

Summary
Background
The signal recognition particle (SRP) is a phylogenetically conserved ribonucleoprotein that mediates cotranslational targeting of secreted and membrane proteins to the membrane. Targeting is regulated by GTP binding and hydrolysis events that require direct interaction between structurally homologous “NG” GTPase domains of the SRP signal recognition subunit and its membrane-associated receptor, SRα. Structures of both the apo and GDP bound NG domains of the prokaryotic SRP54 homolog, Ffh, and the prokaryotic receptor homolog, FtsY, have been determined. The structural basis for the GTP-dependent interaction between the two proteins, however, remains unknown.
Results
We report here two structures of the NG GTPase of Ffh from Thermus aquaticus bound to the nonhydrolyzable GTP analog GMPPNP. Both structures reveal an unexpected binding mode in which the β-phosphate is kinked away from the binding site and magnesium is not bound. Binding of the GTP analog in the canonical conformation found in other GTPase structures is precluded by constriction of the phosphate binding P loop. The structural difference between the Ffh complex and other GTPases suggests a specific conformational change that must accompany movement of the nucleotide from an “inactive” to an “active” binding mode.
Conclusions
Conserved side chains of the GTPase sequence motifs unique to the SRP subfamily may function to gate formation of the active GTP bound conformation. Exposed hydrophobic residues provide an interaction surface that may allow regulation of the GTP binding conformation, and thus activation of the GTPase, during the association of SRP with its receptor.
Keywords: SRP, Ffh, NG domain, GTPase, GMPPNP, X-ray crystallography
Introduction
The structure of the GTPase domain of Ffh (the NG domain) from Thermus aquaticus has been determined at 2.0 Å resolution both in the absence of nucleotide [1] and with bound GDP and Mg2+GDP [2]. The structures of the apo NG domains of Acidianus ambivalens Ffh and Escherichia coli FtsY have also been reported [3, 4]. The protein fold has similarity to other well-known GTPases, such as Ras and EF-Tu, with four well-conserved sequence motifs I–IV defining the GTP binding site. An N-terminal α-helical bundle, called the N domain, and an insertion of the G domain, called the “IBD” subdomain, are both unique to the SRP GTPases and pack against opposite sides of the core GTPase fold [1]. In the presence of magnesium, GDP binds to the T. aquaticus NG domain in a conformation similar to that seen in other GTPases [2]; however, in the absence of magnesium, the β-phosphate of the GDP is turned away from the active site to interact with a glutamine side chain (Gln144), which is universally conserved in the SRP GTPase subfamily. It was proposed that the two conformations reflect different states in the GDP exchange cycle [2].
The binding mode for GTP in other members of the GTPase superfamily has been revealed through numerous structures of complexes with the nonhydrolyzable GTP analogs GTPγS and GMPPNP [5–11]. The conformations of the bound GTP analogs are very similar in the different structures, and both structural [12] and kinetic [13] studies have validated the analogs as tools for understanding the GTP bound state of the proteins. A subset of the interactions between GTP and protein are common to all GTPases. These include hydrogen bonds and packing interactions between the guanine base and motif IV aspartate and lysine side chains, hydrogen bonds between the α-, β-, and γ-phosphate oxygens and main chain and side chain atoms of the motif I P loop, and the coordination of β- and γ-phosphate oxygens to a bound Mg2+ ion (see Figures 1 and 2a). Several direct and water-mediated interactions of motif III with the γ-phosphate group are structurally conserved between different GTPases [14]. Motif III also contributes side chains that are unique to each GTPase and that mediate, with motif II residues unique to each subfamily, the specific conformational and functional properties of each GTPase and thus manifest the “GTPase switch” [15]. It was these structural changes that we sought to identify by crystallization of the NG domain of Ffh in the presence of Mg2+GMPPNP.
Figure 1. GMPPNP Binding to the NG Domain.
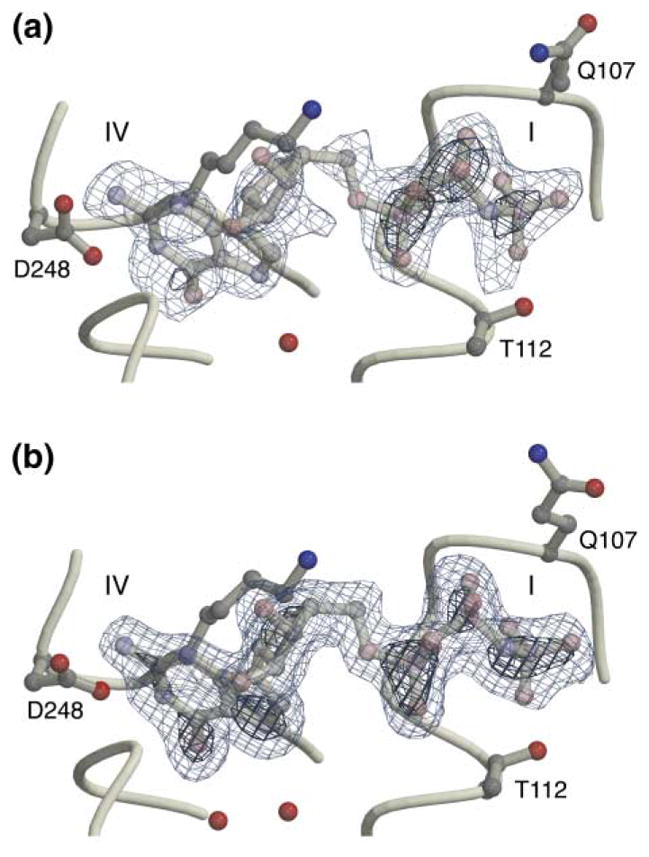
Omit difference (Fo − Fc) electron density maps contoured at 3 σ (light blue) and 6 σ (dark blue) for (a) structure N1 and (b) structure N2a. The triplet of electron-dense peaks to the right in each image indicates the positions of the phosphate groups. Two residues, Gln107 and Thr112, define the top and bottom of the P loop jaws.
Figure 2. Comparison of the Conformation of the GMPPNP with Other Structures.

(a) The GMPPNP conformation in a complex with EF-Tu (1exm [7], representative of other GTPase structures). For clarity, only the side chains of Val20 (which corresponds in position to Gln107 of the SRP GTPase), Lys24, Thr 25, and Thr26 are shown.
(b) A stereo image of the GMPPNP conformation observed in the NG complex. The orientation is approximately perpendicular to that in Figure 1. Only the side chains of Gln107, Lys111, Thr112, and Thr113 are shown.
(c) Three structures (GDP, Mg2+GDP [2], and GMPPNP N2a) are shown superimposed after alignment of the core GTPase domain of each with the other. The two GDP conformations are shown in blue.
Results and Discussion
Two Structures of the GMPPNP Complex Reveal a Novel Binding Mode
Crystals of the complex of NG and the nonhydrolyzable GTP analog GMPPNP were obtained in the presence of magnesium under two different crystallization conditions at 4°C. HPLC analysis of crystallization drops revealed essentially no degradation of the GMPPNP before the crystals were harvested. The structures of the complexes in the two crystal forms, here termed N1 and N2, were determined at 2.3 Å and 1.9 Å, respectively, by using molecular replacement; they have been refined to crystallographic R factors of 20.3% and 19.0%, with corresponding free R factors of 29.9% and 24.0% (Table 1). In structure N2 there are two monomers related by noncrystallographic symmetry (N2a and N2b). Therefore, the two different crystal forms give three independent images of the binding site, and in each case, unbiased difference electron density maps obtained at the initial stages of the work clearly defined the conformation of the bound ligand (Figure 1). Differences between the proteins in the two crystal forms are localized primarily to the N-domain, which is affected by different crystal packing interactions, and the “closing loop”, which in structure N2 is slightly more open (Figure 1b). However, the conformations of the phosphate binding pocket and the IBD change little, and the catalytic residues of the GTPase active site in each structure appear to be in their “empty” conformations [1]. Indeed, the core region of the G domains of structures N1 and N2 (spanning residues 99–219) can be superimposed with the structure of the apo protein with a root-mean-square deviation on α carbons of only 0.25–0.35 Å.
Table 1.
Data Collection Statistics
| N1 | N2 | |
|---|---|---|
| Space group | P212121 | C2 |
| Unit cell (Å) | a = 52.69, b = 60.41, c = 86.06 | a = 108.81, b = 54.53, c = 99.08 β = 97.42° |
| Monomers/AU | 1 | 2 |
| Resolution (Å) | 2.3 | 1.9 |
| Rsyma (%) | 6.4 (17.1)b | 5.1 (7.6) |
| Completeness (%) | 85.3 (95.4) | 100.0 (100.0) |
| Redundancy (%) | 7.6 (8.1) | 10.4 (9.5) |
| Average I/σ(I) | 13.7 | 28.6 |
|
| ||
| Refinement Statistics
| ||
| Number of reflections F > σ(F) (test set) | 10,233 (830) | 40,885 (3,634) |
| Rcrystc (%) | 20.3 | 19.0 |
| Rfreec (%) | 29.9 | 24.0 |
| Number of protein atoms | 2267 | 4549 |
| Number of GMPPNP atoms | 32 | 64 |
| Number of water/solute atoms | 63 | 454 |
| Average B factor (Å2) | ||
| Protein | 23.2 | 22.4 |
| GMPPNP | 17.6 | 42.0 d |
| Water molecules | 20.7 | 34.1 |
Rsym = Σ|Ih − <Ih>|/ΣIh, where <Ih> is the average intensity over symmetry equivalents.
Numbers in parentheses are the high-resolution bin.
Rcryst = Σ|Fo − Fc|/ΣFo, Rfree was calculated for a test set of reflections (8%) omitted from the refinement.
See discussion of N2 ligand B factors in the text.
The presence of GMPPNP stabilizes the closing loop and organizes the binding site such that the interactions of the guanine base and the positions of the ribose and α-phosphate groups are similar to those seen in the GDP and Mg2+GDP complexes of the NG GTPase [2]. Remarkably, however, the phosphate chain is not bound in the extended conformation seen in the structures of other GTPases (Figure 2a) but is instead found in a kinked conformation in which the β-phosphate is rotated away from the P loop and the γ-phosphate is turned back toward it (Figure 2b). To our knowledge, this binding mode is unprecedented in the superfamily of Ras-related GTPases. The position of the β-phosphate is reminiscent of its “flipped out” position in the magnesium-free GDP complex of the NG GTPase (Figure 2c), although, in contrast to the interaction seen in that structure, the β-phosphate group does not appear to hydrogen bond to the side chain of invariant Gln144 extending from the IBD. Interestingly, the phosphate groups of the bound GMPPNP exploit the same backbone and side chain atoms of the P loop that contribute to binding in the canonical mode by forming an alternative pattern of hydrogen bonding. In the canonical conformation, eight hydrogen bonds can be formed between the phosphate oxygens and the backbone and side chain atoms of the P loop (Figure 2a). In the NG complex, an alternative set of seven hydrogen bonds is available to the phosphate groups of the bound GMPPNP (Figure 2b).
Although MgCl2 was present in both crystallization solutions (and was the only available divalent cation in condition N1), there is no evidence of metal ion binding at the canonical magnesium site in either structure. Magnesium was available under the crystallization conditions because crystals of the GDP complex grown under the conditions of form N2 revealed Mg2+ bound as expected (data not shown). Interestingly, in the absence of bound magnesium, the temperature factors of the GMPPNP ligand increase systematically toward the γ-phosphate in each of the binding sites. In structure N1, the difference between the temperature factors of the nucleoside (averaging ~15 Å2) and the phosphate groups (~21 Å2) is relatively small; in structure N2a the difference is somewhat larger, although the positions of the bound phosphate groups are well defined. However, in the second monomer of form N2, N2b, the ligand temperature factors are particularly high (to 70 Å2), and there is evidence for only partial occupancy of the site (presumably due to a different crystal-packing environment). We observe an acetate ion from the crystallization buffer deep in the active site pockets of structures N2a and N2b; it has no direct interaction with the bound ligand but perhaps plays a role in the disorder of the terminal phosphate groups. In the N1 crystal form there is no acetate present, and no other ions are observed in the active site pocket.
The Nucleotide Cannot Bind in the Canonical Conformation
It is not clear from simple inspection why the kinked binding mode for the nucleotide analog is preferred in the T. aquaticus NG GTPase; the active site of the GTPase is relatively open, and the γ-phosphate binding pocket presented by the apo protein is deep [1, 4]. To investigate further, we modeled GMPPNP in the conformation observed in other GTPase structures into the structure of the Mg2+GDP complex of the NG domain [2]. The positions of the Mg2+ ion and two of its coordinating groups (Thr112 OG1 and a β-phosphate oxygen) and the placement of one of the γ-phosphate oxygens at the position of the coordinating water molecule trans to the threonine side chain provided reference points that oriented the modeled GMPPNP molecule relative to the P loop. We optimized the superimposition of the chosen reference points by using LSQMAN [16] and then evaluated the model using computer graphics and packing analysis [17]. In the modeled conformation, the α- and β-phosphate groups of the GMPPNP are in their canonical positions, and the hydrogen atom of the bridging amido group is easily accommodated just outside the P loop (as is seen in the crystal structures of other GTPases). However, an oxygen atom of the modeled γ-phosphate is positioned only 2.5–2.8 Å from the backbone and side chain atoms of Gln107 and, therefore, cannot be accommodated within the P loop (Figure 3a). This clash provides the only apparent constraint on positioning GMPPNP and, presumably, GTP in its canonical conformation in the binding site.
Figure 3. Steric Occlusion of the GTP Binding Site.

(a) The Mg2+GMPPNP ligand complex modeled in the canonical conformation using the structure of the Mg2+GDP complex of NG as a starting point. Packing analysis carried out with Probe [17] illustrates the conflict that prevents movement of the γ-phosphate into the binding site—the spikes represent overlaps between the radii of the adjacent atoms (including hydrogens) [17]. The light dotted lens-shaped features generated by Probe indicate hydrogen bonds.
(b) Conserved residues of the motif I P loop, Leu106, and Gln107, are interdigitated with Leu192 and the main chain backbone of motif III. The interaction constricts the P loop opening. The bound GMPPNP is indicated by the ball-and-stick model, and the positions of motifs IV and II that delimit the binding pocket are labeled. The three structural elements of the NG domain—the N domain helix bundle (left), the G domain, and the IBD insertion (right)—are indicated by distinct tints.
The modeling result suggests that an opening of the P loop, i.e., movement of Gln107, would be necessary to accommodate the γ-phosphate. That this is indeed the case is suggested by inspection of the dimensions of the “jaws” (see Figure 1) of the P loop, which we measure as the distance between residues Gln107 and Thr112 (or their equivalents in other GTPases). This distance ranges from 10.12 to 10.44 Å in five different structures of GTP analog bound complexes of Ras, EF-Tu, and Gα. In the three structures of the GMPPNP complex of the NG domain, the distance between Cα atoms of the two residues is less, ranging from 8.7 to 8.9 Å. Indeed, in all the structures of the T. aquaticus NG, the P loop is somewhat constricted—in the apo and magnesium-free GDP complex structures of NG, the separation averages 8.4 Å, and in the Mg2+GDP complex the opening is 9.5 Å. The latter formed the basis for the modeled structure (Figure 3a); as the jaws of the P loop close a further ~0.6 Å in the GMPPNP complex, the clash identified in our modeling studies is likely to play a role in determining the conformation of the bound nucleotide analog. Thus, while the constriction of the P loop is small (~1.5 Å), it is significant (compare Figures 4a and 4b), and it appears to be sufficient to exclude movement of the terminal phosphate groups into the canonical conformation.
Figure 4. A Mechanism for Gating the NG Active Site.

(a) The EF-Tu GMPPNP complex in the region corresponding to the SRP GTPase motifs I and III. The backbone and selected side chains (labeled in the figure) are shown. The backbone amide groups of motif III that play structurally and functionally similar roles in different GTPases are indicated with an asterisk. The positions of the β- and γ-phosphate groups in the EF-Tu structure are indicated.
(b) The corresponding structure of GMPPNP complex N2a. Again, for clarity, only selected side chains (labeled in the figure) are shown. The hydrogen bond between the backbone amide of Gln107 and the carbonyl oxygen of Arg191 at the center of the interaction is indicated. The carbonyl oxygen of Gly190 (indicated by an arrow) is hydrogen bonded to the side chain of Arg191 [2]. The relative closing of the jaws of the P loop is apparent by comparison of the separation between residues 107 and 112 in (b) with that between the corresponding residues 20 and 25 in (a).
(c) Structural consequences of a hypothesized 180° rotation of the ψ angle of Gly190. The motif III main chain takes on a conformation similar to that seen in other GTPases in the GTP bound state [14]. The conformation of the arginine side chain shown is speculative.
The glycine-rich P loop (sequence GLQGSGKTTT in T. aquaticus Ffh) has few internal constraints on its conformation [18, 19], and in different structures it has been seen to be disordered in the absence of ligand [3, 18] or to have undergone substantial conformational change (e.g., a peptide flip in the EF-Tu/EF-Ts complex that stabilizes an unbound state) [20–22]. Consequently, any restraint on movement of the P loop in the GMPPNP complex of Ffh NG must be due to interaction with moieties external to it. Interestingly, there are a number of interactions between side chains of the P loop and side chains above it that could serve this function. These involve residues of motif I (Leu106 and Gln107) and motif III (Arg191 and Leu192) that are highly conserved in the SRP GTPase family. Most striking is the anomalously solvent-exposed side chain of Leu192 [1], which is positioned just above and extends between the side chains of Leu106 and Gln107 of the motif I P loop (Figure 3b). In the prokaryotic Ffh, only leucine occurs at position 192, while in the homologous eukaryotic SRP54, the corresponding residue only occurs as histidine. Gln107 is invariant in SRP54 and Ffh and is always preceded in sequence by leucine or valine [23]. These three residues (Leu106, Leu192, and Gln107) with the neighboring Arg191 and the main chain atoms of motifs I and III form an interdigitating and largely hydrophobic contact surface above the P loop (Figure 3b) that surrounds a buried hydrogen bond between the carbonyl of Arg191 and the amide nitrogen of Gln107 (see Figure 4b). The polar side chain of Gln107 extends away from the surface and has only weak interactions, primarily with solvent. However, the motif III backbone is fairly rigidly constrained by the network of interactions that spans the binding site (in particular, the interaction between Arg191 of motif III and Asp135 of motif II [1]). These interactions, therefore, serve to orient and stabilize the position of the Leu192 side chain over the P loop and may provide a significant kinetic barrier to conformational change at the interface.
That a solvent-exposed leucine residue contributes to the hydrophobic motif I/III interface is intriguing, and it raises the possibility that the surface plays a role in an intermolecular contact. Hydrophobic residues often occur at protein/protein interaction surfaces, and a functional role for hydrophobic residues associated with motif III of the GTPase fold predicated on conformational changes that expose or obscure protein interaction surfaces has precedence in other GTPases (e.g., the “Switch 2” region of EF-Tu [24] and Ran [25]). It follows from this notion that the underlying interface could act as a “gate” for the active site of the NG domain. We show below that the residues involved in the interface described here are indeed likely to undergo a substantial rearrangement in the active GTP bound conformation. Interestingly, while the well-defined network of interactions among motifs I, III, and II in the T. aquaticus NG domain that positions this interface has been observed under at least six different crystallization conditions [1, 2, 26], the same conformation is not observed in two structures of an SRP GTPase from different species. In the structures of the apo E. coli and A. ambivalens homologs [3, 4], the residue at position 192 (leucine and histidine, respectively) is collapsed toward a hydrophobic pocket between helices α2 and α3, perhaps reflecting a sensitivity of the structurally plastic gating peptide to the particular solution conditions or packing interactions in each crystal. In each, motif III is pulled away from both its apo and active conformations (see below).
Binding to an Empty Site Form
The unexpected binding modes observed with the T. aquaticus protein for both GMPPNP and GDP [2] suggest a complexity to the interaction between nucleotide and the NG domain that may be relevant, in general, to the interpretation of biochemical and kinetics data from the SRP GTPases. It has been reported that binding of both GDP and GTP to the E. coli Ffh can be independent of magnesium [27], and structures of magnesium-independent binding modes for both GDP and GTP have now been determined [2]. Particularly interesting is the observation in studies of the eukaryotic SRP54 that GTP binding to SRP alone does not commit the protein to the active state. In the absence of a receptor, SRP54 rapidly exchanges both GDP and GTP, and it behaves similarly to the apo protein in that its activity is uncommitted until interaction with SR can occur [28]. What is intriguing is that while the SRP GTPases have relatively low (μM) affinity for GDP and GTP and the free proteins rapidly exchange bound nucleotide [29–32], the apparent KDs measured for nucleotide binding are well below the concentration of free GTP in the cell (or in the assays) [28]. Thus, the protein presumably contains bound nucleotide but is not activated; i.e., has not undergone the conformational change that accompanies interaction with receptor, unless its receptor is present. The structures of the GMPPNP complex of the T. aquaticus NG reported here are both consistent with, and help resolve the paradox of, this behavior because they reveal a nucleotide binding mode in which the protein maintains the conformation of the empty state.
On formation of the SRP/receptor complex, the apparent affinity between SRP and nucleotide increases substantially [28, 30], and the rate of nucleotide exchange becomes very slow [33, 34]. In addition, it is in the complex that the two GTPases act to reciprocally stimulate the GTPase activity of the other [30, 34, 35]. The interaction between the SRP and receptor must, therefore, favor positioning the nucleotide in an extended conformation that can undergo hydrolysis catalyzed by the GTPase. The transition between an uncommitted, or “primed,” binding mode, suggested by the structures presented here, and a binding mode that is competent for hydrolysis requires that in the complex the structural elements that limit entry of GTP into the active site must change. We can consider this change to span two well-defined structural states (apo and active) and arrive at a simple model that suggests how this conformational change may be regulated.
A Peptide Flip Allows Concerted Rearrangement of the Active Site
In all GTPases, motif III (sequence DTAGRL in T. aquaticus Ffh) senses and interacts with the γ-phosphate of the bound nucleotide (Figure 4a). It has been shown that despite wide variation of the overall protein structures, the conformation of motif III and its interactions with the nucleotide in the GTP bound state (but not the GDP bound state) are structurally similar and conserved between different GTPases [14, 36]. Two residues of motif III, corresponding to Asp187 and Gly190, are universally conserved, consistent with a common structural and functional role for these residues in the GTPase mechanism [15]. The aspartate side chain positions one of the water molecules coordinating the magnesium ion. The glycine residue is framed by two backbone nitrogens that are directed toward the bound GTP (see Figure 4a); the glycine amide forms a hydrogen bond to the γ-phosphate, and the following amide is often seen to interact with a water molecule that may be catalytically important [5, 37, 38]. Remarkably, however, in the structures of the T. aquaticus NG, and in contrast to the structures of all other GTPases with bound GTP analog, the orientation of the latter peptide bond is reversed (Figure 4b). The position of the carbonyl oxygen, now directed toward the active site, is stabilized by hydrogen bonding with the side chain of the arginine that follows it [2]. If we presume that the main chain atoms of motif III play the role in the SRP GTPase observed in the structures of other GTPases, then at some point during the catalytic cycle, the Gly190/Arg191 peptide bond must reorient in order to direct the amide nitrogen of Arg191 toward the active site. Because the position over the P loop of the Leu192 side chain is closely coupled to the orientation of that peptide bond, the interactions that stabilize the apo conformation of the GTPase can now be understood to both limit access of the ligand to the active site and to maintain elements of the active site in a latent, inactive state.
The structural consequences of the rearrangement that must occur on activation can be shown by simple molecular modeling. The peptide flip, a 180° rotation of the ψ torsion angle of Gly190, must be accompanied by both release of the motif II–III salt bridge and movement of Leu192 from its position above the P loop (Figure 4c). There is no sterically allowable conformation that places the leucine side chain near its original position, and we infer, therefore, that the location of Leu192 is distant from the P loop in the active GTP bound state. Activation of the SRP GTPase by this mechanism implies a number of concurrent structural events: (1) a peptide flip that positions both amide nitrogens to interact with the γ-phosphate and water molecules in the active site, (2) movement of the hydrophobic leucine side chain across the surface of the protein, (3) release of the contact between Leu192 and the P loop, allowing movement of the γ-phosphate into the active site, and (4) reorientation of the side chain of the motif III Arg191 toward the phosphates of the bound GTP (Figure 4c). The latter is intriguing because Arg191, conserved in the SRP subfamily of GTPases, could there function as an internal “arginine finger” in the mechanism of the GTPase [39, 40]. In addition, movement of the Leu192 main chain would have to be accommodated by movement of the α2 helix, which, though too complex to be modeled here, is consistent with the behavior of the corresponding (“Switch 2”) region in other GTPases [41–43]. We propose, therefore, that the Gly190 peptide flip provides a simple structural mechanism that couples entry of GTP into the active site with the activation of the GTPase in the SRP/SRP receptor complex.
Conclusion
The binding mode for GMPPNP we identify in the structure of the T. aquaticus NG domain is unprecedented for a member of the GTPase superfamily. However, the structural data from other GTPases provide a framework for our interpretation of its significance. Thus, we can predict the position of the bound nucleotide with confidence and so identify the constriction of the P loop; we can identify those side chains that play a role in regulating the opening of the P loop; we can show that motif III in the structures of the T. aquaticus NG is in a latent conformation; and further, we can identify the nature of the conformational change that must occur on binding GTP in the active state. If the empty state of the protein is stabilized by the network of interactions across the active site identified previously [1], it follows then that GTP binds first in an inactive conformation and that the subsequent structural transition to the active state must be regulated. It has been proposed that SRP GTPases do not readily undergo the conformational change required for stable GTP binding but that instead they bind GTP in a highly cooperative manner on formation of the GTPase heterodimer consisting of SRP54 and its receptor, SRα [28, 44]. Supporting and extending this notion, our structural results both suggest a specific mechanism by which the transition may be regulated and imply that the two SRP GTPases bring bound nucleotide into the complex in a primed but inactive form.
Why might a preloaded conformation of GTP be functionally important in the SRP GTPases? It has been clear for some time that the mechanism of activation of the SRP GTPases is distinct from the classic GTPase switch and that functional activation requires interaction of SRP with its receptor [28, 30, 35, 45]. Ffh and FtsY (or SRP and SRα) can be trapped as a complex in the presence of GMPPNP [33, 46, 47], and the release of nucleotide from the heterodimer is very slow [33]. Recently, a structural model for the SRP-SR complex was proposed in which the nucleotides are buried between the two proteins [3]. A number of studies suggest that the conformational changes that accompany formation of the active complex allosterically affect interaction with signal sequence that is mediated by the third domain of Ffh (or SRP54), the M domain [30, 44]. If activation couples the two functions—interaction with receptor and release of signal peptide—formation of the active state prior to targeting would, of course, be unfavorable, as it might effect one function but not the other. A requirement that activation occurs on assembly in a protected environment implies that nucleotide would not be exchangeable, and thus the notion that the two proteins are primed by binding their ligands in an inactive conformation is appealing. One might envision, therefore, that it is the concerted transition of the proteins from the primed to the active state that is promoted by their interaction [28, 44, 46]. An interaction between primed complexes prior to the conformational changes that accompany activation is consistent with the observation of low-affinity interaction between the empty states of SRP54 and SR [48]. However, the increase in nucleotide affinity that accompanies interaction with the ribosome-nascent chain complex is more difficult to reconcile with such a model and so remains to be understood [49].
Many well-studied GTPases are regulated at the level of GDP exchange by an external factor that promotes the release of tightly bound GDP to produce a transient empty state competent for GTP binding. Our data suggest a mechanism by which activation of the SRP GTPases may be regulated instead at the level of entry of a preloaded GTP into the active site. This reiterates a theme, first put forth by Miller et al. [30], that it is the transition between the empty state and the GTP bound state that provides the key regulatory switch for the SRP GTPases. The motif I/III gate we identify suggests that the regulatory transition is between latent and active GTP bound states and provides structural insight into the mechanism of this novel GTPase switch.
Biological Implications
A central event in signal-recognition particle-mediated targeting of secreted and membrane proteins is the GTP-dependent interaction between the SRP and its membrane receptor, SR. SRP and SR each contain a structurally homologous NG GTPase domain. It has long been known that the GTPases are essential for function, that they interact directly, and that in vitro they act as reciprocal GTPase-activating proteins (or “GAPs”). What has remained puzzling, however, is the observation that, relative to other members of the GTPase superfamily, the SRP GTPases exhibit low affinity for nucleotide and rapid exchange. Thus, regulation of activity by guanine exchange factors (GEFs), a paradigm relevant to many other GTPase families, is excluded for the SRP GTPases. Also, while the underlying logic for reciprocal GAP activity of the SRP GTPases can be partially understood (because it implies a unidirectionality in the targeting mechanism), the structural basis for the interaction between the two GTPases, the mechanism by which their activity is regulated, and indeed the reason that two GTPases (rather than one) are required in this pathway, remain unknown.
The conformation of bound GMPPNP observed in the two different crystal structures of the T. aquaticus NG domain of Ffh reported here and the identification of the structural elements of the SRP GTPase that favor such a noncanonical binding mode provide new insight. First, the structures are consistent with binding to an empty-site form of the protein observed in biochemical studies of the eukaryotic SRP. GMPPNP binding to the protein does not in itself yield the active form of the GTPase. Second, entry of GTP into what is presumably its active conformation appears to be gated by interactions between highly conserved residues of motifs I and III of the SRP GTPase. This implies that the mechanism by which the SRP GTPase is activated is distinct from that of other GTPases and is consistent with the lack of a requirement for a GEF. Third, we identified a specific conformational change of the motif III peptide that must accompany activation of the SRP GTPase. The proposed peptide flip integrates release of the active site gate, movement of a conserved arginine side chain into proximity with the phosphate groups, and rearrangement of an exposed hydrophobic surface, which could, therefore, provide a site for a regulatory interaction. Finally, we suggest that these structural features imply that the two proteins “dock” GTP in an inactive conformation prior to their interaction and that activation takes place in a sequestered complex.
Experimental Procedures
Crystallization and Data Collection
The NG domain of T. aquaticus Ffh was purified as described previously [1]. Protein was concentrated to 20–30 mgs/ml in 2 mM MgCl2. GMPPNP (Boehringer) was added to 2 mM prior to setting up crystallization experiments at 4°C. Degradation of the GMPPNP in crystallization drops was monitored by anion exchange HPLC. Crystal form N1 was grown from 10% w/v PEG 1000, 10% w/v PEG 6000 (Crystal Screen II, #7; Hampton Research); crystals grew over a month as a cluster of rods. Crystal form N2 was grown from 29% MPD, 100 mM NaAcetate (pH 4.6), and 20 mM CaCl2; crystals grew to ~400 μm over several days. Crystals were not obtained in the absence of divalent cations.
Data were measured from frozen crystals by using a MAR CCD detector at the APS DND-CAT beamline 5ID-B. Both crystal forms N1 and N2 were mounted into a nylon loop directly from the crystallization drop. Identification of a cryoprotectant mother liquor for condition N1 was problematic, and although crystals remained intact when frozen in the mother liquor (at 10% PEG 1000), the loop was somewhat milky, and extremely strong ice diffraction rings were observed. More than 90° of oscillation data were measured from each of two crystals of form N1. The space group was determined with DENZO [50] to be P212121 with a = 52.69 Å, b = 60.41 Å, and c = 86.06 Å. Data were scaled and merged in SCALEPACK by using a −3 σ cutoff. A liberal rejection criterion was used in order to eliminate data affected by ice diffraction; although nearly 95% of the theoretically possible reflections were measured, the final completeness of the data set is only 85%, with the bulk of the rejections being in the 3.4–3.8 Å resolution range (which is therefore only 50% complete). The high multiplicity of the data set made this robust to rejection of good measurements. Data from crystal form N2 were measured from a single 500 μm crystal. After an initial data collection, the crystal was recovered by using cryotongs and later was remounted for measurement of high-resolution data. The space group was determined with DENZO to be C2 with a = 108.81 Å, b = 54.53 Å, c = 99.08 Å, and β = 97.42°, and the data were integrated and scaled by using DENZO and SCALEPACK [50]. This crystal form has 2-fold noncrystallographic symmetry. Data statistics are summarized in Table 1.
Refinement and Analysis
Both structures were solved by molecular replacement with the program AmoRe [51] by using the structure of the apo Ffh (1ffh) as the starting model. The molecular replacement solution of structure N1 yielded a correlation coefficient of 63.8% and an R factor of 36.6%. Two solutions were found for N2; these correspond to the two molecules in the asymmetric unit, and together yielded an initial correlation coefficient of 67.8% and an R factor of 34.9%. Initial rigid-body and slow-cooling molecular dynamics refinement and subsequent positional and temperature factor refinements were carried out with X-PLOR [52]. Midway through each refinement, solvent water molecules were identified and incorporated into the model using the solvent building mode of ARP/wARP [53]. The two monomers in the asymmetric unit of crystal N2 were refined independently. Refinement statistics are summarized in Table 1. The relatively high Rfree for structure N1 is due to problems with the data from ice-ring interference, and we limit our discussion of structure N1 to those parts well defined in the electron density map that bear on our discussion of the ligand binding mode. Atomic models were constructed with O [54].
The program LSQMAN [16] was used to overlap the structures of the NG domains. Coordinates were superposed with the G domains of other GTPases by using the rigid core of the GTPase fold, which in the SRP subfamily comprises the strands β1, β3, β4, β5, and β6 of the core β-sheet plus the αhelix α1. Helix α2, which undergoes structural change, and helices α3 and α4, which are coupled to the movement of the N domain, were not included in the calculation [2]. The PDB codes of the structures compared in the text are: 1exm, 1eft (EF-Tu), 1tnd (Gα), 5p21, and 1rvd (ras) [7, 10, 37, 39, 55]. Figures were generated with Molscript and Raster3d [56, 57].
Acknowledgments
We thank Yi Fan for technical assistance and Pamela Focia for comments on the manuscript. This work was supported by a grant from the National Institutes of Health (GM-58500) and a faculty development grant from the Howard Hughes Medical Institute (76296-549401). The DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) at the Advanced Photon Source is supported by the U.S. National Science Foundation through Grant DMR-9304725 and by the State of Illinois through the Department of Commerce and the Board of Higher Education Grant IBHE HECA NWU 96. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Energy Research under Contract Number W-31-102-Eng-38.
Footnotes
Accession numbers
Atomic coordinates have been deposited in the Protein Data Bank with the accession codes 1JPJ (structure N1) and 1JPN (structure N2).
References
- 1.Freymann DM, Keenan RJ, Stroud RM, Walter P. Structure of the conserved GTPase domain of the signal recognition particle. Nature. 1997;385:361–364. doi: 10.1038/385361a0. [DOI] [PubMed] [Google Scholar]
- 2.Freymann DM, Keenan RJ, Stroud RM, Walter P. Functional changes in the structure of the SRP GTPase on binding GDP and Mg2+GDP. Nature Struct Biol. 1999;6:793–801. doi: 10.1038/11572. [DOI] [PubMed] [Google Scholar]
- 3.Montoya G, Kaat KT, Moll R, Schäfer G, Sinning I. The crystal structure of the conserved GTPase of SRP54 from the archeon Acidianus ambivalens and its comparison with related structures suggests a model for the SRP-SRP receptor complex. Structure. 2000;8:515–525. doi: 10.1016/s0969-2126(00)00131-3. [DOI] [PubMed] [Google Scholar]
- 4.Montoya G, Svensson C, Luirink J, Sinning I. Crystal structure of the NG domain from the signal-recognition particle receptor FtsY. Nature. 1997;385:365–369. doi: 10.1038/385365a0. [DOI] [PubMed] [Google Scholar]
- 5.Hirshberg M, Stockley RW, Dodson G, Webb MR. The crystal structure of human rac1, a member of the rho-family complexed with a GTP analogue. Nature Struct Biol. 1997;4:147–151. doi: 10.1038/nsb0297-147. [DOI] [PubMed] [Google Scholar]
- 6.Ihara K, et al. Hakoshima T. Crystal structure of human RhoA in a dominantly active form complexed with a GTP analogue. J Biol Chem. 1998;273:9656–9666. doi: 10.1074/jbc.273.16.9656. [DOI] [PubMed] [Google Scholar]
- 7.Kjeldgaard M, Nissen P, Thirup S, Nyborg J. The crystal structure of elongation factor EF-Tu from Thermus aquaticus in the GTP conformation. Structure. 1993;1:35–50. doi: 10.1016/0969-2126(93)90007-4. [DOI] [PubMed] [Google Scholar]
- 8.Nassar N, Horn G, Herrmann C, Scherer A, McCormick F, Wittinghofer A. The 2.2 Å crystal structure of the Ras-binding domain of the serine/threonine kinase c-Raf1 and a GTP analogue. Nature. 1995;375:554–560. doi: 10.1038/375554a0. [DOI] [PubMed] [Google Scholar]
- 9.Nissen P, et al. Nyborg J. Crystal structure of the ternary complex of Phe-tRNAphe, EF-Tu, and a GTP analog. Science. 1995;270:1464–1472. doi: 10.1126/science.270.5241.1464. [DOI] [PubMed] [Google Scholar]
- 10.Pai EF, Krengel U, Petsko GA, Goody RS, Kabsch W, Wittinghofer A. Refined crystal structure of the tri-phosphate conformation of H-ras p21 at 1.35 Å resolution: implications for the mechanism of GTP hydrolysis. EMBO J. 1990;9:2351–2359. doi: 10.1002/j.1460-2075.1990.tb07409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prive GG, et al. Kim SH. X-ray crystal structures of transforming p21 ras mutants suggest a transition-state stabilization mechanism for GTP hydrolysis. Proc Natl Acad Sci USA. 1992;89:3649–3653. doi: 10.1073/pnas.89.8.3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scheidig AJ, Burmeister C, Goody RS. The pre-hydrolysis state of p21ras in complex with GTP: new insights into the role of water molecules in the GTP hydrolysis reaction of ras-like proteins. Structure. 1999;7:1311–1324. doi: 10.1016/s0969-2126(00)80021-0. [DOI] [PubMed] [Google Scholar]
- 13.Wagner A, Simon I, Sprinzl M, Goody RS. Interaction of guanosine nucleotides and their analogs with elongation factor Tu from Thermus thermophilus. Biochemistry. 1995;34:12535–12542. doi: 10.1021/bi00039a007. [DOI] [PubMed] [Google Scholar]
- 14.Berghuis AM, Lee E, Raw AS, Gilman AG, Sprang SR. Structure of the GDP-Pi complex of Gly203→Ala gialpha1: a mimic of the ternary product complex of galpha-catalyzed GTP hydrolysis. Structure. 1996;4:1277–1290. doi: 10.1016/s0969-2126(96)00136-0. [DOI] [PubMed] [Google Scholar]
- 15.Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: conserved structure and molecular mechanism. Nature. 1991;349:117–127. doi: 10.1038/349117a0. [DOI] [PubMed] [Google Scholar]
- 16.Kleywegt GJ, Jones TA. Detecting folding motifs and similarities in protein structures. Methods Enzymol. 1997;277:525–545. doi: 10.1016/s0076-6879(97)77029-0. [DOI] [PubMed] [Google Scholar]
- 17.Word JM, et al. Richardson DC. Visualizing and quantifying molecular goodness-of-fit: small-probe contact dots with explicit hydrogen atoms. J Mol Biol. 1999;285:1711–1733. doi: 10.1006/jmbi.1998.2400. [DOI] [PubMed] [Google Scholar]
- 18.al-Karadaghi S, Aevarsson A, Garber M, Zheltonosova J, Liljas A. The structure of elongation factor G in complex with GDP: conformational flexibility and nucleotide exchange. Structure. 1996;4:555–565. doi: 10.1016/s0969-2126(96)00061-5. [DOI] [PubMed] [Google Scholar]
- 19.Cronet P, Bellsolell L, Sander C, Coll M, Serrano L. Investigating the structural determinants of the p21-like triphosphate and Mg2+ binding site. J Mol Biol. 1995;249:654–664. doi: 10.1006/jmbi.1995.0326. [DOI] [PubMed] [Google Scholar]
- 20.Kawashima T, Berthet-Colominas C, Wulff M, Cusack S, Leberman R. The structure of the Escherichia coli EF-Tu·EF-Ts complex at 2.5 Å resolution. Nature. 1996;379:511–518. doi: 10.1038/379511a0. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Jiang Y, Meyering-Voss M, Sprinzl M, Sigler PB. Crystal structure of the EF-Tu·EF-Ts complex from Thermus thermophilus. Nat Struct Biol. 1997;4:650–656. doi: 10.1038/nsb0897-650. [DOI] [PubMed] [Google Scholar]
- 22.Renault L, Kuhlmann J, Henkel A, Wittinghofer A. Structural basis for guanine nucleotide exchange on Ran by the regulator of chromosome condensation (RCC1) Cell. 2001;105:245–255. doi: 10.1016/s0092-8674(01)00315-4. [DOI] [PubMed] [Google Scholar]
- 23.Zwieb C, Samuelsson T. SRPDB (signal recognition particle database) Nucleic Acids Res. 2000;28:171–172. doi: 10.1093/nar/28.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berchtold H, Reshetnikova L, Reiser CO, Schirmer NK, Sprinzl M, Hilgenfeld R. Crystal structure of active elongation factor Tu reveals major domain rearrangements. Nature. 1993;365:126–132. doi: 10.1038/365126a0. [DOI] [PubMed] [Google Scholar]
- 25.Stewart M, Kent HM, McCoy AJ. Structural basis for molecular recognition between nuclear transport factor 2 (NTF2) and the GDP-bound form of the Ras-family GTPase Ran. J Mol Biol. 1998;277:635–646. doi: 10.1006/jmbi.1997.1602. [DOI] [PubMed] [Google Scholar]
- 26.Keenan RJ, Freymann DM, Walter P, Stroud RM. Crystal structure of the signal sequence binding subunit of the signal recognition particle. Cell. 1998;94:181–191. doi: 10.1016/s0092-8674(00)81418-x. [DOI] [PubMed] [Google Scholar]
- 27.Jagath JR, Rodnina MV, Lentzen G, Wintermeyer W. Interaction of guanine nucleotides with the signal recognition particle from Escherichia coli. Biochemistry. 1998;37:15408–15413. doi: 10.1021/bi981523a. [DOI] [PubMed] [Google Scholar]
- 28.Rapiejko PJ, Gilmore R. Empty site forms of the SRP54 and SR alpha GTPases mediate targeting of ribosomenascent chain complexes to the endoplasmic reticulum. Cell. 1997;89:703–713. doi: 10.1016/s0092-8674(00)80253-6. [DOI] [PubMed] [Google Scholar]
- 29.Farmery M, Macao B, Larsson T, Samuelsson T. Binding of GTP and GDP induces a significant conformational change in the GTPase domain of Ffh, a bacterial homologue of the SRP 54 kDa subunit. Biochim Biophys Acta. 1998;1385:61–68. doi: 10.1016/s0167-4838(98)00045-4. [DOI] [PubMed] [Google Scholar]
- 30.Miller JD, Wilhelm H, Gierasch L, Gilmore R, Walter P. GTP binding and hydrolysis by the signal recognition particle during initiation of protein translocation. Nature. 1993;366:351–354. doi: 10.1038/366351a0. [DOI] [PubMed] [Google Scholar]
- 31.Moll R, Schmidtke S, Petersen A, Schäfer G. The signal recognition particle receptor alpha subunit of the hyperthermophilic archaeon Acidianus ambivalens exhibits an intrinsic GTP-hydrolyzing activity. Biochim Biophys Acta. 1997;1335:218–230. doi: 10.1016/s0304-4165(96)00141-9. [DOI] [PubMed] [Google Scholar]
- 32.Moser C, Mol O, Goody RS, Sinning I. The signal recognition particle receptor of Escherichia coli (FtsY) has a nucleotide exchange factor built into the GTPase domain. Proc Natl Acad Sci. 1997;94:11339–11344. doi: 10.1073/pnas.94.21.11339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peluso P, Herschlag D, Nock S, Freymann DM, Johnson AE, Walter P. Role of 4.5S RNA in Assembly of the Bacterial Signal Recognition Particle with Its Receptor. Science. 2000;288:1640–1643. doi: 10.1126/science.288.5471.1640. [DOI] [PubMed] [Google Scholar]
- 34.Song W, Raden D, Mandon EC, Gilmore R. Role of Sec61alpha in the regulated transfer of the ribosome-nascent chain complex from the signal recognition particle to the translocation channel. Cell. 2000;100:333–343. doi: 10.1016/s0092-8674(00)80669-8. [DOI] [PubMed] [Google Scholar]
- 35.Powers T, Walter P. Reciprocal stimulation of GTP hydrolysis by two directly interacting GTPases. Science. 1995;269:1422–1424. doi: 10.1126/science.7660124. [DOI] [PubMed] [Google Scholar]
- 36.Cherfils J, Auzat I, et al. Crystal structures of the small G protein Rap2A in complex with its substrate GTP, with GDP and with GTPγS. EMBO J. 1997;16:5582–5591. doi: 10.1093/emboj/16.18.5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hilgenfeld R, Mesters JR, Hogg T. Insights into the GTPase mechanism of EF-Tu from structural studies. In: Garrett RA, Douthwaite SR, Liljas A, Matheson AT, Moore PB, Noller HF, editors. The Ribosome: Structure, Function, Antibiotics and Cellular Interactions. Vol. 28. Washington, D.C: ASM Press; 2000. pp. 347–357. [Google Scholar]
- 38.Dumas JJ, Zhu Z, Connolly JL, Lambright DG. Structural basis of activation and GTP hydrolysis in Rab proteins. Structure Fold Des. 1999;7:413–423. doi: 10.1016/s0969-2126(99)80054-9. [DOI] [PubMed] [Google Scholar]
- 39.Noel JP, Hamm HE, Sigler PB. The 2.2 Å crystal structure of transducin-alpha complexed with GTP gamma S. Nature. 1993;366:654–663. doi: 10.1038/366654a0. [DOI] [PubMed] [Google Scholar]
- 40.Scheffzek K, et al. Wittinghofer A. The Ras-Ras-GAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science. 1997;277:333–338. doi: 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
- 41.Milburn MV, et al. Kim SH. Molecular switch for signal transduction: structural differences between active and inactive forms of protooncogenic ras proteins. Science. 1990;247:939–945. doi: 10.1126/science.2406906. [DOI] [PubMed] [Google Scholar]
- 42.Sprang SR. G protein mechanisms: insights from structural analysis. Annu Rev Biochem. 1997;66:639–678. doi: 10.1146/annurev.biochem.66.1.639. [DOI] [PubMed] [Google Scholar]
- 43.Wittinghofer A. In: The functioning of molecular switches in three dimensions. Pases GT, Hall A, editors. Oxford: Oxford University Press; 2000. pp. 244–310. [Google Scholar]
- 44.Millman JS, Andrews DW. Switching the model: a concerted mechanism for GTPases in protein targeting. Cell. 1997;89:673–676. doi: 10.1016/s0092-8674(00)80248-2. [DOI] [PubMed] [Google Scholar]
- 45.Connolly T, Gillmore R. GTP hydrolysis by complexes of the signal recognition particle and the signal recognition particle receptor. J Cell Biol. 1993;123:799–807. doi: 10.1083/jcb.123.4.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miller JD, Bernstein HD, Walter P. Interaction of E. coli Ffh/4.5S ribonucleoprotein and FtsY mimics that of mammalian signal recognition particle and its receptor. Nature. 1994;367:657–659. doi: 10.1038/367657a0. [DOI] [PubMed] [Google Scholar]
- 47.Rapiejko PJ, Gilmore R. Protein translocation across the ER requires a functional GTP binding site in the alpha subunit of the signal recognition particle receptor. J Cell Biol. 1992;117:493–503. doi: 10.1083/jcb.117.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rapiejko PJ, Gilmore R. Signal sequence recognition and targeting of ribosomes to the endoplasmic reticulum by the signal recognition particle do not require GTP. Mol Biol Cell. 1994;5:887–897. doi: 10.1091/mbc.5.8.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bacher G, Lütcke H, Jungnickel B, Rapoport TA, Dobberstein B. Regulation by the ribosome of the GTPase of the signal-recognition particle during protein targeting. Nature. 1996;381:248–251. doi: 10.1038/381248a0. [DOI] [PubMed] [Google Scholar]
- 50.Otwinowski Z. Oscillation data reduction program. In: Sawyer L, Isaacs NW, Bailey S, editors. Data Collection and Processing. Warrington, UK: SERC Daresbury Laboratory; 1993. pp. 55–62. [Google Scholar]
- 51.Navaza J. AMoRe: an automated package for molecular replacement. Acta Crystallogr A. 1994;50:157–163. [Google Scholar]
- 52.Brünger AT. X-PLOR: A System for X-Ray Crystallography and NMR. New Haven: Yale University Press; 1992. [Google Scholar]
- 53.Perrakis A, Sixma TK, Wilson KS, Lamzin VS. wARP: improvement and extension of crystallographic phases by weighted averaging of multiple refined dummy atomic models. Acta Crystallogr D. 1997;53:448–455. doi: 10.1107/S0907444997005696. [DOI] [PubMed] [Google Scholar]
- 54.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 55.Ahmadian MR, et al. Scheffzek K. Guanosine triphosphatase stimulation of oncogenic Ras mutants. Proc Natl Acad Sci USA. 1999;96:7065–7070. doi: 10.1073/pnas.96.12.7065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kraulis PJ. Molscript—a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 57.Merritt EA, Bacon DJ. Raster3D photorealistic molecular graphics. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]