

Abstract
The novel three-component domino reactions have been discovered. The reactions are easy to perform simply by mixing three common reactants in HOAc under microwave heating. The reaction proceeds at fast rates and can be finished within 20–36 min, which makes work-up convenient. Most of multiple stereocenters and geometry have been controlled well. The stereochemistry has been unequivocally determined by X-ray structural analysis.
Keywords: Three-component Domino Reaction, Nitrogen heterocycles, Pentcyclic Indeno[2, 1-c]quinoline, Pyrano[4, 3-b]oxepine, Stereochemistry
The search for efficient construction of multicyclic skeletons of chemically and biomedically importance has been an active theme in organic synthesis.[1–3] Among these skeletons, the structurally diverse oxa-azaspiro skeletons commonly exist in nature and represented by daphnilactone A,[1] serratezomine A[2] and nitraramine (Figure 1),[3] that exhibit a broad range of biological activities. These complex architectures have inspired the interest on creating strategies and tactics for total synthesis and methodologies.[4–5]
Figure 1.

Several oxa-azaspiro skeletons
In the past several years, multicomponent domino reactions (MDRs) and related environmentally benign, chemoselective and atom-efficient processes have emerged as powerful tools for the assembly of complex structures with multiple stereocenters in a one-pot operation.[6–10] These reactions can avoid time-consuming and costly purification of various precursors and tedious steps of protection/deprotection of functional groups.[9] There have been several domino strategies for the diverse formation of oxa-azaspiro skeletons. However, the continuing search for more efficient MDRs for this synthesis still remains challenging, and continues to attract interest to synthetic community.
Recently, we and others have developed a series of MDRs that offer easy accesses to multiple functionalized ring structures of chemical and pharmaceutical interest.[11–13] During our continuous study on MDR project,11 we now discovered novel multicomponent annulations of enaminones with o-phthalaldehyde (OPA) and 4-hydroxy-6-methyl-2H-pyran-2-one divergently yielding multifunctionalized pyrano[3′,2′:2,3]indeno[2,1-c]quinolines 4 and ([3,4]furanoimino)benzo[e]pyrano[4,3-b]oxepines 5 (Scheme 1). The great aspects of these domino reactions are shown by the fact that up to five new bonds and three new rings (tricyclic 5-6-6 skeleton including cyclopentene, pyridine, and pyrane) were readily formed in domino fashions that involved [4+1]/[3+2+1]/[5+1] cyclizations; three stereocenters including a quaternary center and geometry were controlled well in a one-pot operation. The latter provided new bicyclic 7-7 skeleton including azepine and oxepine via double [4+3] cyclizations with high stereoselectivity. The present work represents the first example for constructing these special types of oxa-azaspiro 4 and oxa-azabridged 5 skeletons with multiple stereocenters.
Scheme 1.
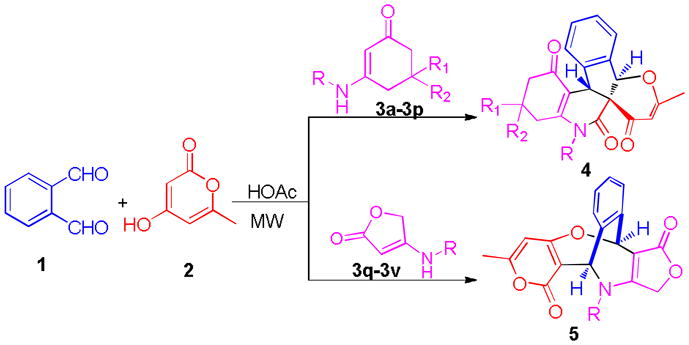
Novel multicomponent domino reactions
The o-phthalaldehyde (OPA), possessing 1,4-biselectrophilic centers, has proven to be important building blocks for the construction of important cyclic skeletons.[14] Our strategy of synthesizing highly functionalized pentacyclic lactams was started from the reaction of o-phthalaldehyde with 4-hydroxy-6-methyl-2H-pyran-2-one and enaminones based on the fact that two formyl groups would undergo double nucleophilic additions and subsequent double nucleophilic substitutions; and pyran-2-ones upon being treated with appropriate nucleophiles can be converted into new nucleophiles through ring cleavage (Scheme 2).
Scheme 2.

Mechanism hypothesis for forming products 4 and 5
Based on the above analysis, the reaction of o-phthalaldehyde 1, 4-hydroxy-6-methyl-2H-pyran-2-one 2 with 5,5-dimethyl-3-(4-chlorophenylamino)cyclohex-2-enone 3a was first carried out in HOAc for 20 min at 80 °C under microwave irradiation condition (Scheme 3). Pleasantly, the white solid 4a was obtained in 68% chemical yield. In contrast, other solvents, such as DMF, toluene, CHCl3 and EtOH, led to poor results. Aprotic solvents ( DMF, toluene and CHCl3) resulted in less that 10% yield; protic solvent, EtOH, gave 12% isolated yield. Since metal triflates were known to show effective Lewis acidity even in the presence of water,[15] three of them, Sc(OTf)3, Cu(OTf)2 and Fe(OTf)2, were then empoyed attempting to enhance yield, but complex mixtures were formed, which made purification difficult.
Scheme 3.
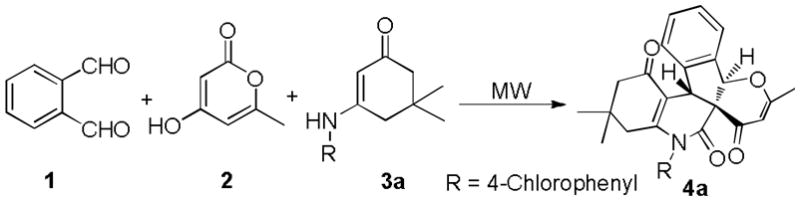
Formation of pentcyclic indeno[2,1-c]quinolines 4a
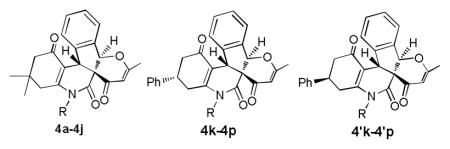
With this acceptable condition in hand, we examined the scope of this synthesis by using various easily available starting materials. As revealed in Table 1, a range of polysubstituted pyrano[3′,2′:2,3]indeno[2,1-c]quinoline derivatives can be generated in moderate to good yields. The reaction is easy to perform simply by subjecting a mixture of o-phthalaldehyde 1, 4-hydroxy-6-methyl-2H-pyran-2-one 2, and enaminones 3 in acetic acids under microwave heating. We also examined the scope of enaminones and found several different N-substituents bearing electron-withdrawing or electron-donating groups were all suitable substrate. Although substrates 3k-3p gave a mixture of two isomers (diastereoselectivity 64:36 to 89:11), most others (3a-3j) led to formation of single diastereoisomers. The structure of 4p was unambiguously confirmed by X-ray crystal structural analysis (Fig. 2). Furthermore, halogen functional groups (Cl, Br and I) were tolerated well and would allow further functional group manipulations via cross-couplings.
Table 1.
Domino Synthesis of Pentcyclic Indeno[2,1-c]quinolines 4
| |||||
|---|---|---|---|---|---|
| Entry | 4a | R | Timeb | Yieldc/% | 4:4′d |
| 1 | 4a | 4-Chlorophenyl (3a) | 20 | 68 | - |
| 2 | 4b | 4-Bromophenyl (3b) | 22 | 62 | - |
| 3 | 4c | 4-Iodophenyl (3c) | 25 | 58 | - |
| 4 | 4d | 3-Fluorophenyl (3d) | 25 | 44 | - |
| 5 | 4e | 3-Chlorophenyl (3e) | 26 | 52 | - |
| 6 | 4f | 3-Bromophenyl (3f) | 26 | 48 | - |
| 7 | 4g | Phenyl (3g) | 30 | 62 | - |
| 8 | 4h | 4-Methylphenyl (3h) | 32 | 68 | - |
| 9 | 4i | 3-Methylphenyl (3i) | 30 | 54 | - |
| 10 | 4j | 3-Bromo-4-methylphenyl (3j) | 28 | 50 | - |
| 11 | 4k | 4-Chlorophenyl (3k) | 28 | 58 | 75:25 |
| 12 | 4l | 4-Bromophenyl (3l) | 30 | 51 | 68:32 |
| 13 | 4m | Phenyl (3m) | 30 | 56 | 81:19 |
| 14 | 4n | 4-Methylphenyl (3n) | 36 | 60 | 89:11 |
| 15 | 4o | 3-Methylphenyl (3o) | 36 | 52 | 86:14 |
| 16 | 4p | 4-Methoxyphenyl (3p) | 30 | 64 | 64:36 |
Conditions: HOAc (1.5 mL), 80 °C, microwave heating.
Time (min).
Isolated yield.
The ratio of isomers was determined by 1H NMR.
Figure 2.

X-ray structural of 4p.
In view of these results, we then turned our attention to investigate several differently substituted enaminones. The reactions of o-phthalaldehyde 1, 4-hydroxy-6-methyl-2H-pyran-2-one 2 with N-substituted 4-aminofuran-2(5H)-ones (3q–3w) were performed under the above conditions for a short period (24–30 min). Surprisingly, the reaction occurred to another direction to form multi-functionalized pentcyclic pyrano[4,3-b]oxepines 5 (Scheme 4). This novel multicomponent domino reaction also exhibit a good scope of enaminone substrates (Table 2) providing a straightforward pathway to construct highly substituted pentcyclic pyrano[4,3-b]oxepines. Similar to the former reaction, the latter also illustrates the remarkable chemo-, stereo-, and regioselectivity starting from very common and easily accessible inexpensive starting materials. The structural elucidation and the attribution of stereoselectivity were unequivocally determined by NMR spectroscopic analysis and X-ray diffraction of single crystals that were obtained by slow evaporation of the solvent, as in the case of product 5d (Fig. 3). Both reactions occurred at a fast speed; in fact, all cases can be finished within 36 minutes. Water is nearly a sole by-product, which makes work-up convenient. In most cases, the products can precipitate out after cold water was poured into the reaction mixture. During these domino processes, up to three new rings and five sigma-bonds were formed and accompanied by cleavage of two C=O of OPA and one C–O bonds of pyran-2-ones 2, and all stereogenetic centers and geometry have been completely controlled including a quaternary center attached on the lactam ring. It would be interesting to make collections of these natural product-like structures for screening.
Scheme 4.
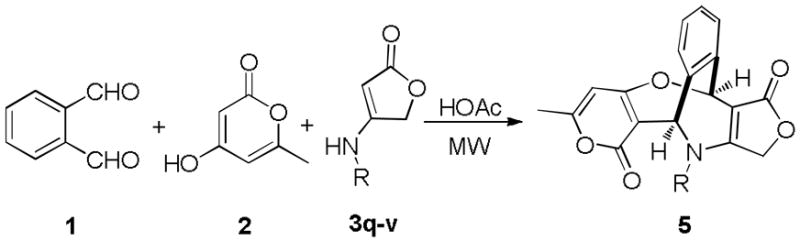
The formation of pentcyclic pyrano[4,3-b]oxepines 5
Table 2.
Domino synthesis of pentcyclic pyrano[4,3-b]oxepines 5
| Entry | 5a | R | Time/min | Yieldb/% |
|---|---|---|---|---|
| 1 | 5a | 4-Chlorophenyl (3q) | 25 | 81 |
| 2 | 5b | 3-Fluorophenyl (3r) | 24 | 72 |
| 3 | 5c | 3-Bromophenyl (3s) | 24 | 73 |
| 4 | 5d | Phenyl (3t) | 28 | 79 |
| 5 | 5e | 4-Methylpheny (3u) | 30 | 84 |
| 6 | 5f | 3-Methylpheny (3v) | 30 | 71 |
| 7 | 5g | 4-Methoxyphenyl (3w) | 26 | 82 |
Conditions: HOAc (1.5 mL), 80 °C, microwave heating.
Isolated yield.
Figure 3.

X-ray structural of product 5d.
The mechanism hypothesis for these novel domino reactions are proposed and shown in Scheme 2. The former involves the ring closure cascade reactions that consist of initial condensation, Michael addition(A to B), intramolecular cyclization and ring-opening of pyran-2-ones 2 (B to C), and the second intramolecular cyclization (C to 4). The latter involves [4+3] cycloaddition to give azepinediols D, which is followed by subsequent intermolecular double nucleophilic substitution (D to 5) leading to thermodynamically stable fused pyrano[4,3-b]oxepines 5.
We reasoned that the divergence in these pathways could be caused by the different nucleophilicity of six-membered and five-membered N-substituted enaminones. With higher nucleophilicity, five-membered N-substituted enaminones favor double nucleophilic additions of two formyl groups on o-phthalaldehyde ring. This would resist o-phthalaldehyde to condensation with pyran-2-ones 2.
In summary, we have successfully established the first domino [4+1]/[3+2+1]/[5+1] and double [4+3] cyclization reactions of o-phthalaldehyde, that led to the novel constructions of pentcyclic pyrano[3′,2′:2,3]indeno[2,1-c]pyridine and ([3,4]furanoimino) benzo[e]pyrano[4,3-b]oxepine skeletons with multiple stereocenters. The present work provides an attractive strategy for construction of structurally diverse pentcyclic oxa-azaspiro and oxa-azabridged skeletons. The ready accessibility of starting materials, the broad compatibility of N-substituted enaminones and generality of these reactions make them important in view of the synthetic and biomedical importance of fused heterocycles. Other features of this tactic include the mild condition, convenient one-pot operation, short periods of 20–36 min and excellent regioselectivity and good to high stereoselectivities. The continuing work on this project will be focused on the development of asymmetric versions of these reactions in due course.
Experimental Section
Example for the synthesis of 4a: 5-(4-Chlorophenyl)-3,3,9-trimethyl-3,4,10a,14b-tetrahydro-1H-pyrano[3′,2′:2,3]indeno[2,1-c]quinoline-1,6,7(2H,5H)-trione
o-Phthalaldehyde (1, 1.1 mmol, 1.1 equiv.) was introduced in a 10-mL vial, 4-hydroxy-6-methyl-2H-pyran-2-one (2, 1.0 mmol, 1.0 equiv.) and 5,5-dimethyl-3-(4-chlorophenylamino)cyclohex-2-enone (3a, 1.0 mmol, 1.0 equiv.) were then added, followed by HOAc (1.5 mL.). The reaction vial was capped and pre-stirring for 20 second. The mixture was irradiated at 80 °C until TLC monitoring (petroleum ether: acetone 3:1) showed that conversion of the starting material 3a was complete (20 min). The reaction mixture was then cooled to room temperature and diluted with cold water (40 ml). The solid product was collected by Büchner filtration and was purified by flash column chromatography (silica gel, mixtures of petroleum ether/acetone, 7:1, v/v) to afford the desired pure products 4a as white solid (Mp: 275–276 °C).
Example for the synthesis of 5a
o-Phthalaldehyde (1, 1.1 mmol) was introduced in a 10-mL vial, 4-hydroxy-6-methyl-2H-pyran-2-one (2, 1.0 mmol), and 4-((4-chlorophenyl)amino)furan-2(5H)-one (3q, 1.0 mmol) were added and followed by adding HOAc (1.5 mL). The reaction vial was capped and pre-stirring for 20 second. The mixture was irradiated at 80 °C until TLC monitoring (petroleum ether: acetone 2:1) showed that conversion of the starting material 3q was complete (25 min). The reaction mixture was cooled to room temperature and diluted with cold water (50 ml). The solid product was collected by Büchner filtration and was purified by flash column chromatography (silica gel, mixtures of petroleum ether/acetone, 4:1, v/v) to afford the desired pure products 5a as white solid (Mp: 247–248 °C).
Supplementary Material
Acknowledgments
We are grateful to financial support from the NSFC (Nos. 20928001, 21072163, 21002083, and 21102124), the Priority Academic Program Development of Jiangsu Higher Education Institutions, Jiangsu Science and Technology Support Program (No. BE2011045), NIH (R21DA031860-01) and Robert A. Welch Foundation (D-1361) for their generous support.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author.
Contributor Information
Prof. Shu-Jiang Tu, Email: laotu@xznu.edu.cn.
Prof. Dr. Guigen Li, Email: guigen.li@ttu.edu.
References
- 1.a) Ruggeri BR, McClure KF, Heathcock CH. J Am Chem Soc. 1989;111:1531–1533. [Google Scholar]; b) Heathcock CH, Ruggeri RB, McClure KF. J Org Chem. 1992;57:2585–2594. [Google Scholar]; c) Ruggeri RB, Heathcock CH. J Org Chem. 1987;52:5746–5749. [Google Scholar]
- 2.a) Chandra A, Pigza JA, Han JS, Mutnick D, Johnston JN. J Am Chem Soc. 2009;131:3470–3471. doi: 10.1021/ja900536d. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Morita H, Arisaka M, Yoshida N, Kobayashi J. J Org Chem. 2000;65:6241–6245. doi: 10.1021/jo000661e. [DOI] [PubMed] [Google Scholar]; c) Morita H, Arisaka M, Yoshida N, Kobayashi J. J Org Chem. 2000;65:6241–6245. doi: 10.1021/jo000661e. [DOI] [PubMed] [Google Scholar]
- 3.a) Wanner MJ, Koomen GJ. J Org Chem. 1995;60:5634–5637. [Google Scholar]; b) Gravel E, Poupon E, Hocquemiller R. Org Lett. 2005;7:2497–2499. doi: 10.1021/ol050849q. [DOI] [PubMed] [Google Scholar]; c) Yu N, Novogorodova, Kh S, Maekh, Yu S, Yunusov Khim Prir Soedin. 1975;11:435–7. [Google Scholar]
- 4.a) Va P, Campbell EL, Robertson WM, Boger DL. J Am Chem Soc. 2010;132:8489–8495. doi: 10.1021/ja1027748. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bettati M, Cavanni P, Di Fabio R, Oliosi B, Perini O, Scheid G, Tedesco G, Zonzini L, Micheli F. Chem Med Chem. 2010;5:361–366. doi: 10.1002/cmdc.200900482. [DOI] [PubMed] [Google Scholar]; c) Morita H, Kobayashi J. J Org Chem. 2002;67:5378–5381. doi: 10.1021/jo025821w. [DOI] [PubMed] [Google Scholar]; d) Schultz AG, Lucci RD, Napier JJ, Kinoshita H, Ravichandran R, Shannon P, Yee YK. J Org Chem. 1985;50:217–31. [Google Scholar]; e) Blowers JW, Saxton JE, Swanson AG. Tetrahedron. 1986;42:6071–6095. [Google Scholar]; f) Toda M, Hirata Y, Yamamura S. Tetrahedron. 1972;28:1477–1484. [Google Scholar]; g) Sasaki K, Hirata Y. Tetrahedron Lett. 1972:1275–1278. [Google Scholar]; h) Sasaki K, Hirata Y. J Chem Soc Perkin Trans II. 1972:1411–1415. [Google Scholar]
- 5.a) Schreiber SL. Science. 2000;287:1964–1969. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]; b) Frederic LM, Thierry C, Jean R. J Am Chem Soc. 2005;127:17176–17177. [Google Scholar]; c) Snyder SA, Breazzano SP, Ross AG, Lin Y, Zografos AL. J Am Chem Soc. 2009;131:1753–1765. doi: 10.1021/ja806183r. [DOI] [PubMed] [Google Scholar]
- 6.a) Tietze LF. Chem Rev. 1996;96:115–136. doi: 10.1021/cr950027e. [DOI] [PubMed] [Google Scholar]; b) Tietze LF, Haunert F. In: Domino reaction in organic synthesis, in Stimulating Concepts in Chemistry. Votle F, Stoddart JF, Shibasaki M, editors. Wiley-VCH; Weinheim: 2000. pp. 39–64. [Google Scholar]; c) Tietze LF, Brasche G, Gericke KM. Domino Reactions in Organic Synthesis. Wiley-VCH; Weinheim: 2006. [Google Scholar]; d) Santra S, Andreana PR. Angew Chem, Int Ed. 2011;50:9418–9422. doi: 10.1002/anie.201103567. [DOI] [PubMed] [Google Scholar]; e) Santra S, Andreana PR. J Org Chem. 2011;76:7632. doi: 10.1021/jo102305q. [DOI] [PubMed] [Google Scholar]
- 7.For step-economy see: Wender PA, Baryza JL, Brenner SE, Clarke MO, Gamber GG, Horan JC, Jessop TC, Kan C, Pattabiraman K, Williams TJ. Pure Appl Chem. 2003;75:143–155.Wender PA, Gamber GG, Hubbard RD, Pham SM, Zhang L. J Am Chem Soc. 2005;127:2836–2837. doi: 10.1021/ja042728b.
- 8.For atom-economy see: Trost BM. Science. 1991;254:1471–1477. doi: 10.1126/science.1962206.Trost BM. Angew Chem, Int Ed. 1995;34:258–281.Trost BM. Acc Chem Res. 2002;35:695–705. doi: 10.1021/ar010068z.
- 9.For asymmetric catalytic domino reactions see: Huang Y, Waljji AM, Larsen CH, MacMillan DWC. J Am Chem Soc. 2005;127:15051–15053. doi: 10.1021/ja055545d.Yang JW, Fonseca HMT, List B. J Am Chem Soc. 2005;127:15036–15037. doi: 10.1021/ja055735o.Lu M, Zhu D, Lu Y, Hou B, Tan B, Zhong G. Angew Chem, Int Ed. 2008;47:10187–10191. doi: 10.1002/anie.200803731.
- 10.a) Groenendaal B, Ruijter E, Orru RVA. Chem Commun. 2008:5474–5489. doi: 10.1039/b809206k. [DOI] [PubMed] [Google Scholar]; b) Ismabery N, Lavila R. Chem-Eur J. 2008;14:8444–8454. [Google Scholar]; c) Sunderhaus JD, Martin SF. Chem-Eur J. 2009;15:1300–1308. doi: 10.1002/chem.200802140. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Ruijter E, Scheffelaar R, Orru RVA. Angew Chem, Int Ed. 2011;50:6234–6246. doi: 10.1002/anie.201006515. [DOI] [PubMed] [Google Scholar]; e) Ganem B. Acc Chem Res. 2009;42:463–472. doi: 10.1021/ar800214s. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Tietze LF, Kinzel T, Brazel CC. Acc Chem Res. 2009;42:367–378. doi: 10.1021/ar800170y. [DOI] [PubMed] [Google Scholar]
- 11.a) Jiang B, Li C, Shi F, Tu SJ, Kaur P, Wever W, Li G. J Org Chem. 2010;75:2962–2965. doi: 10.1021/jo1002278. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jiang B, Tu SJ, Kaur P, Wever W, Li G. J Am Chem Soc. 2009;131:11660–11661. doi: 10.1021/ja904011s. [DOI] [PubMed] [Google Scholar]; c) Jiang B, Wang X, Shi F, Tu SJ, Ai T, Ballew A, Li G. J Org Chem. 2009;74:9486–9489. doi: 10.1021/jo902204s. [DOI] [PubMed] [Google Scholar]; d) Li G, Wei HX, Kim SH, Carducci MD. Angew Chem, Int Ed. 2001;40:4277–4280. doi: 10.1002/1521-3773(20011119)40:22<4277::AID-ANIE4277>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]; e) Ma N, Jiang B, Zhang G, Tu SJ, Wever W, Li G. Green Chem. 2010;12:1357–1361. [Google Scholar]; f) Jiang B, Yi MS, Shi F, Tu SJ, Pindi S, McDowell P, Li G. Chem Commun. 2012:808–810. doi: 10.1039/c1cc15913e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For domino reactions and atom economic synthesis see 12–13: Tietze LF, Brazel CC, Hoelsken S, Magull J, Ringe A. Angew Chem, Int Ed. 2008;47:5246–5249. doi: 10.1002/anie.200800626.Padwa A. Chem Soc Rev. 2009;38:3072–3081. doi: 10.1039/b816701j.Padwa A, Bur SK. Tetrahedron. 2007;63:5341–5378. doi: 10.1016/j.tet.2007.03.158.
- 13.a) Huang Y, Walji AM, Larsen CH, MacMillan DWC. J Am Chem Soc. 2005;127:15051–15053. doi: 10.1021/ja055545d. [DOI] [PubMed] [Google Scholar]; b) Lu M, Zhu D, Lu Y, Hou Y, Tan B, Zhong G. Angew Chem, Int Ed. 2008;47:10187–10191. doi: 10.1002/anie.200803731. [DOI] [PubMed] [Google Scholar]; c) Snyder SA, Breazzano SP, Ross AG, Lin Y, Zografos AL. J Am Chem Soc. 2009;131:1753–1765. doi: 10.1021/ja806183r. [DOI] [PubMed] [Google Scholar]; d) Yang JW, Fonseca MTH, List B. J Am Chem Soc. 2005;127:15036–15037. doi: 10.1021/ja055735o. [DOI] [PubMed] [Google Scholar]
- 14.For a review on the reactions of o-phthalaldehyde with nucleophiles, see: Zuman P. Chem Rev. 2004;104:3217–3238. doi: 10.1021/cr0304424.For some recent papers on o-phthalaldehyde reactions, see: Petrignet J, Roisnel T, Gree R. Chem Eur J. 2007;13:7374–7384. doi: 10.1002/chem.200700613.Ramana CV, Reddy CN, Gonnade RG. Chem Commun. 2008;3151:3153. doi: 10.1039/b801755g.Bornhoeft J, Siegwarth J, Nather C, Herges RE. J Org Chem. 2008;73:1619–1624.Navarro C, Csaky AG. Org Lett. 2008;10:217–219. doi: 10.1021/ol702571c.For aldol reactions of o-phthalaldehyde with mono/dicarbonyl compounds, see: Tarbell DS, Wargotz B. J Am Chem Soc. 1954;76:5761–5767.Dimroth K, Freyschlag H. Angew Chem. 1957;69:95–96.Meuche D, Strauss H, Heilbronner E. Helv Chim Acta. 1958;41:2220–2229.Davey W, Gottfried H. J Org Chem. 1961;26:3699–3705.Gazit A. J Chem Soc, Chem Commun. 1986;6:445–446.Langer P, Albrecht U. Synlett. 2001;4:526–528.Yadav JS, Syamala M. Chem Lett. 2002;7:688–689.Albrecht U, Nguyen THV, Langer P. J Org Chem. 2004;69:3417–3424. doi: 10.1021/jo049736v.
- 15.a) Kobayashi S, Hachiya I, Araki M, Ishitani H. Tetrahedron Lett. 1993;34:3755–3758. [Google Scholar]; b) Kobayashi S, Nagayama S, Busujima T. J Am Chem Soc. 1998;120:8287–8288. [Google Scholar]; c) Nishina Y, Kida T, Ureshino T. Org Lett. 2011;13:3960–3963. doi: 10.1021/ol201479p. [DOI] [PubMed] [Google Scholar]
- 16.Single crystals of product 4p and 5d were obtained via careful evaporation of co-solvent of DMF and ethanol solvent. For crystal data, see Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.