

Abstract
Nitrous oxide (N2O) is a key atmospheric greenhouse gas that contributes to global climatic change through radiative warming and depletion of stratospheric ozone. In this report, N2O flux was monitored simultaneously with photosynthetic CO2 and O2 exchanges from intact canopies of 12 wheat seedlings. The rates of N2O-N emitted ranged from <2 pmol⋅m−2⋅s−1 when NH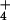 was the N source, to 25.6 ± 1.7 pmol⋅m−2⋅s−1 (mean ± SE, n = 13) when the N source was shifted to NO
was the N source, to 25.6 ± 1.7 pmol⋅m−2⋅s−1 (mean ± SE, n = 13) when the N source was shifted to NO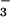 . Such fluxes are among the smallest reported for any trace gas emitted by a higher plant. Leaf N2O emissions were correlated with leaf nitrate assimilation activity, as measured by using the assimilation quotient, the ratio of CO2 assimilated to O2 evolved. 15N isotopic signatures on N2O emitted from leaves supported direct N2O production by plant NO
. Such fluxes are among the smallest reported for any trace gas emitted by a higher plant. Leaf N2O emissions were correlated with leaf nitrate assimilation activity, as measured by using the assimilation quotient, the ratio of CO2 assimilated to O2 evolved. 15N isotopic signatures on N2O emitted from leaves supported direct N2O production by plant NO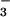 assimilation and not N2O produced by microorganisms on root surfaces and emitted in the transpiration stream. In vitro production of N2O by both intact chloroplasts and nitrite reductase, but not by nitrate reductase, indicated that N2O produced by leaves occurred during photoassimilation of NO
assimilation and not N2O produced by microorganisms on root surfaces and emitted in the transpiration stream. In vitro production of N2O by both intact chloroplasts and nitrite reductase, but not by nitrate reductase, indicated that N2O produced by leaves occurred during photoassimilation of NO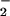 in the chloroplast. Given the large quantities of NO
in the chloroplast. Given the large quantities of NO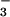 assimilated by plants in the terrestrial biosphere, these observations suggest that formation of N2O during NO
assimilated by plants in the terrestrial biosphere, these observations suggest that formation of N2O during NO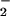 photoassimilation could be an important global biogenic N2O source.
photoassimilation could be an important global biogenic N2O source.
Plants play a critical role in regulating the chemical and physical state of the atmosphere through the exchange of biogenic greenhouse gases. Most notable are plant–atmosphere exchanges of CO2, O2, and H2O, but leaves also emit a variety of carbon- and nitrogen-based trace gases involved in climate alteration processes (1). One such trace gas is nitrous oxide (N2O). Plants—either aerenchymous (2) or nonaerenchymous (3, 4)—can serve as conduits for N2O between the soil and atmosphere. They transpire significant quantities of N2O when its concentration in the soil solution greatly exceeds the solution equilibrium concentration with ambient N2O, currently at ≈315 nmol⋅mol−1 (5).
The global N2O budget is beset by uncertainty, and sources of N2O have historically fallen short of the primary sink, photolysis in the upper atmosphere (6–9). The primary biogenic N2O sources are from soils (70%) and involve the microbial nitrogen transformations brought about by nitrification and denitrification (6). Although nitrification and denitrification are the major N2O sources, several microbial organisms that do not nitrify or denitrify can also produce N2O during NO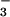 assimilation (10, 11). These observations have led to the general hypothesis that any enzymatic nitrogen transformation through the +2 to +1 oxidation state may generate N2O (12). One such transformation in higher plants is NO
assimilation (10, 11). These observations have led to the general hypothesis that any enzymatic nitrogen transformation through the +2 to +1 oxidation state may generate N2O (12). One such transformation in higher plants is NO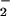 assimilation in chloroplasts. Nitrite assimilation in chloroplasts can generate intermediates capable of reacting to produce N2O, including NO
assimilation in chloroplasts. Nitrite assimilation in chloroplasts can generate intermediates capable of reacting to produce N2O, including NO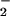 (as HNO2) with hydroxylamine (13) or reaction of NO released during NO
(as HNO2) with hydroxylamine (13) or reaction of NO released during NO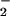 reduction (14, 15) with ascorbate (16). Nonetheless, early attempts to observe N2O production by higher plant tissues were not successful (10) and were probably limited by lack of an analytical method capable of detecting plant N2O emission at the exceptionally slow rates reported here. We developed an analytical approach by using cryogenic trapping (17) and gas chromatography coupled to high-precision isotope ratio mass spectrometry (18). This approach resolved leaf N2O emissions at more than six orders of magnitude lower than photosynthetic gas exchanges of CO2 and O2 (Table 1), placing such fluxes among the smallest ever reported for any trace gas emitted by a higher plant (1).
reduction (14, 15) with ascorbate (16). Nonetheless, early attempts to observe N2O production by higher plant tissues were not successful (10) and were probably limited by lack of an analytical method capable of detecting plant N2O emission at the exceptionally slow rates reported here. We developed an analytical approach by using cryogenic trapping (17) and gas chromatography coupled to high-precision isotope ratio mass spectrometry (18). This approach resolved leaf N2O emissions at more than six orders of magnitude lower than photosynthetic gas exchanges of CO2 and O2 (Table 1), placing such fluxes among the smallest ever reported for any trace gas emitted by a higher plant (1).
Table 1.
Shoot and rhizosphere gas exchange rates for 14-day-old wheat seedlings (T. aestivum L. cv. Veery 10)
| N source | Photosynthesis
|
N2O-N emissions
|
|||
|---|---|---|---|---|---|
| CO2, μmol⋅m−2⋅s−1 | O2, μmol⋅m−2⋅s−1 | AQ, CO2/O2 | Shoot, pmol⋅m−2⋅s−1 | Rhizosphere, pmol⋅g−1⋅s−1 | |
| 50 μM (15NH4)2SO4 | 14.31 ± 1.32 | 12.06 ± 0.77 | 1.21 ± 0.06 | ND | 40.4 ± 2.1 |
| 100 μM K15NO3 | 15.32 ± 1.48 | 13.63 ± 0.84 | 1.13 ± 0.05 | 25.59 ± 1.68 | 304.1 ± 93.8 |
Shown for shoots are net photosynthetic CO2 assimilation rates, net photosynthetic O2 evolved, AQ (the ratio of CO2 assimilated to O2 evolved), and shoot N2O emissions (mean ± SE, n = 11). Shown for the rhizosphere are N2O production rates (n = 7).
Identifying and quantifying plant N2O exchange is important. Atmospheric N2O concentration is increasing at a rate of about 0.27% per year (19), and each mol of N2O has ≈290 times the radiative forcing potential of CO2 (20). Consequently, N2O will account for as much as 7% of projected atmospheric warming (21). In addition to its greenhouse gas properties, photolytic reaction with excited oxygen [O(1D)] in the upper atmosphere produces nitric oxide (NO), and NO, in turn, consumes stratospheric ozone (22). Biogenic and anthropogenic sources of N2O are poorly constrained (23) and often do not account for the quantity of N2O known to undergo photolysis in the upper atmosphere (6). Extreme heterogeneity of soil N2O emissions largely contributes to such uncertainty, but unidentified hydrologic or biogenic sources may also play a role (7, 8). Kroeze et al. (24) have argued for closure of the global N2O budget, but the theoretical uncertainty range for estimates of N2O from agricultural soils is extreme [0.6–14.8 Tg N2O-N y−1 (6)], and new evidence suggests that N2O emissions from agricultural soils may be in the lower range of recent Intergovernmental Panel on Climate Change (IPCC) estimates (25). To moderate the atmospheric increases of N2O and to better understand the role of the biosphere in its production, it is critical to identify all major N2O sources and exchange pathways, including any potential contributions by plants.
Methods
Wheat seeds (Triticum aestivum L. cv. Veery 10) were surface sterilized, germinated on rolled moist germination paper, and transferred to opaque hydroponics tanks containing dilute nutrient solutions (26). The hydroponics systems were kept in a controlled environment chamber (PGV36, Conviron, Winnipeg, MB, Canada) with a 25°C, 16-h, 600 μmol⋅m−2⋅s−1 photosynthetic photon flux density day and a 20°C, 8-h night. After 10 days, when the plants had three true leaves, 12 individuals were transferred into a gas exchange chamber where their shoots produced a canopy with a leaf surface area of about 0.02 m2. Roots were sealed into 12 individual gas-tight cuvettes that were connected to the lower surface of a platform over which the canopy chamber was sealed (27).
Net canopy CO2 and O2 exchanges were measured under steady-state conditions. A differential infrared gas analyzer (VIA-500R, Horiba, Sunnyvale, CA) monitored net CO2 exchange. From a second parallel gas stream, a custom oxygen analyzer that resolves a 2 μmol⋅mol−1 O2 partial pressure difference on a background of 209,460 μmol⋅mol−1 monitored net O2 exchange (28). Before passing through the O2 electrodes, gases that can interfere with O2 measurements, like water vapor and plant secondary carbon compounds, were cryogenically condensed to constant trace levels by using a liquid argon trap. The CO2 concentration during the gas exchange measurements was sustained at 330 μmol⋅mol−1 and the N2O concentration at 280 nmol⋅mol−1. A third parallel gas stream exiting the plant chamber flowed at 1.0–1.5 dm3 s−1 through an ascarite filter to remove CO2, a drierite filter to remove H2O, and then a second cryogenic trap cooled with liquid argon to condense N2O (17). This trap concentrated up to 10 μmol⋅mol−1 N2O in 15 min when the N2O concentration in the gas stream was at 280 nmol⋅mol−1. Gas trapped in the condenser was injected into a mass spectrometer tuned to determine the ratio of ion currents m/z at 45/44 and 46/44 (18). The rate of N2O exchange by shoots (mol⋅m−2 s−1) was calculated as FN2O = (Jl⋅dl⋅Cl/A) (29). Jl is the flow rate of air through the plant chamber (mol⋅s−1). dl is the mol fraction of N2O proportional to canopy N2O emission, calculated according to the isotopic enrichments in masses 45N2O and 46N2O (30). Cl is the concentration of N2O in the gas stream exiting the chamber. A is leaf area (m2). The theoretical detection limit for leaf N2O emission by the mass spectrometer, on the basis of the observed variation in the mass ratios reported for a 1.5 μmol⋅mol−1 N2O standard, was ≈2 pmol⋅m−2⋅s−1.
During the first 24 h of our experiments, roots received a nitrogen source containing 50 μM (15NH4)2SO4 (99.6 atom % 15N), and shoot gas fluxes of CO2, O2 and N2O were assessed during the final 6 h. This pretreatment allowed us to purge the xylem stream of NO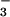 and establish a baseline value for assimilation quotient (AQ, CO2/O2) when little NO
and establish a baseline value for assimilation quotient (AQ, CO2/O2) when little NO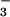 was undergoing assimilation in leaves. The nitrogen source was then shifted to 100 μM K15NO3 (99.6 atom % 15N) for 24 h, and shoot gas fluxes were again assessed during the final 6 h.
was undergoing assimilation in leaves. The nitrogen source was then shifted to 100 μM K15NO3 (99.6 atom % 15N) for 24 h, and shoot gas fluxes were again assessed during the final 6 h.
For rhizosphere N2O production, the nutrient solution was delivered to roots by using a gas tight, continuous flow system (31). Fluxes of N2O from the root cuvettes represent the production of N2O by roots and any microbial organisms on the root surface capable of generating N2O (32). Before passing through the root cuvettes, the solution concentrations of O2 and N2O were brought to their respective saturation concentrations. Dissolved oxygen concentration in the nutrient solution passing through the root cuvettes declined by only about 20% and the solutions were well stirred, so denitrification in the rhizosphere was minimized. During the experiments, two 5-ml samples of nutrient solution were collected into 15-ml septum bottles. One sample was collected before the nutrient solution entered the root cuvette and another after it exited the root cuvette. The head space gases from the septum bottles were injected into the mass spectrometer after equilibration at 22.5°C to determine the concentration of N2O and the ratio of ion currents m/z at 45/44 and 46/44. The rate of N2O production by the rhizosphere (mol⋅g−1⋅s−1) was calculated as FN2O = (Jr⋅dr⋅Cr/W). Jr is the flow rate of nutrient solution through the root cuvette (liter⋅s−1). dr is the mol fraction of N2O in the nutrient solution proportional to rhizosphere N2O production, and calculated according to the isotopic enrichments in masses 45N2O and 46N2O (30). Cr is the total concentration of N2O (mol⋅liter−1) in the nutrient solution (33). W is root dry mass.
For chloroplast assays, ≈40 g of fresh leaves from 2-week-old hydroponics-grown wheat plants was blended in a buffer solution containing 0.05 M K-Hepes (pH 7.3), 0.33 M Sorbitol, 1 mM MgCl2, 1 mM MnCl2, 2 mM Na2EDTA, and 0.1% BSA. The extract was centrifuged at 3,000 × g for 5 min, resuspended in a 50/50 Percol gradient, and centrifuged at 7,000 × g for 10 min. Intact chloroplasts were then collected and washed with 0.05 M K-Tricine (pH 8.0) and 0.33 M Sorbitol. The washed chloroplasts were introduced into 5 ml of an incubation medium consisting of 0.3 mM K15NO2 (99.6 atom % 15N) in 0.05 M K-Tricine (pH 8.0), 0.33 M sorbitol, and 0.3 mM NaHCO3. The chloroplasts were incubated for 40 min in 15-ml septum bottles at 25°C and a light intensity of 600 μmol⋅m−2⋅s−1 photosynthetic photon flux density. At the end of the incubation period, the head-space gases were collected and injected into the mass spectrometer. The concentration of N2O and ratio of ion currents m/z at 45/44 and 46/44 were used to calculate the quantity of N2O produced, as previously described for the dynamic flow measurements.
For nitrate and nitrite reductase assays (34), 2 g of fresh leaves from 2-week-old hydroponics-grown wheat plants was ground in a mortar and pestle with cold-purified N-free sand in 8 ml of a chilled buffer solution. The buffer solution consisted of 0.05 M Tris (pH 8.5), 1 mM EDTA, 1 μM NaMoO4, 10 μM FAD, 1 mM DTT, 10 μM leupeptin, and 1 μg ml−1 pepstatin. The extract was centrifuged at 30,000 × g for 20 min. Then 0.05 ml of the supernatant was added to 0.95 ml of an assay solution containing 62.5 mM potassium phosphate buffer (pH 7.5), 0.7 mM K15NO2 (99.6 atom % 15N), and 0.052 g ml−1 of methyl viologen in a 15-ml septum bottle. The reaction was initiated by injecting 0.2 ml of a solution containing 8.3 mg ml−1 Na2S2O4, and incubated at 30°C for 15 min. For nitrate reductase, 0.05 ml of the supernatant was added to 0.95 ml of assay buffer containing 1.4 mM K15NO3 (99.6 atom % 15N). The reaction was initiated by adding 0.2 ml of a solution containing 2 mg ml−1 NADH and incubated at 30°C for 15 min. The head-space gases from the nitrite and nitrate reductase assays were collected and analyzed on the mass spectrometer as previously described.
Results and Discussion
This is the first study, to our knowledge, to report quantitative leaf N2O emissions under normal physiological conditions for an intact plant by using steady-state gas exchange methods. Earlier investigations have examined detached leaves or used static chamber methods, where CO2, H2O, and temperature change rapidly (3, 4, 10), and nitrogen source (NH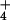 and NO
and NO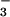 ) cannot be controlled. These preliminary studies were valuable in that they clearly demonstrated N2O can move from soil to atmosphere via the plant transpiration stream, but they did not resolve the question of N2O production by plant nitrogen metabolism.
) cannot be controlled. These preliminary studies were valuable in that they clearly demonstrated N2O can move from soil to atmosphere via the plant transpiration stream, but they did not resolve the question of N2O production by plant nitrogen metabolism.
In our investigation, leaves did not emit N2O at a detectable rate while metabolizing 15NH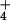 . The AQ during 15NH
. The AQ during 15NH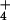 exposure averaged 1.21 ± 0.05 units (Table 1). When the N source was shifted to 15NO
exposure averaged 1.21 ± 0.05 units (Table 1). When the N source was shifted to 15NO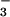 , canopy leaves emitted N2O-N at an average rate of 25.6 ± 1.7 pmol⋅m−2⋅s−1, and the AQ declined to 1.13 ± 0.06 units (Table 1). Both observations are important. The change in AQ (ΔAQ) gives a nondestructive measure of NO
, canopy leaves emitted N2O-N at an average rate of 25.6 ± 1.7 pmol⋅m−2⋅s−1, and the AQ declined to 1.13 ± 0.06 units (Table 1). Both observations are important. The change in AQ (ΔAQ) gives a nondestructive measure of NO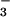 assimilation under steady-state conditions. Net O2 exchange provides a measure of photosynthetic electron transport, and either CO2 or NO
assimilation under steady-state conditions. Net O2 exchange provides a measure of photosynthetic electron transport, and either CO2 or NO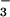 reduction can be coupled to such electron transfer (35). As NO
reduction can be coupled to such electron transfer (35). As NO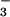 photoassimilation increases, the AQ declines because reductant produced by photosynthetic electron transport increases to support NO
photoassimilation increases, the AQ declines because reductant produced by photosynthetic electron transport increases to support NO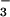 and NO
and NO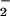 reduction in addition to CO2 fixation (28, 36). Thus, not only did shoot N2O emission become detectable when the N source was shifted to 15NO
reduction in addition to CO2 fixation (28, 36). Thus, not only did shoot N2O emission become detectable when the N source was shifted to 15NO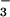 , but the strong correlation with ΔAQ (Fig. 1A) suggested that leaf N2O flux was driven by leaf NO
, but the strong correlation with ΔAQ (Fig. 1A) suggested that leaf N2O flux was driven by leaf NO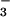 assimilation.
assimilation.
Figure 1.

The relationship between N2O emission from wheat leaves (pmol N2O-N m−2⋅s−1) and (A) the change in assimilatory quotient (ΔAQ) when the nitrogen source was shifted from 15NH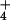 to 15NO
to 15NO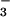 , or (B) N2O production in the rhizosphere (pmol N2O-N g−1⋅s−1). Shown are the regression lines and the R2 statistic. The rates of N2O emission were normalized by using the log10 transformation.
, or (B) N2O production in the rhizosphere (pmol N2O-N g−1⋅s−1). Shown are the regression lines and the R2 statistic. The rates of N2O emission were normalized by using the log10 transformation.
On the other hand, rhizosphere N2O production also increased more than 7-fold (Table 1) with the change in N source from 15NH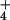 to 15NO
to 15NO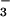 (Table 1), raising N2O concentration in the nutrient solution around roots to an average of 89.6 nmol⋅liter−1. Such a concentration is an order of magnitude higher than the solution equilibrium concentration when N2O in the shoot chamber is near ambient, so that movement of N2O in the transpiration stream could have been significant. At least two experimental observations did not support this scenario. First, shoot N2O emission during exposure to 15NO
(Table 1), raising N2O concentration in the nutrient solution around roots to an average of 89.6 nmol⋅liter−1. Such a concentration is an order of magnitude higher than the solution equilibrium concentration when N2O in the shoot chamber is near ambient, so that movement of N2O in the transpiration stream could have been significant. At least two experimental observations did not support this scenario. First, shoot N2O emission during exposure to 15NO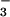 was not correlated with root zone N2O production (Fig. 1B). Second, leaf N2O flux fell below detectable limits during exposure to 15NH
was not correlated with root zone N2O production (Fig. 1B). Second, leaf N2O flux fell below detectable limits during exposure to 15NH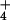 despite high rates of N2O production by the rhizosphere (Table 1). The production of N2O during 15NH
despite high rates of N2O production by the rhizosphere (Table 1). The production of N2O during 15NH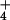 exposure comes from the activity of nitrifying bacteria on the root surface (37), an activity we were able to completely shut down by using the nitrification inhibitor nitrapyrin. Thus, the absence of a correlation between shoot N2O emission and rhizosphere N2O production provides further evidence that photoassimilation of NO
exposure comes from the activity of nitrifying bacteria on the root surface (37), an activity we were able to completely shut down by using the nitrification inhibitor nitrapyrin. Thus, the absence of a correlation between shoot N2O emission and rhizosphere N2O production provides further evidence that photoassimilation of NO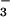 was the major source of N2O emitted from leaves in this investigation. For investigations where N2O transpiration was responsible for leaf N2O flux (6), the concentrations of N2O in the soil solution were nearly four orders of magnitude higher, at ≈326 μmol⋅liter−1, than N2O concentrations observed in our nutrient solutions. This may help to explain why N2O transpiration was not a factor in this investigation.
was the major source of N2O emitted from leaves in this investigation. For investigations where N2O transpiration was responsible for leaf N2O flux (6), the concentrations of N2O in the soil solution were nearly four orders of magnitude higher, at ≈326 μmol⋅liter−1, than N2O concentrations observed in our nutrient solutions. This may help to explain why N2O transpiration was not a factor in this investigation.
The isotopic composition of N2O emitted from leaves provided further verification that shoot NO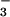 assimilation was largely responsible for the observed N2O flux. In the course of the experiments, we used nitrogen sources in nutrient solutions highly enriched in 15N (>99.6 atom % 15N as NH
assimilation was largely responsible for the observed N2O flux. In the course of the experiments, we used nitrogen sources in nutrient solutions highly enriched in 15N (>99.6 atom % 15N as NH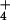 or NO
or NO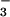 ). Thus, any N2O produced by microbial metabolism in the rhizosphere, nitrification or denitrification, and subsequently transpired, would be predominantly composed of the 15N15N16O or 46N2O isoform. We grew our wheat plants in a nutrient solution with 200 μM NH
). Thus, any N2O produced by microbial metabolism in the rhizosphere, nitrification or denitrification, and subsequently transpired, would be predominantly composed of the 15N15N16O or 46N2O isoform. We grew our wheat plants in a nutrient solution with 200 μM NH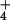 plus 200 μM NO
plus 200 μM NO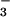 containing 15N close to natural abundance levels (0.3674% 15NH
containing 15N close to natural abundance levels (0.3674% 15NH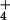 and 0.3665% 15NO
and 0.3665% 15NO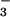 , or > 99.6 atom % 14N). These plants stored relatively large quantities of NO
, or > 99.6 atom % 14N). These plants stored relatively large quantities of NO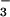 in their tissues (8.76 ± 2.23 mM, mean ± SE, n = 6), of which 2.25 ± 1.65 mM was withdrawn and assimilated during the first 24-h exposure to 15NH
in their tissues (8.76 ± 2.23 mM, mean ± SE, n = 6), of which 2.25 ± 1.65 mM was withdrawn and assimilated during the first 24-h exposure to 15NH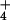 . N2O emitted during the assimilation of this internal NO
. N2O emitted during the assimilation of this internal NO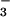 would not be detected during exposure of roots to 15NH
would not be detected during exposure of roots to 15NH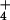 , because the isotopic signature on such stored NO
, because the isotopic signature on such stored NO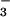 was very similar to that of N2O in the background air. However, if stored NO
was very similar to that of N2O in the background air. However, if stored NO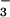 were being mobilized and assimilated in leaves during the subsequent period of root exposure to 15NO
were being mobilized and assimilated in leaves during the subsequent period of root exposure to 15NO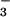 , or chloroplast reactants were participating in N2O production, then some of the N2O emitted would contain the 45N2O isoforms of 15N14N16O and 14N15N16O. Indeed, 45N2O was detected in the N2O stream emitted from wheat leaves during exposure to 15NO
, or chloroplast reactants were participating in N2O production, then some of the N2O emitted would contain the 45N2O isoforms of 15N14N16O and 14N15N16O. Indeed, 45N2O was detected in the N2O stream emitted from wheat leaves during exposure to 15NO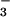 . From the mass ratios of 44N2O, 45N2O, and 46N2O collected during such experiments, we estimated (30) that 5.19 ± 0.92 pmol⋅m−2⋅s−1 (mean ± SE, n = 13), or about 20%, of the N2O-N emitted by leaves came from the 14N stored in the plant. It is unlikely that bacterial mineralization of organic-N in the rhizosphere followed by nitrification and denitrification contributed to the 45N2O emitted. Roots treated with antibiotic cocktails (38) and nitrapyrin to eliminate rhizosphere microbial nitrogen transformations still emitted detectable quantities of 45N2O. Nonetheless, the largest fraction of the observed leaf N2O flux was clearly generated by plant NO
. From the mass ratios of 44N2O, 45N2O, and 46N2O collected during such experiments, we estimated (30) that 5.19 ± 0.92 pmol⋅m−2⋅s−1 (mean ± SE, n = 13), or about 20%, of the N2O-N emitted by leaves came from the 14N stored in the plant. It is unlikely that bacterial mineralization of organic-N in the rhizosphere followed by nitrification and denitrification contributed to the 45N2O emitted. Roots treated with antibiotic cocktails (38) and nitrapyrin to eliminate rhizosphere microbial nitrogen transformations still emitted detectable quantities of 45N2O. Nonetheless, the largest fraction of the observed leaf N2O flux was clearly generated by plant NO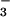 assimilation and not from N2O produced by microbial processes in the rhizosphere.
assimilation and not from N2O produced by microbial processes in the rhizosphere.
The two enzymes responsible for plant NO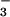 assimilation, NO
assimilation, NO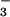 reductase (NR) and NO
reductase (NR) and NO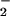 reductase (NiR), are located in the cytoplasm and chloroplasts, respectively. To determine whether NO
reductase (NiR), are located in the cytoplasm and chloroplasts, respectively. To determine whether NO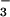 or NO
or NO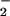 reduction was responsible for the N2O emitted, we extracted NR, NiR, and intact chloroplasts from wheat leaves and assayed for N2O production in vitro as described. Whereas NR assays did not produce any detectable N2O, NiR assays and intact chloroplasts did (Fig. 2). These experiments provide in vitro evidence that the N2O emitted from wheat leaves was generated by NO
reduction was responsible for the N2O emitted, we extracted NR, NiR, and intact chloroplasts from wheat leaves and assayed for N2O production in vitro as described. Whereas NR assays did not produce any detectable N2O, NiR assays and intact chloroplasts did (Fig. 2). These experiments provide in vitro evidence that the N2O emitted from wheat leaves was generated by NO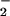 reduction to NH3 in the chloroplasts, where NO
reduction to NH3 in the chloroplasts, where NO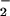 undergoes transformation through the +2 to +1 oxidation state, as predicted.
undergoes transformation through the +2 to +1 oxidation state, as predicted.
Figure 2.

N2O production in vitro from (A) intact chloroplasts (nmol N2O-N g−1 chlorophyll s−1), (B) nitrate reductase (nmol N2O-N g−1 protein s−1), and (C) nitrite reductase (nmol N2O-N g−1 protein s−1). Intact chloroplasts, nitrate reductase, and nitrite reductase were extracted from fully expanded leaves of 2- to 3-week-old wheat plants (T. aestivum L. cv. Veery 10).
The leaf N2O emissions we measured were small (Table 1), and it is not currently known how important fluxes of this magnitude are in relation to the global N2O budget. Some recent micrometeorological N2O flux measurements over grass swards failed to detect a higher N2O emission rate than that observed by using static chambers over soil alone (39). However, the error in such measurements was too large, with coefficients of variation for the chamber data ranging from 0.42 to 1.83 (40, 41), to resolve plant N2O emissions of the size measured in this investigation. In support of a role for plant N2O emission, Hutchinson and Mosier (42) found that aerodynamic flux measurements over an irrigated corn field were always higher than, although never exceeding twice the mean of, fluxes measured by using soil chambers. Some recent chamber studies found that ryegrass canopies were responsible for 21.1% of the total N2O emissions (4), and that Linum perenne canopies contributed to as much as 50% of N2O flux to the atmosphere (43). Our results indicated that 0.02–0.2% of the NO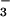 -N assimilated by wheat was released as N2O-N. Current estimates are that terrestrial land plants assimilate 1,200 Tg of N annually (44). Nearly half of this N is thought to be absorbed and assimilated as NO
-N assimilated by wheat was released as N2O-N. Current estimates are that terrestrial land plants assimilate 1,200 Tg of N annually (44). Nearly half of this N is thought to be absorbed and assimilated as NO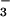 (45), of which 25–75% is assimilated in leaves (46). The NR and NiR enzymes involved are highly conserved among higher plants (47). Thus, if other terrestrial land plants behave as wheat does, we calculate that NO
(45), of which 25–75% is assimilated in leaves (46). The NR and NiR enzymes involved are highly conserved among higher plants (47). Thus, if other terrestrial land plants behave as wheat does, we calculate that NO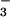 photoassimilation alone could produce from 0.03 to 0.9 Tg N2O-N yr−1. The uncertainty in the IPCC's estimates of global N2O source emissions is substantial, with nearly two orders of magnitude in the range of estimated emission of N2O from soils [0.6–14.8 Tg N y−1 (1)]. Robertson et al. (25) tracked N2O emissions for 9 years from soils of six cropping systems, including a nitrogen-intensive corn rotation and four successional communities, and found emissions to be at the lower end of the IPCC's calculated emission factor, which is based on fertilizer application. Our estimates of plant N2O emissions represent ≈5–6% of the total amount of N2O-N thought to be emitted by agricultural plant–soil systems alone (1, 44). These approximations do not include the quantity of N2O that might be conducted to the atmosphere via the plant transpiration stream. Thus, our results suggest that higher plants could play an intriguing role in N2O exchange not previously considered in biosphere–atmosphere interactions.
photoassimilation alone could produce from 0.03 to 0.9 Tg N2O-N yr−1. The uncertainty in the IPCC's estimates of global N2O source emissions is substantial, with nearly two orders of magnitude in the range of estimated emission of N2O from soils [0.6–14.8 Tg N y−1 (1)]. Robertson et al. (25) tracked N2O emissions for 9 years from soils of six cropping systems, including a nitrogen-intensive corn rotation and four successional communities, and found emissions to be at the lower end of the IPCC's calculated emission factor, which is based on fertilizer application. Our estimates of plant N2O emissions represent ≈5–6% of the total amount of N2O-N thought to be emitted by agricultural plant–soil systems alone (1, 44). These approximations do not include the quantity of N2O that might be conducted to the atmosphere via the plant transpiration stream. Thus, our results suggest that higher plants could play an intriguing role in N2O exchange not previously considered in biosphere–atmosphere interactions.
Acknowledgments
We thank Na Trinh for assistance in many phases of these experiments and Duy Nguyen for assistance with chloroplast and enzyme preparations. This study was supported by the Department of Energy under Grant 95ER62128 TECO and the National Science Foundation under Grant IBN-99–74927.
Abbreviation
- IPCC
Intergovernmental Panel on Climate Change
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Sharkey T D, Holland E A, Mooney H A. Trace Gas Emissions by Plants. San Diego: Academic; 1991. [Google Scholar]
- 2.Mosier A R, Mohanty S K, Bhadrachalam A, Chakravorti S P. Biol Fertil Soils. 1990;9:61–67. [Google Scholar]
- 3.Chang C, Janzen H H, Cho C M, Nakonechny E M. Soil Sci Soc Am J. 1998;62:35–38. [Google Scholar]
- 4.Chen X, Boeckx P, Shen S, Van Cleemput O. Biol Fertil Soils. 1999;28:393–396. [Google Scholar]
- 5.Battle M, Bender M, Sowers T, Tans P P, Butler J H, Elkins J W, Ellis J T, Conway T, Ahang N, Lang P, Clarke A D. Nature (London) 1996;383:231–235. [Google Scholar]
- 6.Mosier A C, Kroeze C, Nevison C, Oenema O, Seitzinger S, van Cleemput O. Nutr Cycl Agroecosyst. 1998;52:225–248. [Google Scholar]
- 7.Naqvi S W A, Yoshinari T, Jayakumar D A, Altabet M A, Narvekar P V, Devol A H, Brandes J A, Codispoti L A. Nature (London) 1998;394:462–464. [Google Scholar]
- 8.Bouwman A F, Van der Hoek K W, Olivier J G J. J Geophys Res. 1995;100:2785–2800. [Google Scholar]
- 9.Houghton J T, Callander B A, Varney S K, editors. Climate Change. The Supplementary Report to the IPCC Scientific Assessment. Cambridge, U.K.: Cambridge Univ. Press; 1992. [Google Scholar]
- 10.Bleakley B H, Tiedje J M. Appl Environ Microbiol. 1982;44:1342–1348. doi: 10.1128/aem.44.6.1342-1348.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hutchinson G L, Davidson E A. In: Agricultural Ecosystem Effects on Trace Gases and Global Climate Change, ASA, Special Publication No. 55. Harper L A, Mosier A R, Duxbury J M, Rolston D E, editors. Madison WI: ASA/CSSA/SSSA; 1993. pp. 79–94. [Google Scholar]
- 12.Firestone M K, Davidson E. In: Exchange of Trace Gases Between the Terrestrial Ecosystems and the Atmosphere: Report of the Dahlem Workshop on Exchange of Trace Gases between Terrestrial Ecosystems and the Atmosphere, Berlin 1989, February 19–24. Andreae M O, Shimel D S, editors. New York: Wiley; 1989. pp. 7–21. [Google Scholar]
- 13.Bothner-By A, Friedman J. J Chem Phys. 1952;20:459–462. [Google Scholar]
- 14.Aparicio P J, Knaff D B, Malkin R. Arch Biochem Biophys. 1975;169:102–107. doi: 10.1016/0003-9861(75)90321-5. [DOI] [PubMed] [Google Scholar]
- 15.Dean J V, Harper J E. Plant Physiol. 1986;82:718–723. doi: 10.1104/pp.82.3.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brudvig G W, Stevens T H, Chan S I. Biochemistry. 1980;19:5275–5285. doi: 10.1021/bi00564a020. [DOI] [PubMed] [Google Scholar]
- 17.Huff A K, Cliff S S, Thiemens M H. AnalChem. 1997;69:4267–4270. [Google Scholar]
- 18.Brooks P D, Herman D J, Atkins G J, Prosser S J, Barrie A. In: Agricultural Ecosystem Effects on Trace Gases and Global Climate Change, ASA Special Publication No. 55. Harper L A, Mosier A R, Duxbury J M, Rolston D E, editors. Madison, WI: ASA/CSSA/SSSA; 1993. pp. 193–202. [Google Scholar]
- 19.Ramanathan V, Cicerone R J, Singh H B, Kiehl J T. J Geophys Res. 1985;90:5547–5566. [Google Scholar]
- 20.Houghton J T, Meira Filho L G, Callander B A, Harris N, Kattenberg A, Maskell K, editors. IPCC. Climate Change 1995, The Science of Climate Change. Cambridge, U.K.: Cambridge Univ. Press; 1996. pp. 9–50. [Google Scholar]
- 21.Robertson G P. In: Agricultural Ecosystem Effects on Trace Gases and Global Climate Change, ASA Special Publication No. 55. Harper L E, Mosier A R, Duxbury J M, Rolston D E, editors. Madison, WI: ASA/CSSA/SSSA; 1993. pp. 96–108. [Google Scholar]
- 22.Cicerone R J. Science. 1987;237:35–42. doi: 10.1126/science.237.4810.35. [DOI] [PubMed] [Google Scholar]
- 23.IPCC. Intergovernmental Panel on Climate Change Guidelines for National Greenhouse Gas Inventories. Chapter 4. Agriculture: Nitrous Oxide from Agricultural Soils and Manure Management, Paris, France, 1997. Paris: Organization for Economic Cooperative Development; 1997. [Google Scholar]
- 24.Kroeze C, Mosier A R, Bouwman L. Global Biogeochem Cycles. 1999;13:1–8. [Google Scholar]
- 25.Robertson G P, Paul E A, Harwood R R. Science. 2000;289:1922–1925. doi: 10.1126/science.289.5486.1922. [DOI] [PubMed] [Google Scholar]
- 26.Smart D R, Ritchie K, Bloom A J, Bugbee B. Plant Cell Environ. 1998;21:753–763. doi: 10.1046/j.1365-3040.1998.00315.x. [DOI] [PubMed] [Google Scholar]
- 27.Kosola K R, Bloom A J. Plant Physiol. 1994;104:435–442. doi: 10.1104/pp.105.1.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bloom A J, Caldwell R M, Finazzo J, Warner R L, Weissbart J. Plant Physiol. 1989;91:352–356. doi: 10.1104/pp.91.1.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sestak Z, Catsky J, Jarvis P G. Plant Photosynthetic Production: Manual of Methods. The Hague, The Netherlands: W. Junk; 1971. [Google Scholar]
- 30.Mulvaney R L, Boast C W. Soil Sci Soc Am J. 1986;50:360–363. [Google Scholar]
- 31.Bloom A J. In: Application of Continuous and Steady State Methods to Root Biology. Torrey J G, Winship L J, editors. Dordrecht, The Netherlands: Kluwer; 1989. pp. 147–163. [Google Scholar]
- 32.Smart D R, Ritchie K, Stark J M, Bugbee B. Appl Environ Microbiol. 1997;63:4621–4624. doi: 10.1128/aem.63.11.4621-4624.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilhelm E, Battino R, Wilcock R J. Chem Rev. 1977;77:219–248. [Google Scholar]
- 34.Aslam M, Huffaker R C. Plant Physiol. 1989;91:1152–1156. doi: 10.1104/pp.91.3.1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knaff D B. In: Oxygenic Photosynthesis: The Light Reactions. Ort D R, Yocum C F, editors. Vol. 4. Dordrecht, The Netherlands: Kluwer; 1996. pp. 333–361. [Google Scholar]
- 36.Cramer M, Myers J. J Gen Physiol. 1948;32:93–102. doi: 10.1085/jgp.32.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Padgett P E, Leonard R T. Plant Physiol. 1993;101:141–146. doi: 10.1104/pp.101.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smart D R, Ferro A, Ritchie K, Bugbee B B. Physiol Plant. 1995;95:533–540. [PubMed] [Google Scholar]
- 39.Hargreaves K J, Skiba U, Fowler D, Arah J, Wienhold F G, Klemedtsson L, Galle B. J Geophys Res. 1994;99:16569–16574. [Google Scholar]
- 40.Wienhold F G, Frahm H, Harris G W. J Geophys Res. 1994;99:16557–16567. [Google Scholar]
- 41.Clayton H, Arah J R M, Smith K A. J Geophys Res. 1994;99:16599–16607. [Google Scholar]
- 42.Hutchinson G L, Mosier A R. Science. 1979;205:1125–1127. doi: 10.1126/science.205.4411.1125. [DOI] [PubMed] [Google Scholar]
- 43.Anderson L, Hopkins D W. In: Gaseous Nitrogen Emissions from Grasslands. Jarvis S C, Pain B F, editors. New York: CAB International; 1997. pp. 222–224. [Google Scholar]
- 44.Schlesinger W H. Biogeochemistry: An Analysis of Global Change. San Diego: Academic; 1997. [Google Scholar]
- 45.Raven J A, Wollenweber B, Handley L L. Physiol Plant. 1993;89:512–518. [Google Scholar]
- 46.Andrews M. Plant Cell Environ. 1986;9:511–519. [Google Scholar]
- 47.Caboche M, Rouze P. Trends Genet. 1990;6:187–192. doi: 10.1016/0168-9525(90)90175-6. [DOI] [PubMed] [Google Scholar]