized in situ by Agilent Technologies (Santa Clara, CA) to create an 8x15K microarray with the randomized feature layout option selected. Since this array format contains more than 15K spots, some of the probes were duplicated to fill in the whole array. The final array design consisted of 15,208 probes representing 3,126 gene clusters of A. actinomycetemcomitans and 536 control probes.
The performance of individual probes in hybridization was not expected to be uniform across all strains due to sequence variation in orthologous genes across the strains. Therefore, in silico analyses were performed to filter out the probes that spanned highly variable regions. The filtering process started by comparing each probe sequence to all orthologous gene sequences that were found in each gene cluster using the NCBI blast package. A probe was considered acceptable if it matched all members of the target homologous gene cluster with at least 80% sequence identity over the probe length (60 bp) and did not match genes belonging to a different gene cluster. In addition, homologous gene clusters that were not represented by at least 3 probes were excluded from subsequent analysis. This filtering process resulted in a set of 10,934 probes representing 2,676 gene clusters, including 1,762 core genes (i.e., shared by all strains) and 914 accessory genes (i.e., present in one or more but not all strains).
Comparative genomic hybridization (CGH) by microarray
The genomic DNA was labeled and hybridized to the pan-genome microarray of A. actinomycetemcomitans according to the protocol recommended by Agilent Oligonucleotide Array-Based CGH for Genomic DNA (Agilent Technologies, Palo Alto, CA). Briefly, 0.5 μg of genomic DNA was labeled with cyanine3. DNA denaturation and fragmentation was done using a thermocycler by mixing 2.5 μl of Random Primers (Agilent) and 13 μl of genomic DNA at 94 °C for 10 minutes. The mixture was cooled to 4 °C and 9.5 μl of labeling master mix (Agilent) was added to make a total volume of 25 μl. DNA labeling was done using a thermocycler following the program: 37 °C for 2 hours, 65 °C for 10 minutes, and holding at 4 °C. Labeled genomic DNA was purified using individual Amicon 30 kDA filters (Millipore) following the manufacturer’s protocol. The yield of labeled DNA was determined by measuring the absorbance at A260nm (DNA) and A550nm (cyanine 3) using a Nano-Drop (ND-1000). Then, 40μl of hybridization sample mixture was loaded onto the glass slide containing the array and incubated at 65 °C for 24 hours with rotation. The arrays were then washed with Agilent CGH wash buffer and scanned using an Agilent scanner 6000C with the manufacturer’s recommended settings for CGH arrays (scan area 61 × 21.6 mm, 5 μm resolution, 100% PMT R&G). Data was extracted from the scanner using Agilent Feature Extractor v10.5 software using protocol CGH_105_Dec08.
The signal data obtained from the Agilent Feature Extraction Software were processed to extract the gProcessedSignal value, which is a background subtracted signal, for each probe. Since the labeling process involved incorporating Cyanine 3-dUTP into the genomic sample, it is possible that probes that are rich in nucleotide A have a higher signal relative to probes with lower A content and may not accurately reflect the true abundance of the target sequence. For this reason, the signal values were normalized by dividing them by the total number of A nucleotides in the probe sequence. Finally, this value was transformed using log2. Processed signal values from probes that are targeting the same gene cluster were consolidated into a single value by averaging the signals. A specific cutoff point was then selected for declaring gene absence or presence. The comparative genomic hybridization data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE39143 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE39143).
The performance of gene detection by CGH was first evaluated using four completed genome sequences and 7 draft genomes. The sensitivity for gene detection by CGH was calculated as (number of genes in the genome detected by CGH)/(number of genes identified in the genome by WGS). The specificity for gene detection was calculated as (number of genes in the genome not detected by CGH)/(number of genes not found in the genome by WGS).
PCR analysis of gene absence/presence
Six genes found to be distinct between JP2 and non-JP2 genotypes by CGH were confirmed by PCR analysis. A gene present in all strains was used as a positive control. The primers were designed based on published genomic data (Kittichotirat et al., 2011)(see supplemental Table S2 for primers sequences). The 25 μl PCR mixture included 50–100 ng of genomic DNA, 0.3 μM concentration of each primer, 2.5 μl of 2mM dNTPs, and 1unit of Taq DNA polymerase in 1X Taq DNA polymerase buffer. The PCR amplification was performed with the following thermocycling profile: 2 minutes at 94 °C for denaturation followed by 30 cycles of denaturation, annealing, and extension at 94 °C for 30 seconds, 64 °C to 68 °C for 1 minute, and 72 °C for 1 minute, respectively; and then a final extension of 8 minutes at 72 °C. The resultant amplicons were analyzed in 1% agarose gels.
Hierarchical clustering analysis of A. actinomycetemcomitans strains by genomic content
The software MeV (Saeed et al., 2006) was used to perform a hierarchical clustering of binary data representing the present/absent profiles of all genes (as determined by CGH) using the Euclidean distance metric. The Jstree tool (http://lh3lh3.users.sourceforge.net/jstree.shtml) was then used to generate the cladogram.
Transcriptome analysis
Three strains of JP2 genotype (HK1651, D28S-1 and D41S-1) and two strains of non-JP2 genotype (SCC1398 and ANH9381) were examined (Table 1). The transcriptome analysis was performed for each strain in biological duplicates. A starter culture was prepared by inoculating the bacteria as a single cell suspension (Karched et al., 2007) in tryptic soy broth with 0.6% yeast extract and incubated overnight at 37°C in an atmosphere supplemented with 5% CO2. The starter culture (OD650 of 0.11–0.59) was then diluted with fresh broth to ~OD650=0.1 and incubated further for 4 hours. The OD650 was determined again to assure the continued growth of the bacteria (OD650 in the range of 0.12–0.35). Aliquots of the bacterial cultures were also used to determine colony forming unit/ml and checked for contamination.
The bacterial RNA was isolated using RiboPure™-Bacteria Kit (Life technology). The resultant RNAs were checked for DNA contamination by PCR using 16S rRNA primers. The RNAs were then stored at −70 °C until use. MessageAmp™ II-Bacteria kit (Life technology) was used to label the RNA(500 ng for each sample) following the protocol recommended by the manufacturer, with a slight modification in the IVT reaction in which a combination of 2.6 μL T7 CTP and 1.4 μL of cy-3 labeled CTP were used for the master mix. The labeled samples were hybridized to the custom Agilent slides following the recommended protocol. A change to the protocol was made to use a nitrogen gun for drying the arrays after washing (instead of centrifugation or drying solution). RNA Yield and Quality was assessed by Nanodrop-1000 spectrometer and Agilent bioanalyzer. When dry, the slides were immediately scanned using the Agilent Scanner 6000C according to the manufacturer’s recommended settings for 1-color gene expression. For the 8 × 15K custom arrays, we scanned at 5 μm resolution, dual pass at 100% and 10% PMT in one channel only (green, or cy-3). The data was then Feature-extracted using Agilent’s Feature Extractor software v10.5 running the extraction protocol GE1_105_Dec08.
Expression data obtained from the Agilent Feature Extraction Software were processed using the following steps. First, the gProcessedSignal value for all arrays, a background subtracted signal, was extracted for each probe. Quantile normalization was then applied across all the array data being compared to adjust for differences in the probe intensity distribution using the limma package (Smyth & Speed, 2003). Since we were interested in comparison of expression levels of the same genes among different A. actinomycetemcomitans strains, no adjustment was made for the signals based on the numbers of G nucleotides in the probe sequences. Finally, the normalized signal values from probes targeting the same gene cluster were consolidated into a single median value. A total of 1,952 genes shared between JP2 and non-JP2 genotypes were analyzed for their expression. The data for each replicate were treated individually in statistical analysis. Differentially expressed genes between genotypes were identified by t-test in MeV at P<0.05. The expression data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002)and are accessible through GEO Series accession number GSE38943 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE38943).”
Identification of gene operons
Operons in the genome of A. actinomycetemcomitans strain HK1651 were identified via Database of prOkaryotic OpeRons (DOOR) tool, which predicts bacterial gene operons using a classifier algorithm based on features such as intergenic distance, neighborhood conservation, phylogenetic distance, information from short DNA motifs, similarity score between GO terms of gene pairs, and length ratio between a pair of genes(Mao et al., 2009).
Quantitative Real-Time PCR (qRT-PCR)
Five of the same RNA samples used in transcriptome analysis (one sample per strain) were also used to assess the transcript levels of selected genes (p-cluster 00978 tolR, p-cluster 00011 lktA, p-cluster 01456 napD, p-cluster 00459 metF, p-cluster 00078 nrfE) by qRT-PCR using BioRad iCycler iQ® Real-Time PCR Detection System. Aconstitutively expressed house-keeping gene clpX (p-cluster 00289) was used as a reference to compare the expression levels (Ramsey & Whiteley, 2009). For each sample, 1 μg of RNA in a 20 μl reaction mixture was reverse transcribed into first strand cDNA using SuperScript VILO kit (Life technology). Reactions without reverse transcriptase or RNA template were included as controls. The first strand cDNA synthesis was performed at 25 °C for 10 minutes, 42 °C for 60 minutes, 85 °C for 5 minutes. The 20 μl volume containing the cDNA was then diluted to 200 μl using sterile water. For qRT-PCR, a volume of 2 μl of the diluted cDNA from each sample was used following the protocol described by the manufacturer. Briefly, the reaction mixture included 2.5 μl of each primer (3 μM), 12.5 μl of 2X Supermix, 2 μl of cDNA, and water to 25 μl. The thermocycling profile consisted of four cycles as follows: Cycle 1: (1X) Step 1: 95.0 °C for 3 minutes. Cycle 2: (40X) Step 1: 95.0 °C for 10 seconds Step 2: 55.0 °C for 30 seconds. Cycle 3: (1X) Step 1: 95.0 °C for 1 minute. Cycle 4: (1X) Step 1: 55.0 °C for 1 minute. For the melting curves, the final DNA products were denatured at 95 °C for 1 minute and then incubated at 5 °C below the annealing temperature for 1 minute before the temperature was increased to 95 °C at a ramp rate of 0.5 °C/10 seconds. For each sample, both target gene and reference gene were done in triplicate. Additional controls include samples without cDNA for each target gene. Data analysis was performed based on the protocol provided by BioRad. The transcript levels determined by qRT-PCR were expressed as ratios to the sample with the lowest transcript level for each assessed gene.
RESULTS
Criteria for calling gene presence or absence by CGH
A total of 38 hybridizations for 19 strains (two replicates for each strain) were obtained. The distribution of the processed signals for each gene cluster for each A. actinomycetemcomitans strain showed a bimodal pattern, suggesting that the two peaks represented signals for the absent or the present genes (Fig 1). The signal cutoff for determining gene presence or absence in CGH was taken to be the minimum count found between the two peaks. The cutoff point was determined individually for each CGH analysis (see supplemental Table S3 for complete dataset of the distribution of hybridization signals and cutoff points).
Figure 1. Distribution of processed hybridization signals for gene clusters in A. actinomycetemcomitans D7S-1.
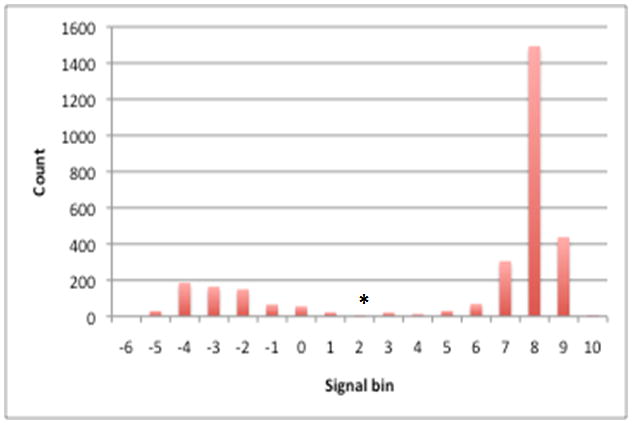
Processed hybridization signals from an array hybridized with D7S-1 genomic DNA were plotted where the X-axis represents the processed signal bins (e.g. 1 represents processed signals ranging from >0 to 1) and the Y-axis represents the count of data points in each bin. The distribution shows a bimodal shape where the right and left peaks represent processed signal values of genes that are present and absent, respectively. A minimum count found between the two peaks (denoted as an asterisk in the figure) was chosen as a cutoff point for gene present/absent status (e.g. a processed signal above this value is considered present and a signal below this value is considered absent.
Assessing the performance of gene detection by CGH
The results from two hybridizations failed to form bimodal distribution patterns (for strain D11S-1 and I63B) and were not analyzed. The whole genome sequence (WGS) information was used as the standard to test the performance of the pan-genome microarray in gene detection. The sensitivity for gene detection of the four complete genomes (D7S-1, HK1651, ANH9381 and D11S-1) was in the range of 0.948 to 0.986, while the specificity was between 0.867 to 0.976 (Table 2). The relatively low specificity (0.867) for one of the duplicates of D7S-1 was due to a few false positive results with hybridization signals just above the break point for gene detection. For the 7 draft genomes, the sensitivity was 0.974–0.989 while the specificity was 0.900–0.986. The specificity for replicates for strain I23C was relatively low (0.900 and 0.902), which may be attributed to genes not detected by WGS but nevertheless detected by CGH. This interpretation is supported by the relatively poor WGS quality for I23C, with the lowest coverage (16X), the highest number of contigs (400) and the highest number of Q39 bases (33,125) in comparison with the other strains listed in Table 2 (Kittichotirat et al., 2011, Chen et al., 2010a, Chen et al., 2009).
Table 2.
Sensitivity and specificity for gene detection by microarray among 11 sequenced A. actinomycetemcomitans strains
| Complete Genome | ||
|---|---|---|
| Strain | Sensitivity | Specificity |
| D7S-1 | 0.986 | 0.976 |
| D7S-1 | 0.986 | 0.867 |
| HK1651 | 0.984 | 0.986 |
| HK1651 | 0.985 | 0.986 |
| ANH9381 | 0.948 | 0.978 |
| ANH9381 | 0.986 | 0.981 |
| D11S-1 | 0.979 | 0.967 |
| Draft Genome | ||
| Strain | Sensitivity | Specificity |
| I63B | 0.989 | 0.980 |
| SCC393 | 0.983 | 0.977 |
| SCC393 | 0.986 | 0.977 |
| D18P-1 | 0.981 | 0.971 |
| D18P-1 | 0.988 | 0.977 |
| SCC1398 | 0.984 | 0.979 |
| SCC1398 | 0.985 | 0.981 |
| I23C | 0.983 | 0.900 |
| I23C | 0.983 | 0.902 |
| D17P-2 | 0.986 | 0.977 |
| D17P-2 | 0.984 | 0.977 |
| SCC2302 | 0.977 | 0.912 |
| SCC2302 | 0.974 | 0.986 |
Sensitivity=(number of genes in the genome detected by CGH)/(number of genes identified in the genome by WGS). Specificity= (number of genes in the genome not detected by CGH)/(number of genes not found in the genome by WGS).
Duplicates of the CGH results for 9 of the sequenced strains and 8 clinical isolates were further examined to evaluate the consistency in gene detection between duplicates. As shown in Table 3, the percentage of shared genes detected in the replicates ranged from 95.95%–100%. In some instances the basis for the differences between duplicates could be identified. For strain D7S-1, the discrepancy between replicates can be attributed to the relativley low specificity (0.867) in one of the replicates (Table 2), while for strain ANH9381 the differences in detection between replicates were likely due to the relatively low sensitivity (0.948) in one of the replicates (Table 2). The information was used to help in selecting one of the duplicates from each strain for further analysis below.
Table 3.
Gene detection between the replicates for each of the 17 A. actinomycetemcomitans strains
| Strain | No. genes in replicate #1 | No. genes in replicate #2 | No. genes found only in replicate #1 | No. genes found only in replicate #2 | No. of genes detected in both replicates (% of genes) |
|---|---|---|---|---|---|
| HK1651 | 2003 | 2005 | 0 | 2 | 2003 (99.90) |
| D7S-1 | 2113 | 2173 | 4 | 64 | 2109 (96.88) |
| SCC1398 | 1991 | 1992 | 0 | 1 | 1991 (99.95) |
| SCC393 | 2164 | 2171 | 2 | 9 | 2162 (99.49) |
| D17P-2 | 2101 | 2105 | 0 | 4 | 2101 (99.81) |
| D18P-1 | 2133 | 2145 | 4 | 16 | 2129 (99.07) |
| ANH9381 | 1957 | 2033 | 2 | 78 | 1955 (96.07) |
| SCC2302 | 1987 | 1977 | 0 | 10 | 1977 (99.50) |
| I23C | 2055 | 2055 | 0 | 0 | 2055 (100) |
| A26 | 2036 | 2037 | 0 | 1 | 2036 (99.95) |
| ATCC29524 | 2035 | 2032 | 4 | 1 | 2031 (99.75) |
| 194 | 1992 | 2037 | 0 | 45 | 1992 (97.79) |
| G104-2 | 2062 | 2059 | 3 | 0 | 2059 (99.85) |
| S067 | 1993 | 2003 | 5 | 15 | 1988 (99.00) |
| G111-1 | 2011 | 2014 | 0 | 3 | 2011 (99.85) |
| G121-2 | 2040 | 2038 | 2 | 0 | 2038 (99.90) |
| D41S-1 | 1989 | 2073 | 0 | 84 | 1989 (95.95) |
The % of genes detected was calculated by (no. of detected genes in both replicates)/(total unique genes identified by both replicates)
Clustering analysis of genome content of A. actinomycetemcomitans serotype b and c strains
Based on the CGH results, a hierarchical clustering analysis of the genomic content (based on the presence and absence of genes) was performed for strains of serotypes a–f (Fig 2). Serotypes a, d, e and f formed a major branch separate from serotypes b and c. The six serotype b JP2 genotype strains formed a unique group separated from six other serotype b non-JP2 genotype strains and three serotype c strains.
Figure 2. Hierarchical clustering analysis of A. actinomycetemcomitans strains by genomic content.
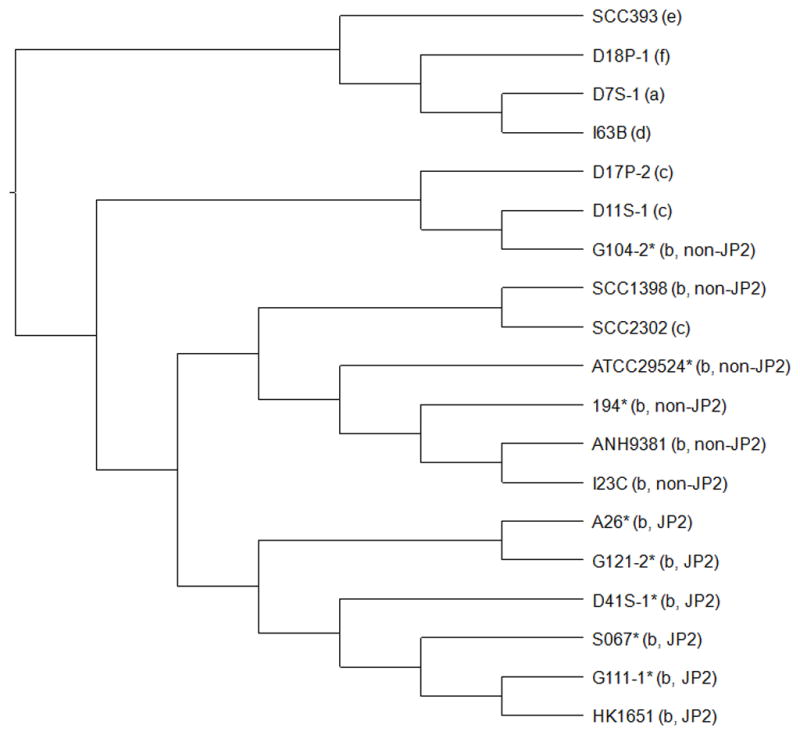
The MeV software was used to perform the hierarchical clustering of binary data representing the present/absent profiles of genes using Euclidean distance metric. The Jstree tool was then used to create the cladogram. The serotype and genotype information is provided in parenthesis. Asterisks indicate clinical strains without genome sequence information. The results showed that strains of serotypes a, d, e and f formed a major branch. The six serotype b JP2 genotype strains formed a unique cluster but the six serotype b non-JP2 genotype strains were mixed together with three serotype c strains
Genome content variation between JP2 and non-JP2 genotypes
Among the 2,044 gene clusters found in any of the six strains of JP2 genotype, 1968 gene clusters (96%) were shared by all strains. In comparison, a total of 2,201 gene clusters were present among the 6 strains of non-JP2 genotype with 1986 gene clusters (90%) shared by all six non-JP2 strains.
Nine genes were found exclusively in JP2 genotype strains while 12 genes were found only in non-JP2 genotype strains (Table 4). The genotype-specificity of 5 gene clusters (p-cluster03717, p-cluster03907, and p-cluster03273 of JP2 strains, and p-cluster01731p-cluster02176 of non-JP2 strains) were selected and confirmed by PCR analysis in the 12 serotype b strains (data not shown). For the purpose of clarity, these 21 JP2 and non-JP2 specific genes are further described below in the context of the genomes of the fully sequenced HK1651 and ANH9381 strains.
Table 4.
JP2 and Non-JP2 specific genes
| Genes specific to JP2 genotype | ||||
|---|---|---|---|---|
| p-Cluster | Gene ID (nucleotide coordinates) | Length (bp) | Function | Island |
| 03231 | AA00018 (10690–11817) | 1128 | hypothetical protein | iAAI-1 |
| 03273 | AA00019 (11839–12882) | 1044 | phage protein | iAAI-1 |
| 03132 | AA00020 (12939–14546) | 1608 | phage protein | iAAI-1 |
| 03717 | AA01904(1284588–1285109) | 522 | hypothetical protein | iAAI-3 |
| 03907 | AA01911(1287931–1288344) | 414 | hypothetical protein | iAAI-3 |
| 03979 | AA00785(539732–539361) | 372 | phage integrase | Non-Island |
| 05260 | AA00666 (457610–457714) | 105 | hypothetical protein | Non-Island |
| 04245 | AA01318(892473–892222) | 252 | hypothetical protein | Non-Island |
| 03340 | AA01449(971629–970709) | 921 | prophage integrase | Non-Island |
| Genes specific non-JP2 genotype | ||||
| p-Cluster | Gene ID (nucleotide coordinates) | Length (bp) | Function | |
| 01473 | ANH-1698 (1601972–1602181) | 210 | TOBE domain protein | Non-Island |
| 05754 | ANH_1697 (1600856–1601890) | 1035 | iron chelatin ABC transporter | Non-Island |
| 08744 | N/Aa (816767–816909) | 141 | hypothetical protein | Non-Island |
| 01210 | ANH9381_0165 (137315–136116) | 1200 | flavoproteins | Non-Island |
| N/A | N/Aa (1378798–1378947) | 150 | hypothetical protein | iANH-1 |
| 01585 | ANH9381_1475 (1378521–1378814) | 294 | CRISPR-associated protein Cas2 | iANH-1 |
| 01731 | ANH9381_1474 (1377573–1378517) | 945 | CRISPR-associated protein Cas1 | iANH-1 |
| 02176 | ANH9381_1473 (1375030–1377450) | 2421 | ATP-dependent OLD family endonuclease | iANH-1 |
| 02878 | N/Aa (1140063–1139875) | 189 | antirestriction protein | Non-Island |
| 04532 | ANH9381_1521 (1414996–1414814) | 183 | YcfA family protein | iANH-2 |
| 03415 | ANH9381_1526 (1418261–1417428) | 834 | prophage integrase | iANH-2 |
| 04299 | ANH9381_2215 (2116174–2115722) | 453 | hypothetical protein | Non-Island |
N/A: Genes were not detected by the annotation programs, but were identified by BLAST search against the genome.
Insertion of an island “iAAI-1” in the genome of HK1651
JP2-specific genes, which are identified by p-cluster03231, p-cluster03273, and p-cluster03132 (see Table 4), were found to be contiguous and located on a 23,394 bp genomic island (here renamed iAAI-1; HK1651 nucleotide coordinates 6573–29966) comprising 20 genes reported previously (Chen et al., 2005, Kittichotirat et al., 2011). The iAAI-1 island was flanked by a truncated malate dehydrogenase and a truncated flavoprotein homolog, followed by an intact cytochrome c protein homolog (Chen et al., 2005). Strain ANH9381 did not harbor iAAI-1 and instead contained a full-length malate dehydrogenase gene and flavoprotein gene in the same locus (Table 4; non-JP2 specific p-cluster1210). The same genetic arrangement (i.e., having an intact malate dehydrogenase and flavoprotein) was also found in the genomes of serotype b non-JP2 genotype strains SCC1398 and I23C (data not shown). While homologs of the genes on iAAI-1 were detected by CGH in all of the 5 clinical JP2 strains, the presence of iAAI-1 (as a contiguous island) in these strains was not verified.
Insertion of an island “iANH-1” in the genome of ANH9381
The genes p-cluster02176, p-cluster01731, p-cluster01585, and p-cluster09851 were detected in the non-JP2 genotype but not in the JP2 genotype by CGH (Table 4). These four genes were found to be contiguous and located on a genomic island (designated iANH-1) in ANH9381 (Fig 3). A homolog of the iANH-1 island was also found in the same locus in strain SCC1398 (annotated as genes SCC1398_0850 to SCC1398_0858)(Kittichotirat et al., 2011).
Figure 3. Genetic maps of the genomic island iANH-1 in ANH9381 and the corresponding region in strain HK1651.
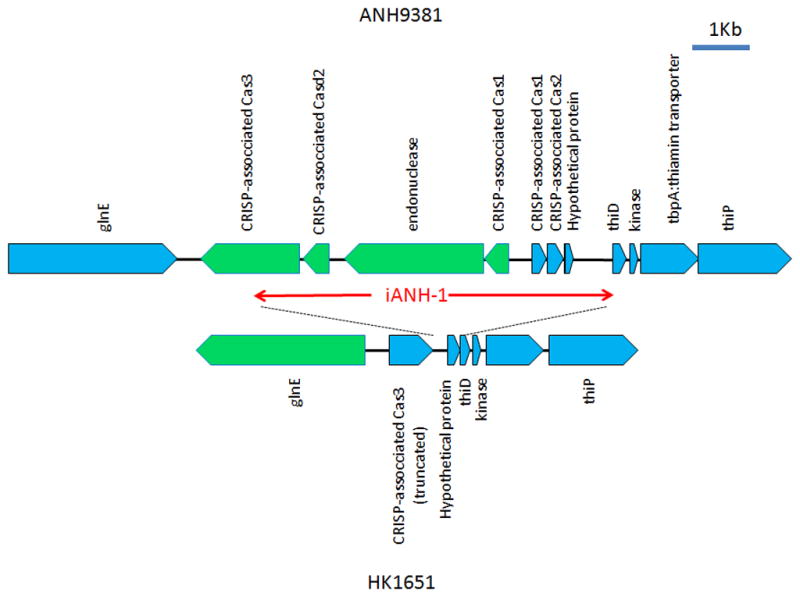
All ORFs are noted and indicated by blue (forward) or green (reverse) pentagons. The gene encoding CRISP-associated Cas3 is truncated at the 3′ end by more than 50% of its length in HK1651. In addition, the glnE and the genes for Cas3 were transcribed in opposite directions between these strains, suggesting a genomic reversion in conjunction with the insertion/deletion of the island iANH-1. HP: hypothetical protein.
Different islands identified in the same locus in the genomes of HK1651 and ANH9381
JP2-specific p-cluster03717 and p-cluster03907 were located on a 7,244 bp genomic island designated as iAAI-3 (HK1651 nucleotide coordinates 1,281,101–1,288,344) flanked by guaA and a gene encoding hypothetical protein (AA01895)(Fig 4). Adifferent genomic island of 9,392 bp was identified (here designated as iANH-2; strain ANH9389 nucleotide coordinates 1408870–1418261) in the same locus in ANH9381 (Fig 3)(Chen et al., 2012). While homologous genes of iANH-2 were identified in I23C, they were located in two contigs that may or may not be a homolog of iANH-2. A distinct 5,682 bp genomic island comprising 13 genes that demonstrated partial homology to both iAAI-3 and iANH-2 was found in the same locus in strain I23C. This island was also identified in the previous study (I23C contig00245, nucleotide coordinates 2082–7763)(Kittichotirat et al., 2011).
Figure 4. Genetic map of the genomic island iAAI-3 in strain HK1651 and genomic island iANH-2 in strain ANH9381.
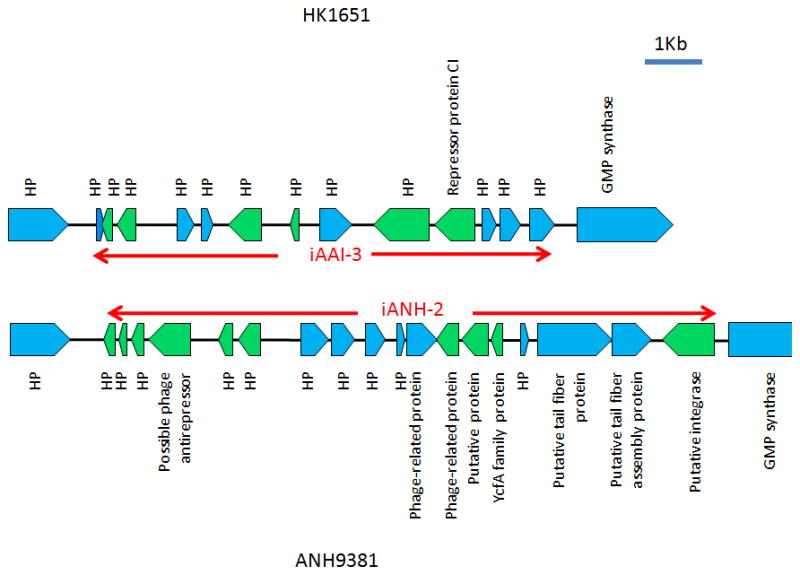
Red arrows delineate the boundary of the islands. ORFs are noted and indicated by blue (forward) or green (reverse) pentagons. HP: hypothetical protein. The islands are flanked by a hypothetical protein and guaA (GMP synthase).
For the remaining JP2-specific and non-JP2-specific genes, none was found to be on genomic islands. In one instance, the insertion of a p-cluster04245 within radC (DNA repair protein) divided the gene into two apparently nonfunctional pseudogenes in HK1651, while ANH9381 (as well as SCC1398 and I23C) has an intact radC.
Transcriptome profiles of log phase A. actinomycetemcomitans
The results for two biological duplicates per strain for A. actinomycetemcomitans JP2 genotype strains HK1651, D28S-1 and D41S-1 and for non-JP2 genotype strains ANH9381 and SCC1398 are presented in supplemental information (Table S4. The R values for the linear correlation between duplicates were 0.64 for HK1651, 0.95 for D41S-1, 0.87 for D28S-1, 0.97 for SCC1398 and 0.72 for ANH9381.
One sample from each of the five strains was used to verify expression of each of five selected genes. The results were compared to the same samples used for gene expression analysis with the microarray. The correlations between the results by qRT-PCR and hybridization with the microarray are shown in Fig 5. Three of the five genes demonstrated excellent correlations between results obtained by qRT-PCR and microarray analysis (R=0.89–0.93). For the other two genes, the overall trend in the transcript levels among samples was consistent between the two quantification methods.
Figure 5. Correlation of the gene expression levels determined by qRT-PCR and by hybridization with microarray.
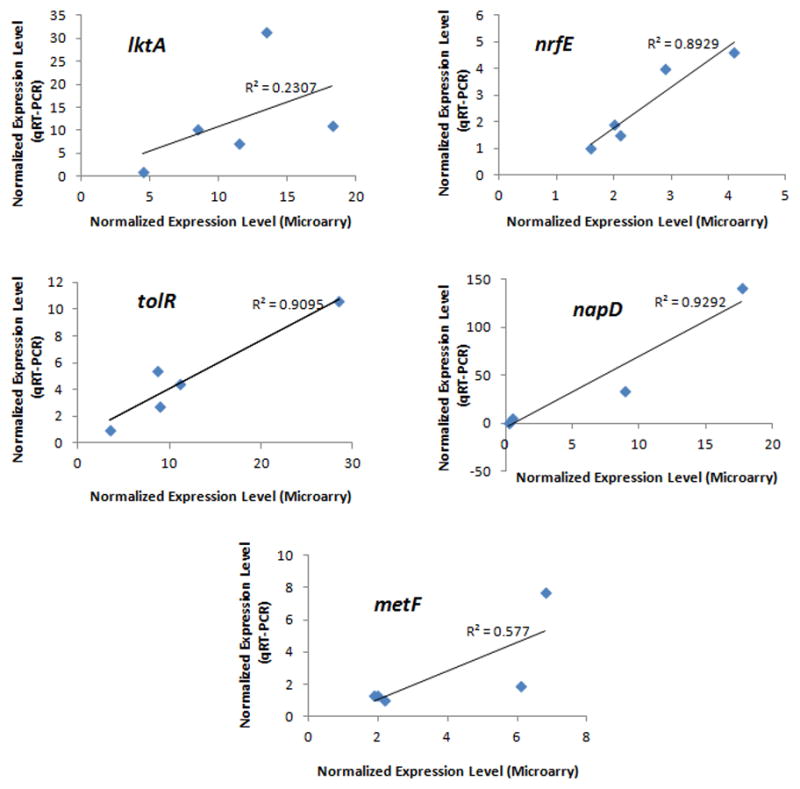
Identical samples (3 strains of JP2 genotype and 2 strains of non-JP2 genotype) were used in both quantification methods to determine the expression levels of the 5 selected genes: lktA, tolR, nrfE, napD and metF. The expression level of the housekeeping gene clpX was used to normalize the levels of the target genes in each sample. For qRT-PCR, the sample with the lowest transcript level was arbitrarily set as 1. Linear trend and R2 value are provided for each graph. Excellent correlations were found for tolR, nrfE, napD. For lktA and metF, the trend in the expression levels was consistent between the two quantification methods.
The 150 genes with significantly different transcript levels between JP2 and non-JP2 genotypes (P<0.05 by t-test) and their expression ratios are shown in Supplemental Table S5. We further used prOkaryotic OpeRons (DOOR) tool to predict the operons of A. actinomycetemcomitans HK1651 (see supplemental Table S6), and identified operons with at least two genes with a 2-fold difference or greater in expression levels between JP2 and non-JP2 genotypes. Five operons were found to meet the criteria and are listed in Table 5. As expected, the expression levels of the leukotoxin operon were higher in JP2 genotype than in non-JP2 genotype. For the remaining 4 operons, two were more highly expressed in the JP2 genotype, and the others were more highly expressed in the non-JP2 genotype.
Table 5.
Differentially expressed operons between A. actinomycetemcomitans JP2 and non-JP2 genotypes
| Cluster ID | HK1651 Gene ID | Length (bp) | Product Description | JP2/nonJP2 ratio |
|---|---|---|---|---|
| p-cluster00813 | AA00401 | 594 | slyD: FKBP-type peptidyl-prolyl cis-trans isomerase | 1.721 |
| p-cluster00710 | AA00402 | 717 | periplasmic/secreted protein | 2.634a |
| p-cluster00965 | AA00404 | 432 | COG3076: Uncharacterized protein conserved in bacteria | 3.276a |
| p-cluster00993 | AA00406 | 393 | mscL: large-conductance mechanosensitive channel | 2.416a |
| p-cluster00218 | AA00407 | 1374 | trkA: trk system potassium uptake protein | 3.008 |
| p-cluster00233 | AA00408 | 1353 | sun, fmu, fmv, rsmB: SUN protein (FMU protein) | 3.502a |
| p-cluster01270 | AA00409 | 954 | fmt: methionyl-tRNA formyltransferase | 2.662 |
| p-cluster01400 | AA00410 | 510 | def: peptide deformylase | 2.656a |
|
| ||||
| p-cluster00778 | AA00546 | 633 | napC: denitrification system component | 0.091a |
| p-cluster00946 | AA00547 | 447 | napB: periplasmic nitrate reductase | 0.089a |
| p-cluster00545 | AA00549 | 879 | napH: ferredoxin-type protein | 0.06a |
| p-cluster06328 | AA00550 | 678 | napG: ferredoxin-type protein | 0.054a |
| p-cluster01495 | AA00552 | 2484 | napA: periplasmic nitrate reductase precursor | 0.046 |
| p-cluster01456 | AA00553 | 282 | napD: NapD component of periplasmic nitrate reductase | 0.034 |
| p-cluster01596 | AA00555 | 162 | hypothetical protein D11S_0211 | 1.157 |
|
| ||||
| p-cluster00484 | AA02563 | 942 | tkt: transketolase C-terminal section | 0.593 |
| p-cluster01310 | AA02564 | 531 | tkt: transketolase N-terminal section | 0.348a |
| p-cluster06816 | AA02566 | 1092 | PTS system, IIC component | 0.401a |
| p-cluster09728 | AA02568 | 267 | sgaB: sugar phosphotransferase component II B | 0.606 |
|
| ||||
| p-cluster00192 | AA02803 | 1431 | lktD | 1.656 |
| p-cluster00056 | AA02805 | 2121 | lktB | 3.996a |
| p-cluster00011 | AA02806 | 3165 | lktA | 2.925a |
| p-cluster13459 | AA02807 | 504 | lktC | 6.1a |
|
| ||||
| p-cluster05272 | AA02828 | 105 | Hypothetical protein | 1.449 |
| p-cluster00938 | AA02829 | 465 | pal: peptidoglycan-associated outer membrane | 1.183 |
| p-cluster00276 | AA02830 | 1278 | tolB: colicin tolerance protein | 2.768a |
| p-cluster01209 | AA02832 | 1206 | tolA: outer membrane integrity protein | N/Ab |
| p-cluster00978 | AA02834 | 420 | tolR: colicin transport protein | 2.862a |
| p-cluster05329 | AA02835 | 96 | Hypothetical protein | 2.11a |
| p-cluster00717 | AA02836 | 687 | tolQ: colicin transport protein | 2.008a |
| p-cluster00991 | AA02837 | 402 | tol-pal system-associated acyl-CoA thioesterase | 1.731a |
| p-cluster01904 | AA02838 | 276 | hypothetical protein APJL_0311 | 1.891a |
| p-cluster05330 | AA02839 | 96 | Hypothetical protein | 1.028 |
| p-cluster00338 | AA02840 | 1134 | cydB: cytochrome D ubiquinol oxidase, subunit II | 0.762 |
| p-cluster01161 | AA02841 | 1563 | cydA: cytochrome D ubiquinol oxidase, subunit I | 0.838 |
Significantly different between JP2 and non-JP2 genotypes by t-test at P<0.05
No analysis was performed due to problems in probe design
Analysis of promoters of 4 differentially expressed operons
Differential gene expression could possibly be attributed to different promoter structures in the two genotypes. Therefore, the upstream regions of the 4 operons differentially expressed between HK1651 and ANH9381 were analyzed for sequence variations. The sgaB/tkt operon and its upstream region in HK1651 and ANH9381 are depicted in Fig 6. A 223-bp insert (including the 93 bp AA02569 sequence encoding a hypothetical protein annotated by OralGen) flanked by 48-bp and 49-bp inverted repeated elements was found in HK1651, while only the 49-bp element was found in the same locus in ANH9381. However, the same 223-bp insert was found in the non-JP2 strain SCC1398. Therefore, the 223-bp insertion is not JP2-specific and cannot be the cause of differential expression of the operon. For the other 3 operons, no differences were found in the promoters between JP2 and non-JP2 genotypes. It was noted that the napG gene of HK1651 is 816-bp in length as found in all other A. actinomycetemcomitans strains, not 678-bp as reported by OralGen.
Figure 6. Genetic map of sagB/tkt operon in HK1651 and ANH9381.

The dash-lines indicate the homologous regions. ORFs are noted and indicated by blue (forward) or green (reverse) pentagons. An insertion of a 223 bp fragment flanked by 48-bp/49-bp repeats (indicated by light blue boxes) is found in HK1651, while only the 49-bp element was found in the comparable region in ANH9381. It was noted that the tkt (transketolase N-terminal section) was truncated in HK1651 (annotated at OralGen as two overlapping genes of AA02564 and AA02565) but intact in ANH8381. Here is the location of each repeat in HK1651 and ANH9381 genomes
>hk1651-repeat-1 (1788949-1788997)
TCCACGCTTGGACCGACACAAGCAAAAGCGCGGATGCTTGCGCTATCAT
>hk1651-repeat-2 (1789221-1789268)
TCCACGCTTGGACCGGATAAGCACTAGCGCGGACGCTTGCGCTATCAT
>anh9381-repeat-1 (1922975-1923023)
TCCACGCTTGGACCGACACAAGCAAAAGCGCGGATGCTTGCGCTATCAT
DISCUSSION
A. actinomycetemcomitans demonstrates remarkable strain-to-strain variation in genome content. While the pan-genome of A. actinomycetemcomitans is open-ended (Kittichotirat et al., 2011), 16 or fewer new genes are expected with every additional strain included for genome sequencing in the future. The pan-genome microarray described in this study was designed based on the genomes of 18 diverse A. actinomycetemcomitans strains. It will provide a good degree of gene coverage and can serve as a cost-effective tool for interrogating gene contents of A. actinomycetemcomitans strains. Also, this microarray is applicable for transcriptome analysis of diverse A. actinomycetemcomitans strains. While alternatives for genomic comparison or transcriptome analysis are available (such as high throughput sequencing), the costs may be higher than using the microarray.
Gene detection with the pan-genome microarray was evaluated with both sequenced strains and clinical isolates of A. actinomycetemcomitans. Overall, the performance for gene detection was excellent and demonstrated high degrees of sensitivity, specificity, and reproducibility between biological duplicates. Some of the probes on the pan-genome microarray were excluded in our analysis because they may provide inconsistent hybridization results across the strains due to sequence variations. Nearly all the excluded probes were designed for genes that encode small hypothetical proteins and may not represent critical genetic elements of the strains. Nevertheless, we are currently redesigning the pan-genome microarray to further improve its utility.
The results of cluster analysis in this study confirmed our previous finding that A. actinomycetmcomitans strains were divided into two major branches. One comprised strains of serotypes a, d, e, f, and the other comprised strains of serotypes b and c. This study further focused on assessing the variation in genomes and transcriptomes of JP2 and non-JP2 genotypes. Haubek et al (Haubek et al., 2007) suggested that strains of JP2 genotype arose relatively recently as a distinct clone derived from an ancestral strain of serotype b non-JP2. Moreover, serotype b JP2 strains could be distinguished into two groups based on a mutation in the hbpA pseudogene. One group was found in individuals from North Africa of distinct ethnic group (Arabs) and the other was associated with individuals from West Africa (Africans)(Haubek et al., 2007). The results obtained in this study are consistent with this interpretation since it also shows that JP2 and non-JP2 strains constitute separate clusters in the dendrogram based on the analysis of genome content of the strains (i.e., presence and absence of 2,676 genes). As expected, the genomic diversity of non-JP2 strains was greater than JP2 strains. To the best of our knowledge, this is the first study to demonstrate the differences between JP2 and non-JP2 genotypes by full genome content analysis. Other than strain HK1651, no sequence information of hbpA pseudogene was available for the other 4 JP2 strains. However, the 4 JP2 strains in this study likely belong to the West Africa subgroup of JP2 genotype because they were identified from ethnic groups of African and Hispanic in US and Brazil, as suggested by Haubek et al (Haubek et al., 2007).
One unique aspect of the JP2 genotype is its apparent high virulence (Haubek et al., 2008, DiRienzo et al., 1994b). A premise of this study is that other virulence factors, e.g., genomic islands, may be the basis for the high virulence of JP2 genotype. The acquisition of genomic islands is thought to play a crucial role in bacterial evolution (Ochman et al., 2000). This study identified several genomic islands specific to JP2 and non-JP2 genotypes. It was also noted that the insertion of genomic islands may interrupt the function of housekeeping genes. Moreover, different genomic islands may be inserted into the same locus in different genomes, suggesting an insertion hot-spot for genomic islands in A. actinomycetemcomitans. Our laboratory is currently assessing the functions of these genomic islands and the effects of their genomic insertion in A. actinomycetemcomitans.
Variable virulence may also arise due to differential regulation of virulence determinants. Therefore, transcriptome analysis was performed for A. actinomycetemcomitans strains grown to log phase in an enriched liquid medium. The expression levels of 5 genes determined by microarray hybridization and by qRT-PCR were also compared. The gene expression levels determined by these two methods were relatively consistent except for lktA. This could be due to experimental variables that were difficult to replicate between experiments. It was noted that the transcriptomes of stran HK1651 demonstrated the least correlation between the biological duplicates than other strains. The correlations between gene expression by microarray hybridization and qRT-PCR improved (R2 values of 0.5732, 0.9924, 0.851. 0.923 and 0.8168 for lktA, tolR, nrfE, napD and metF, respectively) if the data for strain HK1651 were removed from the analysis.
In our transcriptome analysis, we found that the expression levels of 150 genes (7.7%) were significantly different between JP2 and non-JP2 genotypes (t-test, P<0.05) while 98 genes (1,952 × 0.05) were expected. We reasoned that the overall expression patterns of operons may be less likely to be subjected to experimental variations than those of individual genes. Therefore, we identified 5 operons each with two or more genes demonstrating 2-fold or greater expression ratios between JP2 and non-JP2 genotypes. The leukotoxin operon was one of the 5 operons identified, suggesting the validity of our approach. The expression patterns of the other 4 operons were not known to be genotype-specific. The differences in the expression levels of the nap operon between JP2 and non-JP2 genotypes were particularly pronounced, reaching 10-fold or greater differences between genotypes.
The genotype-specific differential expression pattern of the leukotoxin operon correlated with the promoter structure of the operon as expected (Brogan et al., 1994). While a 223-bp insertion was found in the putative promoter the tkt operon in strain HK1651, this insertion was not JP2-specific. No evidence of promoter structure variation was found for the remaining 3 operons. These results suggest that the observed differential gene expression may be due to distinct gene regulation mechanisms in JP2 and non-JP2 genotypes.
Testing the role of these 4 operons in the pathogenesis of A. actinomycetemcomitans in periodontitis is beyond the scope of this study. Nevertheless, the results may be used to design experiments to test the influence of these operons on the virulence of A. actinomycetemcomitans. The differentially expressed nap operon is an interesting choice for further testing. The membrane bound nitrate reductase has been shown to reduce nitrate (terminal electron acceptor) to nitrite for anaerobic growth in E. coli (Potter et al., 1999) and may provide a similar function for A. actinomycetemcomitans. Another operon of interest encodes genes of the Tol-Pal system, which is essential for maintaining the integrity of cell envelopes in many Gram-negative bacterial species (Godlewska et al., 2009). Pal protein is one of the pathogen-associated molecular patterns and modulates host immune response via TLR2 (Godlewska et al., 2009). In A. actinomycetemcomitans, Pal is released as a soluble protein and may enhance local and systemic immune response in periodontitis (Karched et al., 2008, Paul-Satyaseela et al., 2006, Oscarsson et al., 2008). Whether JP2 genotype releases greater amounts of Pal protein than non-JP2 genotype remains to be investigated.
In conclusion, we have created a custom-designed pan-genomic microarray and validated it for applications in comparative genomic analysis and transcriptome analysis of A. actinomycetemcomitans. New genotype-specific genes or genomic islands of unknown functions were identified in strains of JP2 and non-JP2 genotypes. Some genomic islands appeared to be inserted into specific locations in the genomes in strains of JP2 and non-JP2 genotypes. Transcriptome analysis confirmed the up-regulation of the leukotoxin operon in the JP2 genotype, and further identified 4 operons whose expression patterns appeared to be genotype-specific. The information can be used to formulate hypothesis-driven experiments to examine the molecular basis of the difference in virulence between JP2 and non-JP2 genotypes of A. actinomycetemcomitans.
Supplementary Material
Acknowledgments
This study was supported by NIDCR grant R01 DE12212. We are grateful to Bruce Roe, Fares Najar, Sandy Clifton, Tom Ducey, Lisa Lewis, and Dave Dyer for the use of unpublished nucleotide sequence data from the A. actinomycetemcomitans Genome Sequencing Project at The University of Oklahoma.
References
- Asikainen S, Chen C, Slots J. Actinobacillus actinomycetemcomitans genotypes in relation to serotypes and periodontal status. Oral Microbiol Immunol. 1995;10:65–68. doi: 10.1111/j.1399-302x.1995.tb00120.x. [DOI] [PubMed] [Google Scholar]
- Bozdech Z, Zhu J, Joachimiak MP, Cohen FE, Pulliam B, DeRisi JL. Expression profiling of the schizont and trophozoite stages of Plasmodium falciparum with a long-oligonucleotide microarray. Genome Biol. 2003;4:R9. doi: 10.1186/gb-2003-4-2-r9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogan JM, Lally ET, Poulsen K, Kilian M, Demuth DR. Regulation of Actinobacillus actinomycetemcomitans leukotoxin expression: analysis of the promoter regions of leukotoxic and minimally leukotoxic strains. Infect Immun. 1994;62:501–508. doi: 10.1128/iai.62.2.501-508.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Kittichotirat W, Chen W, Downey JS, Bumgarner R. Genome sequence of a serotype b non-JP2 Aggregatibacter actinomycetemcomitans strain, ANH9381, from a periodontally healthy individual. J Bacteriol. 2012;194:1837. doi: 10.1128/JB.06770-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Kittichotirat W, Chen W, Downey JS, Si Y, Bumgarner R. Genome sequence of naturally competent Aggregatibacter actinomycetemcomitans serotype a strain D7S-1. J Bacteriol. 2010a;192:2643–2644. doi: 10.1128/JB.00157-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Kittichotirat W, Si Y, Bumgarner R. Genome sequence of Aggregatibacter actinomycetemcomitans serotype c strain D11S-1. J Bacteriol. 2009;191:7378–7379. doi: 10.1128/JB.01203-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Wang T, Chen W. Occurrence of Aggregatibacter actinomycetemcomitans serotypes in subgingival plaque from United States subjects. Molecular Oral Microbiology. 2010b;25:207–214. doi: 10.1111/j.2041-1014.2010.00567.x. [DOI] [PubMed] [Google Scholar]
- Chen W, Wang Y, Chen C. Identification of a genomic island of Actinobacillus actinomycetemcomitans. J Periodontol. 2005;76:2052–2060. doi: 10.1902/jop.2005.76.11-S.2052. [DOI] [PubMed] [Google Scholar]
- DiRienzo JM, Slots J, Sixou M, Sol MA, Harmon R, McKay TL. Specific genetic variants of Actinobacillus actinomycetemcomitans correlate with disease and health in a regional population of families with localized juvenile periodontitis. Infect Immun. 1994a;62:3058–3065. doi: 10.1128/iai.62.8.3058-3065.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiRienzo JM, Slots J, Sixou M, Sol MA, Harmon R, McKay TL. Specific genetic variants of Actinobacillus actinomycetemcomitans correlate with disease and health in a regional population of families with localized juvenile periodontitis. Infect Immun. 1994b;62:3058–3065. doi: 10.1128/iai.62.8.3058-3065.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godlewska R, Wisniewska K, Pietras Z, Jagusztyn-Krynicka EK. Peptidoglycan-associated lipoprotein (Pal) of Gram-negative bacteria: function, structure, role in pathogenesis and potential application in immunoprophylaxis. FEMS Microbiol Lett. 2009;298:1–11. doi: 10.1111/j.1574-6968.2009.01659.x. [DOI] [PubMed] [Google Scholar]
- Haubek D, Dirienzo JM, Tinoco EM, Westergaard J, Lopez NJ, Chung CP, Poulsen K, Kilian M. Racial tropism of a highly toxic clone of Actinobacillus actinomycetemcomitans associated with juvenile periodontitis. J Clin Microbiol. 1997;35:3037–3042. doi: 10.1128/jcm.35.12.3037-3042.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubek D, Ennibi OK, Poulsen K, Vaeth M, Poulsen S, Kilian M. Risk of aggressive periodontitis in adolescent carriers of the JP2 clone of Aggregatibacter (Actinobacillus) actinomycetemcomitans in Morocco: a prospective longitudinal cohort study. Lancet. 2008;371:237–242. doi: 10.1016/S0140-6736(08)60135-X. [DOI] [PubMed] [Google Scholar]
- Haubek D, Poulsen K, Kilian M. Microevolution and patterns of dissemination of the JP2 clone of Aggregatibacter (Actinobacillus) actinomycetemcomitans. Infect Immun. 2007;75:3080–3088. doi: 10.1128/IAI.01734-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan JB, Schreiner HC, Furgang D, Fine DH. Population structure and genetic diversity of Actinobacillus actinomycetemcomitans strains isolated from localized juvenile periodontitis patients. J Clin Microbiol. 2002;40:1181–1187. doi: 10.1128/JCM.40.4.1181-1187.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karched M, Ihalin R, Eneslatt K, Zhong D, Oscarsson J, Wai SN, Chen C, Asikainen SE. Vesicle-independent extracellular release of a proinflammatory outer membrane lipoprotein in free-soluble form. BMC Microbiol. 2008;8:18. doi: 10.1186/1471-2180-8-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karched M, Paul-Satyaseela M, Asikainen S. A simple viability-maintaining method produces homogenic cell suspensions of autoaggregating wild-typeActinobacillus actinomycetemcomitans. J Microbiol Methods. 2007;68:46–51. doi: 10.1016/j.mimet.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Kittichotirat W, Bumgarner R, Chen C. Markedly different genome arrangements between serotype a strains and serotypes b or c strains ofAggregatibacter actinomycetemcomitans. BMC Genomics. 2010;11:489. doi: 10.1186/1471-2164-11-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittichotirat W, Bumgarner RE, Asikainen S, Chen C. Identification of the pangenome and its components in 14 distinct Aggregatibacter actinomycetemcomitans strains by comparative genomic analysis. PLoS One. 2011;6:e22420. doi: 10.1371/journal.pone.0022420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao F, Dam P, Chou J, Olman V, Xu Y. DOOR: a database for prokaryotic operons. Nucleic Acids Res. 2009;37:D459–463. doi: 10.1093/nar/gkn757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H, Lawrence JG, Groisman EA. Lateral gene transfer and the nature of bacterial innovation. Nature. 2000;405:299–304. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- Oscarsson J, Karched M, Thay B, Chen C, Asikainen S. Proinflammatory effect in whole blood by free soluble bacterial components released from planktonic and biofilm cells. BMC Microbiol. 2008;8:206. doi: 10.1186/1471-2180-8-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paju S, Carlson P, Jousimies-Somer H, Asikainen S. Heterogeneity of Actinobacillus actinomycetemcomitans strains in various human infections and relationships between serotype, genotype, and antimicrobial susceptibility. J Clin Microbiol. 2000;38:79–84. doi: 10.1128/jcm.38.1.79-84.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul-Satyaseela M, Karched M, Bian Z, Ihalin R, Boren T, Arnqvist A, Chen C, Asikainen S. Immunoproteomics of Actinobacillus actinomycetemcomitans outer-membrane proteins reveal a highly immunoreactive peptidoglycan-associated lipoprotein. J Med Microbiol. 2006;55:931–942. doi: 10.1099/jmm.0.46470-0. [DOI] [PubMed] [Google Scholar]
- Potter LC, Millington P, Griffiths L, Thomas GH, Cole JA. Competition between Escherichia coli strains expressing either a periplasmic or a membrane-bound nitrate reductase: does Nap confer a selective advantage during nitrate-limited growth? Biochem J. 1999;344(Pt 1):77–84. [PMC free article] [PubMed] [Google Scholar]
- Ramsey MM, Whiteley M. Polymicrobial interactions stimulate resistance to host innate immunity through metabolite perception. Proc Natl Acad Sci U S A. 2009;106:1578–1583. doi: 10.1073/pnas.0809533106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rylev M, Kilian M. Prevalence and distribution of principal periodontal pathogens worldwide. J Clin Periodontol. 2008;35:346–361. doi: 10.1111/j.1600-051X.2008.01280.x. [DOI] [PubMed] [Google Scholar]
- Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, Li J, Thiagarajan M, White JA, Quackenbush J. TM4 microarray software suite. Methods Enzymol. 2006;411:134–193. doi: 10.1016/S0076-6879(06)11009-5. [DOI] [PubMed] [Google Scholar]
- Slots J. Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis in periodontal disease: introduction. Periodontology 2000. 1999;20:7–13. doi: 10.1111/j.1600-0757.1999.tb00155.x. [DOI] [PubMed] [Google Scholar]
- Smyth GK, Speed T. Normalization of cDNA microarray data. Methods. 2003;31:265–273. doi: 10.1016/s1046-2023(03)00155-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.