

Abstract
Apolipoprotein E (apoE) is an anti-atherogenic protein that plays a critical role in maintaining plasma cholesterol and triglyceride homeostasis by virtue of its ability to act as a ligand for the low-density lipoprotein receptor (LDLr) family of proteins. In this study, we characterized the biochemical and biophysical features of recombinant rat apoE, in comparison with those of human apoE3. Rat apoE was overexpressed in Escherichia coli using a codon optimized system and purified by affinity chromatography. SDS-PAGE and RP-HPLC of rat apoE confirmed the purity, while immunoblot verified the identity and cross-reactivity with the LDLr-binding region of apoE3. The α-helical content was calculated to be ~45% by circular dichroism spectroscopy. The protein exists in a predominantly tetrameric form in lipid-free state. Chemical denaturation studies reveal that the unfolding pattern is biphasic with mid points of denaturation corresponding to 0.8 and 2.2 M guanidine hydrochloride, suggesting the presence of two domains. Rat apoE converts DMPC vesicles to smaller DMPC/apoE complexes with a first order rate constant of 0.12 min−1. It has the ability to bind the LDLr and to heparin. Our studies indicate that although its sequence resembles apoE4, an isoform of apoE3, rat apoE displays the biophysical behavior of apoE3.
Keywords: Rat apolipoprotein E, apolipoprotein E3, codon-optimization, denaturation, LDL receptor binding, heparin binding
INTRODUCTION
Apolipoprotein E (apoE) plays a key role in lipid transport, lipoprotein metabolism and cholesterol homeostasis in the plasma and the central nervous system in humans. It lowers plasma lipid levels primarily by acting as a ligand for the low-density lipoprotein (LDL) receptor and by promoting cholesterol efflux from atherosclerotic lesions [1, 2]. In humans, apoE is polymorphic, with three common alleles, APOE ε2, ε3 and ε4, encoding the three isoforms, apoE2, apoE3 and apoE4, represented with frequencies of 8%, 77% and 15%, respectively, in the population. Several biochemical, structural and physiological studies have been directed at human apoE due to its dominant role in disease states such as cardiovascular and Alzheimer’s disease [3–8]. ApoE3 is considered atheroprotective, while apoE4 is associated with hyperlipoproteinemia, cardiovascular disease and Alzheimer’s disease. However, much less is known about apoE from commonly used animal models such as rats and mice, which do not have isoforms. In this study, we investigate the biochemical and biophysical features of rat apoE in comparison with apoE3, the common isoform in humans.
The 3 human isoforms differ in amino acids at positions 112 and 158; apoE2 has Cys, while apoE4 has Arg in both locations; apoE3, however, bears a Cys at 112 and Arg at 158. APOE ε4 is considered the ancestral gene among primates, based on its similarities with primate APOE [9]. Figure 1 shows the protein sequence alignment of rat apoE and human apoE3 (and apoE4). The overall sequence identity between rat apoE and apoE3 is 73.5% (73.9% with apoE4). A segment between residues 1 to 183 around the N-terminal portion had higher extent of identity (78.7%) compared to the segment between 193 and 294 around the C-terminal portion of the protein (65.0%) suggesting conservation of key structural and functional features related to the N-terminal portion of apoE [2]. In apoE3, residues 1–191 and 201–299 are designated as the N-terminal (NT) and C-terminal (CT) domains, respectively; the two domains are linked via a protease-sensitive segment [10, 11]. The availability of high resolution structural information for apoE3(1-191) [12, 13], and full length monomeric apoE3 [14] helps us to draw parallels and understand the conformation of rat apoE.
Figure 1. Sequence alignment of rat apoE and human apoE3 and apoE4.

The primary sequence of rat apoE was aligned with those of human apoE3 and apoE4 using the LALIGN program ((http://embnet.vital-it.ch/software/LALIGN_form.html) [45], which is available in ExPASy, the Swiss Institute of Bioinformatics Resource Portal (www.expasy.org). The only difference between the two human isoforms lies at position 112, which is a Cys in apoE3 and an Arg in apoE4 (bold). The corresponding site in rat apoE (position 104) is an Arg (bold). In apoE4, Arg61 (bold) is proposed to form a salt bridge with Glu255 (bold); in rat apoE, the corresponding position bears a Thr residue (bold) (Thr53). The sequence shown in blue for rat apoE is predicted to be the LDLr binding region, based on the known sites for human apoE3 and apoE4 (shown in red) [2]. Similarly, the predicted HSPG binding site in rat apoE (Lys135 and Lys138) are shown in bold, based on the known sites in human isoforms (Lys143 and Lys146).
Despite the 99.7% identity between apoE3 and apoE4, there are significant differences in biophysical and physiological behavior between the two; indeed individuals bearing apoE4 are at a higher risk for developing cardiovascular and Alzheimer’s disease [5]. While there is no single feature that can satisfactorily explain the vast difference, the concept of domain interaction in apoE4 offers a possible explanation at the molecular level [15, 16]. This interaction was attributed to the presence of a salt bridge between Arg61 in the NT domain and Glu255 in the CT domain of apoE4, and is believed to be the consequence of a subtle structural feature in the microenvironment of Arg112 [15]. It was proposed that apoE3, bearing a Cys at position 112, lacks the domain interaction, and therefore differs significantly from apoE4. Interestingly, of all mammalian apoE sequences identified so far, only human apoE has an Arg at position 61; all others, including rats, bear a Thr at the corresponding position (Thr53 in rat apoE) and display functional features of apoE3 [2]. Thus, throughout this study, we compare the biochemical and biophysical properties of the recombinant forms of rat apoE and human apoE3, citing apoE4 where relevant.
MATERIALS AND METHODS
Materials
HiTrap heparin-Sepharose and nickel-affinity column (HiTrap Chelating column), Amersham High Molecular Weight Standard and the enhanced chemiluminescence (ECL) were obtained from GE Healthcare (Uppsala, Sweden). The Snap i.d. Protein Detection System and 4–20% acrylamide gradient SDS-PAGE were obtained from Millipore Corporation (Billerica, MA) and Invitrogen (Carlsbad, CA) respectively; 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) was obtained from Avanti Polar Lipids (Alabaster, AL). Anti-human apoE polyclonal antibody (goat anti-apoE-HRP) was obtained from BIODESIGN International (Saco, ME), and monoclonal antibodies 1D7 and 3H1 were from Ottawa Heart Institute Research Corporation (Ottawa, Canada). All chemical reagents were of analytical grade.
Codon optimization, expression, isolation and purification of rat apoE
A pET-20b(+) expression vector bearing rat apoE sequence was custom-designed with a hexa-His tag linked via a tobacco etch virus (TEV) protease cleavage site (underlined) (AcTEV™ Protease, Invitrogen) at the N-terminal end (HHHHHHENLYFQGDP) (Eurofins Scientific, Huntsville, AL). The hexa-His tag facilitates isolation and purification, while TEV protease cleavage site allows us to cleave the His-tag when necessary. The recombinant protein was over-expressed in Escherichia coli BL21(DE3)pLysS Gold (Stratagene, La Jolla, CA) in the presence of ampicillin (50 μg/ml) and chloramphenicol (50 μg/ml), as described previously [17]. Protein expression was induced with 0.4 mM IPTG. Following expression, the resuspended cells were lyzed using a microfluidizer (Microfluidics, Newton, MA) and the recombinant proteins purified using a Ni-affinity matrix (Hi-Trap chelating column, GE Healthcare, Uppsala, Sweden). The purity of the protein was verified by SDS-PAGE and reversed phase high-performance liquid chromatographic (RP-HPLC) analyses. SDS-PAGE was carried out using a 4–20% acrylamide gradient under reducing and non-reducing conditions. RP-HPLC analysis was carried out using a Zorbax C8 column (Zorbax 300SB-C8, 2.1 mm × 15 cm), with a linear AB gradient of 2% B/min, where solvent A and B were 0.05% trifluoroacetic acid (TFA) in water and 0.05% TFA in acetonitrile, respectively. The His-tag was removed by treating with AcTEV™ protease. ApoE (45 μg protein) was incubated with AcTEV™ protease (1 unit enzyme per 3 μg of protein) at 24 °C or 30 °C for 0–4 hr. The proteolytic digestion was visualized by SDS-PAGE using a 4–20% acrylamide gradient. Protein concentration was determined in a Nano-Drop 2000/2000c spectrometer (Thermo Fischer Scientific, Wilmington, DE) using the molar extinction coefficient at 280 nm of 34,950 M−1 cm−1.
Immunoblot analysis
Recombinant rat apoE was probed with polyclonal apoE-HRP antibody (1:1000 dilution), or monoclonal antibodies 1D7 (1:1000) and 3H1 (1:1000), and horseradish peroxidase-conjugated anti-mouse IgG (1:10,000) (Chemicon, Millipore, Billerica, MA) in a Snap i.d. system, using the ECL detection system (GE Healthcare, Uppsala, Sweden).
Circular dichroism (CD) spectroscopy
The secondary structure of rat apoE was examined by CD spectroscopy on a Jasco J-810-150S spectropolarimeter at 24 °C [18]. Far-UV CD scans were recorded between 185 nm and 260 nm in 10 mM ammonium bicarbonate buffer, pH 7.4 using protein concentrations of 0.2 mg/ml in a 0.1 cm path length cuvette. Far-UV CD profiles were the average of four independent scans recorded with a response time of 1s and bandwidth of 1 nm. The molar ellipticity ([θ]) in deg. cm2dmol−1 at 222 nm was obtained using the equation:
where MRW is the mean residue weight (obtained by dividing molecular weight by the number of residues) calculated to be 115.64, θ is the measured ellipticity in degrees at 222 nm, l is the cuvette path-length (in cm) and c is the protein concentration (in g/ml). The percent α-helix content was calculated as described by others [19]:
GdnHCl-induced unfolding
Unfolding of rat apoE was assessed by following changes in molar ellipticity at 222 nm as a function of increasing concentration of GdnHCl as described earlier [17]. The samples (0.2 mg/ml) were treated with 0–8 M GdnHCl in 10mM sodium phosphate buffer, pH 7.4 for 18 h at 37 °C. The % maximal change was calculated from the ellipticity values at 222 nm as:
The concentration of GdnHCl required to cause a 50% decrease in the maximal change ([GdnHCl]1/2) was calculated.
Gel filtration chromatography
Gel filtration chromatography of recombinant rat apoE (1–2 mg/ml) was carried out on an 80 ml column packed with Sepharose CL-6B (Sigma Aldrich, St. Louis, MO) in 10 mM sodium phosphate, pH 7.4, 150 mM NaCl with a flow rate of 0.5 ml/min. The fractions (1.2 ml) were monitored at 280 and 210 nm. The void volume (V0) was calculated using blue dextran; the elution volume (Ve) for the protein standards and apoE samples were calculated at the maximal absorbance of the peak.
Lipid binding assay
The ability of apolipoproteins to bind lipids was determined as described previously [20, 21], wherein the assay is performed in a Perkin Elmer UV/VIS spectrophotometer equipped with a Peltier controlled (PTP-6) cell holder. DMPC MLV (500 μg) was pre-incubated at 24°C in PBS in the cuvette followed by addition of 100 μg apoE. The decrease in absorbance was measured at 325 nm. Data were normalized to initial absorbance at 325 nm prior to addition of protein. The time required for initial absorbance to decrease by 50% (T1/2) and the rate constant (K), (reciprocal of T1/2), were determined.
Preparation of DMPC/apoE complex
DMPC/apoE complexes were prepared as described earlier [22] using rat apoE or apoE3(1-191). ApoE was incubated with DMPC vesicles (2.5:1 weight ratio or 5:2 molar ratio) at 24 °C for 16 h; lipid-bound protein was separated from the unbound protein by density gradient ultracentrifugation using a KBr gradient. Fractions containing both protein and lipid were pooled and concentrated. Protein assay was carried out by the bicinchoninic acid method (Pierce Biotechnology, Rockford, IL), and phospholipid assay using the phospholipid assay kit (Wako Chemicals USA, Inc., Richmond, VA) on each fraction. The complexes were characterized in terms of particle size and diameter, lipid and protein composition as described earlier [23, 24]. The protein and lipid contents of the complexes were estimated using the DC™ protein (BioRad Laboratories, Hercules, CA) and phospholipid assay. Non-denaturing PAGE of the isolated lipoprotein complexes was carried out to evaluate the molecular mass and average particle size on a 4–20% gradient gel for 24 h at 110 V and stained with Amido Black.
LDLr binding assay
To examine the ability of rat apoE to bind to the LDLr, a co-immunoprecipitation (co-IP) assay was performed [25]. A construct bearing the LDLr ligand binding domains LA3-LA6 with a c-Myc epitope was employed. This construct represents the essential ligand binding elements of the extra cellular soluble portion of the mature LDLr and is represented as sLDLr. DMPC/apoE complex (10 μg protein) was incubated with 10 μg of sLDLr in the presence of 2 mM Ca+2, or 2 mM EDTA, in PBS at 4°C for 1 h. This was followed by co-IP with an anti-c-Myc antibody-linked agarose (Sigma-Aldrich, St. Louis, MO) to capture the DMPC/apoE/sLDLr complexes. ApoE was detected by Western blot analysis using HRP-conjugated polyclonal apoE antibody. A replica experiment was conducted wherein an anti-c-Myc antibody (9E10) was utilized to identify the presence of LDLr in each reaction.
Heparin binding assay
A typical functional feature of apoE is its ability to interact with cell surface-localized heparan sulfate proteoglycans (HSPG) in the blood vessels. In vitro, this is followed using the HiTrap heparin-Sepharose column appended to ÄKTA FPLC system (Amersham Pharmacia Biotech, Uppsala, Sweden) as described by us previously [26, 27]. Purified rat apoE was dialyzed against 20 mM sodium phosphate buffer, pH 7.4, and injected onto the column at a flow rate of 1.0 ml/min. The bound protein was eluted with a salt gradient (0 to 1.0 M NaCl) and monitored at 280 nm.
RESULTS
A pET-20b(+) expression vector bearing the coding sequence of rat apoE optimized for expression in E. coli was used. The protein was over-expressed, isolated, purified by affinity chromatography and visualized by SDS-PAGE analysis, Figure 2A. Rat apoE migrates with a molecular mass corresponding to about 34 kDa (calculated mass is 33, 844 Da). For comparison, apoE expressed using a plasmid bearing non-optimized codons is also shown. A significantly higher yield of protein was obtained using the optimized-codon system (50 mg/L culture medium versus 10 mg/L), in addition to increased purity. The overexpressed protein was significantly more stable during storage at −20 °C; apoE expressed using non-optimized codons displayed increased degradation. The identity of apoE was confirmed by Western blot using the anti-human apoE monoclonal antibodies 1D7 and 3H1 (the epitopes for which lie between residues 139 and 169 at the N-terminal portion, and, between 243 and 272 at the C-terminal portion of human apoE, respectively), Figure 2B. While 1D7 cross reacts with rat apoE, 3H1 does not under the conditions employed, suggesting immunogenic similarity and conservation between the two around the LDLr binding site, but not around the C-terminal end. Figure 2C shows analytical RP-HPLC profiles of rat apoE prepared using the optimized- and non-optimized codon system; the profile confirms the purity of the protein using the optimized expression system.
Figure 2. SDS-PAGE, Western blot and RP-HPLC profile of rat apoE.

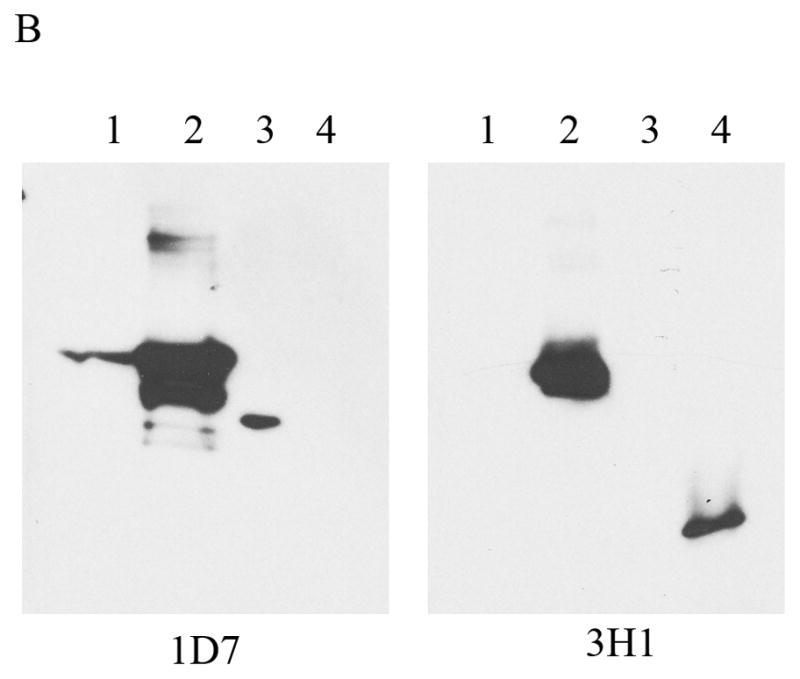


A. SDS-PAGE analysis (4–20 % acrylamide gradient) under reducing conditions of recombinant rat apoE (5 μg) obtained using a plasmid bearing optimized (Left) or non-optimized (Right) codons. The molecular mass standards are shown in the first lane. B. Western blot analysis of recombinant rat apoE (0.2 μg) using anti-apoE antibody mAb1D7 (Left) and mAb3H1 (Right); lanes 1 and 5, rat apoE; lanes 2 and 6, human apoE3; lanes 3 and 7, human apoE3(1-191); lanes 4 and 8, human apoE(201-299). C. Analytical RP-HPLC analysis of recombinant rat apoE (100 μg protein) obtained using a plasmid bearing optimized (Left) or non-optimized (Right) codons. Following the Ni-affinity chromatography step, the samples were loaded on a Zorbax C-8 HPLC column and eluted at 0.25 ml/min with a linear AB gradient of 2% B/min, where A is 0.05% aqueous TFA and B is acetonitrile containing 0.05% TFA.
Purified apoE (10 μg) was treated with 3U of AcTEV™ protease at 24 °C or 30 °C for 0–4 h to determine the conditions required to cause complete cleavage of the His-tag at the N-terminal end of the protein. Following incubation, the samples were analyzed by SDS-PAGE. A shift in the mobility is indicative of the loss of the His-tag. Figure 3 shows that ~90% cleavage was achieved in about 3 h regardless of the temperature of incubation.
Figure 3. Cleavage of hexa-His tag from purified rat apoE.
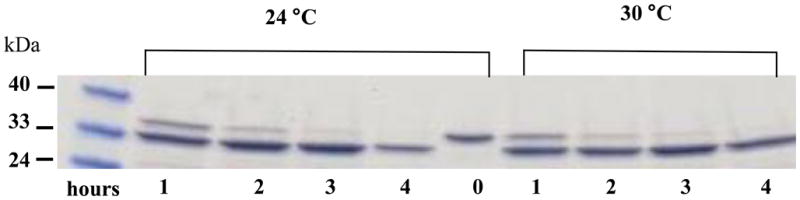
Following purification, 4–20% acrylamide gradient SDS-PAGE was performed of recombinant rat apoE (10 μg) that was treated with 3U of AcTEV™ protease at 24 °C (left) or 30 °C (right) for 0, 1, 2, 3 or 4 h. Following incubation, the reaction was stopped by addition of SDS-PAGE sample treatment buffer and the samples directly electrophoresced on a 4–20% acrylamide gradient SDS-PAGE gel.
In the next step, far-UV CD spectra were recorded for recombinant rat apoE and human apoE3 to assess the overall secondary structure of the protein, Figure 4A. The spectra revealed features of a highly helical protein, characterized by troughs at 208 and 222 nm. The α-helical content was calculated from the molar ellipticity values at 222 nm to be 45 ± 5%, which is comparable to that reported for human apoE3 (~50%) [28]. Further, GdnHCl-induced unfolding studies were performed by following changes in the molar ellipticity at 222 nm as a function of varying GdnHCl concentration, Figure 4B. Rat apoE undergoes a two-phase unfolding process with a distinct plateau between the two phases, revealing two midpoints of denaturation around 0.8 and 2.2 M GdnHCl.
Figure 4. Secondary structure and GdnHCl-induced unfolding of rat apoE.


A. Far-UV CD spectra of apoE. The spectra of rat (solid line) and human apoE3 (dashed line) (0.2 mg/ml) were recorded in 20 mM sodium phosphate buffer, pH 7.4. The spectra were recorded from 185 to 260 nm using a 0.1 cm path length cell, scan speed of 20 nm per minute, and response time of 1 second (average of 3 scans shown). B. GdnHCl-induced unfolding of apoE. The % maximal change in ellipticity of 0.2 mg/ml rat apoE (closed circles) and human apoE3 (open circles) at 222 nm was plotted as a function of increasing GdnHCl concentration. ApoE3 was pre-incubated with 5-fold molar excess of reducing agent, dithiothreitol, to reduce existing disulfide bonds.
To assess the self-associated state of apoE, size exclusion chromatography was performed using a protein concentration of 1.5 mg/ml Figure 5. A prominent peak between 46.6 and 47.9 ml (average: 47.3 ml) was noted. From the plot of log molecular weight versus Ve/V0 for the standards (Figure 5, Inset), the molecular mass was calculated to be 124,000 Da for the peak.
Figure 5.

Size exclusion chromatography of apoE. ApoE (1.5 mg/ml) was loaded on a Sepharose CL-6B size exclusion column operating at a flow rate of 0.5 ml/min in PBS, with 1.2 ml per fraction. The elution profile shows a prominent peak, the molecular mass of which was calculated based on the plot of log molecular weight versus Ve/V0 for the following standards (Inset): α-lactalbumin from bovine milk (14,200 Da), carbonic anhydrase from bovine erythrocytes (29,000 Da), human serum albumin (66,400 Da), urease from Jack Bean (272,000 Da). The arrow corresponds to the peak elution position of blue dextran.
The functional characteristics of recombinant rat apoE were then examined by following its ability to bind: (i) lipids, (ii) LDLr, and, (iii) heparin. A characteristic feature of apolipoproteins is their ability to bind and transform MLV composed of DMPC to discoidal bilayer structures at the gel-crystalline transition temperature (24 °C). Conversion of the MLV (~200 nm diameter) to the smaller lipid/protein complexes (diameter ~ 20 nm) is accompanied by a decrease in turbidity that can be followed as change in absorbance at 325 nm. Figure 6A shows changes in absorbance obtained when DMPC MLV (500 μg lipid) was treated with PBS (curve a) or rat apoE (100 μg) (curve b). While the absorbance of DMPC vesicles remained mostly unchanged in absence of protein, it decreased rapidly upon addition of apoE. The time required for the absorbance to decrease to 50% of the initial value (T1/2) and the rate constant, K (reciprocal of T1/2) were determined be 8.8 ± 1.0 min and 0.12 ± 0.01 min−1, respectively. The rate constant is identical to that reported for human apoE3 by others [29], even though the latter assay was carried with DMPC MLV: apoE 7:1 w/w ratio, in the presence of 0.1 M GdnHCl. The DMPC/apoE complexes formed are ~ 14 nm in diameter and ~550 kDa in size as noted by non-denaturing PAGE, Figure 6B. The lipid:protein ratio of the complexes was found to be about 50:1 (mole/mole). A minor band was also noted with a particle diameter of 15 nm and mass of ~630 kDa.
Figure 6.

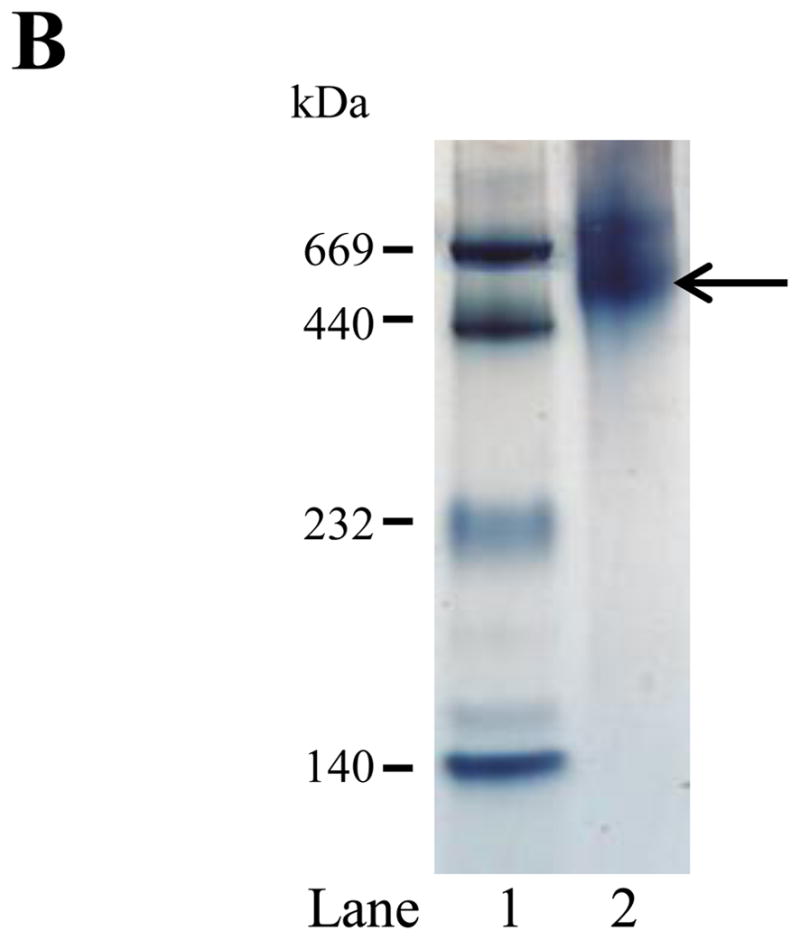
Lipid-binding ability of rat apoE. A. Transformation of DMPC MLV to smaller lipid/protein complexes by apoE. DMPC MLV (500 μg) was treated with PBS (curve a) or rat apoE (100 μg) (curve b) (5:1 w/w ratio for lipid:protein) and the change in absorbance at 325 nm was followed as a function of time at 24 °C. The data are normalized to absorbance at 0 min. Representative curves from 3 different experiments are shown. B. Native PAGE analysis of DMPC/apoE complexes. About 50 μg of DMPC/apoE complexes were loaded on a 4–20% acrylamide gradient PAGE gel and electrophoresced in 10 mM Tris-glycine, pH 8.4 for 24 h at 110V and stained with Amido Black. Lane 1, high molecular weight standard; lane 2, DMPC/apoE complex (arrow draws attention to the major band). The molecular mass and average particle sizes were calculated from a calibration curve using the following standards and their corresponding molecular masses and Stokes diameters: thyroglobulin (669 kDa, 17 nm); ferritin (440 kDa, 12.2 nm); catalase (232 kDa, 9.2 nm); lactate dehydrogenase (140 kDa, 8.2 nm).
In its lipid-associated state, apoE serves as a ligand and binds to the LDLr, which mediates internalization of the entire lipoprotein particle by receptor mediated endocytosis. The LDLr binding ability of rat apoE was followed by a co-IP assay using sLDLr/LA3-LA6/Myc bound to anti-Myc agarose. Following incubation of DMPC/apoE complexes with sLDLr/LA3-LA6/Myc at 4 °C for 4 h, the receptor-bound complexes were captured by co-IP with anti-c-Myc-agarose and detected by HRP-conjugated polyclonal apoE antibody, Figure 7 (top panel) or anti-Myc antibody (bottom panel). DMPC/rat apoE elicits the ability to bind the sLDLr/LA3-LA6/Myc in the presence of Ca2+ (lane 2). The binding ability was abolished when Ca2+ was omitted and 2 mM EDTA included in order to chelate residual Ca2+ in the incubation mixture, lane 3. This indicates that the interaction of rat apoE with sLDLr/LA3-LA6/Myc is specific, as Ca2+ is essential for maintaining the structural and functional integrity of the latter. The lipid-bound form of human apoE3 (1-191) (10 μg) bearing the LDLr binding segment was used as a positive control, lane 4.
Figure 7.
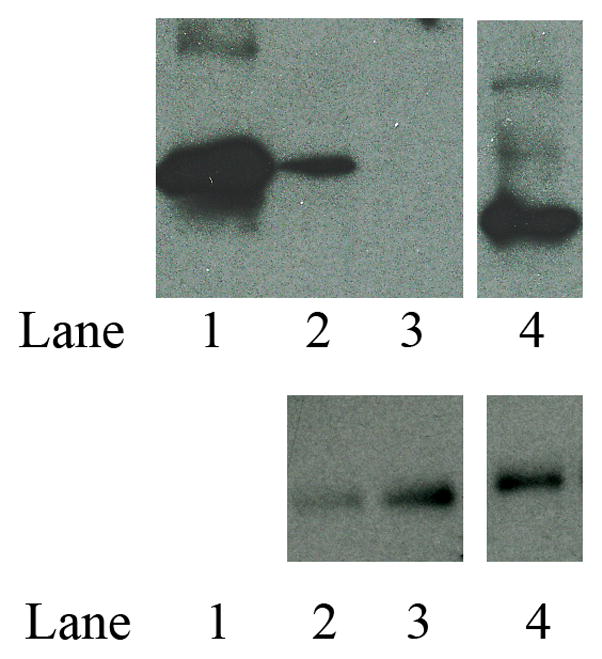
LDLr binding ability of lipid-associated rat apoE. DMPC/apoE complexes were incubated with 1 μg of sLDLr/LA3-LA6/Myc in 25 mM Tris-HCl, pH 7.4, 140 mM NaCl, 27 mM KCl, 2 mM CaCl2 for 2 h at 4 °C, followed by co-IP with anti-c-Myc antibody. LDLr-bound apoE was detected by Western blot using HRP-conjugated polyclonal apoE antibody (top panel). Incubations where Ca2+ were omitted included 2 mM EDTA to chelate residual Ca2+ in the mixture. The lane assignments were as follows: lane 1, lipid-free rat apoE used as control for Western blot (0.5 μg protein); lane 2, DMPC/rat apoE (10 μg) in the presence of Ca2+; lane 3, DMPC/rat apoE (10 μg) in the absence of Ca2+ (and in the presence of 2 mM EDTA); lane 4, DMPC/human apoE3/(1-191) (10 μg). Western blot with anti-Myc antibody (bottom panel) to confirm the presence of sLDLr/LA3-LA6/Myc in each reaction. The assignments for the bottom panel in lanes 2, 3 and 4 are the same as those for the top panel.
Lastly, the heparin binding ability of rat apoE was assessed using heparin-Sepharose columns. A solution of 300 μg rat apoE was injected onto a heparin column and the bound protein eluted using a 0 – 1 M NaCl gradient, Figure 8. Rat apoE eluted with a retention time of 28 min, corresponding to ~ 450 mM NaCl.
Figure 8.

Heparin binding ability of rat apoE. Lipid free apoE (100 μg) was loaded onto a heparin-Sepharose column in 20 mM sodium phosphate pH 7.4 in an ÄKTA FPLC system. The flow rate was maintained at 1 ml/min. A salt gradient of 0 – 1M NaCl was used to elute the bound protein; the elution of the protein was monitored at 280 nm. The arrow represents the start of the gradient.
DISCUSSION
The biochemical and biophysical characteristics of recombinant rat apoE resemble those of human apoE3, with which it bears 73.5% sequence identity. The coding sequence of rat apoE was optimized for over-expression in E. coli. Rat apoE contains some codons that were not utilized by bacterial transcriptional and translational mechanisms, which resulted in low yield and high degradation when expressed in E. coli [30] under our conditions. The synthetic sequence had 24 optimized and 13 endogenous codons. The codon optimization significantly improved the yield and purity of the protein. The plasmid was also designed to bear an AcTEV™ protease cleavage site; the TEV protease is an endoprotease that specifically cleaves between the Q and G of the underlined segment (shown under Materials and Methods) [31], thereby removing the His-tag. The protease is a recombinant protein that also bears a His-tag at its N-terminal end. This is an advantageous feature as it allows effective removal of the protease following proteolysis of rat apoE. Following treatment, the incubation mixture is passed over a Ni2+-affinity column, which binds the cleaved tag, un-cleaved apoE bearing His-tag, and the His-tag bearing protease; apoE without the tag is recovered in the flow-through fraction. In subsequent studies, we used the recombinant protein as such without protease treatment, since our earlier experience showed that the presence of the tag does not affect the overall biochemical or biophysical behavior of apolipoproteins [17, 32].
Size-exclusion chromatography indicates that rat apoE is predominantly tetrameric at 1–2 mg/ml concentration; the broad peak suggests that it likely exists as an equilibrium mixture of tetramers, dimers and monomers. A small fraction of higher state oligomers was also noted eluting around V0. A similar heterogeneous mixture has been observed for apoE3 and apoE4 in lipid-free environments [28, 33]. Secondary structural analysis indicates that rat apoE adopts a highly helical structure as expected based on its identity with human apoE, which has a helical content of 50% (for both apoE3 [10]and apoE4) [28]. Consistent with the CD data, high resolution analyses of various forms of human apoE show a highly helical structure: apoE2 (1-191) [34], apoE3 (1-191) [12, 13], apoE4 (1-191) [35], monomeric apoE3 [14]. The NT domain is comprised of a helix bundle of 4 long amphipathic α-helices, wherein the hydrophobic face of each helix is oriented towards the protein interior and the polar face towards the aqueous environment. The CT domain is comprised of amphipathic α-helical segments as well. Although the high-resolution structure of tetrameric apoE3 is not known, biochemical and biophysical studies indicate that the CT domain promotes apoE3 (or apoE4) self-association by mediating inter-molecular helix-helix contacts leading to formation of dimers, tetramers and higher state oligomers [4, 10, 17, 36, 37]. C-terminal truncation analysis [4, 38] and site-directed mutagenesis of specified residues in the CT domain of apoE3 (F257A/W264R/V269A/L279Q/V287E) [14] show loss of tetramerization behavior, supporting the role of this domain in self-association.
The unfolding pattern of rat apoE induced by GdnHCl was biphasic, similar to that for human apoE3, which displays two plateaus with midpoints of denaturation ranging from 0.7–0.9 and 2.4–2.5 M GdnHCl [4, 10, 19, 36, 39]. Each phase represents a two-state denaturation equilibrium, and corresponds to unfolding of the CT and NT domains. In concurrence, isolated human apoE3 NT and CT domains displayed midpoints of denaturation of 0.9 and 2.4 M GdnHCl, respectively. Compared to the CT domain, the NT domain reveals a higher midpoint of denaturation due to the extensive intra molecular salt bridge and hydrophobic interactions that stabilize the compact 4-helix bundle structure. Previous studies show that apoE4 displays a unique denaturation profile that is distinctly different from that of apoE3: it lacks the two plateaus corresponding to unfolding of the two domains. Further, isolated apoE4 NT domain displays a midpoint of denaturation of 2.0 M GdnHCl [19, 40]. The former observation has been attributed to the possible formation of a molten globule by apoE4, with a compact structure that retains the overall stable secondary structure but bearing less stable tertiary structural conformation and lacking the tightly packed interior [40, 41]. This behavior was attributed to the presence of a salt bridge between Arg61 and Glu255 in apoE4 [15]. In apoE4, an intra-helical salt bridge between Arg112 and Glu109 displaces Arg61, making it available for inter-domain salt bridge formation with Glu255 in the CT domain [15]. Lacking high-resolution information of full-length apoE4, this interaction needs to be confirmed from a structural perspective. Nevertheless, it has been proposed that this domain interaction may define the unique physiological/pathological behavior of apoE4 in humans. In contrast, apoE3 (lacking the intra-helical salt bridge) is able to accommodate Arg61 in the vicinity of Cys112 [35]. Interestingly, mouse apoE, bearing Arg and Thr at positions corresponding to 112 and 61 in helix 3 and helix 2, respectively of apoE4, behaves like human apoE3 in terms of lipoprotein binding preference for HDL; substituting Arg in helix 2 introduced apoE4-like behavior, i.e., lipoprotein binding preference for VLDL [42]. Similarly, we propose that although rat apoE bears Arg at a position corresponding to 112 in human apoE, it elicits the biophysical behavior of apoE3 rather than apoE4 due to the presence of a Thr53 (corresponding to Arg61).
Rat apoE displays all the characteristic functional features of human apoE: from an in vitro functional standpoint, there are no significant differences between apoE3 and apoE4. Like apoE3, it shows rapid and efficient lipid binding as observed by its ability to transform DMPC vesicles to smaller DMPC/apoE complexes, and likely adopts a similar lipid-bound conformation. Several lines of evidence indicate that apolipoproteins undergo a dramatic lipid-induced conformational change, which involves opening of the helix bundle and interaction of the hydrophobic face of the amphipathic helices with phospholipid side chains [4, 7, 19, 22, 23, 43]. In this lipid-bound conformation, the helices of apoE3 circumscribe a cluster of phospholipids in a bilayer configuration forming a discoidal complex [32].
In the lipid-associated state, recombinant rat apoE recapitulates the functional features of apoE3, specifically its ability to act as a ligand for the LDLr. The sequence identity between rat apoE and human apoE3 in the LDLr-binding region (122–142 in rat apoE and 130–150 in human apoE4) is ~76%. The LDLr belongs to a superfamily of receptors that are characterized by five domains: an extra-cellular ligand-binding domain, an epidermal growth factor precursor homology domain, an O-linked glycosylation domain, a transmembrane domain and an intra-cellular C-terminal domain [44]. The ligand-binding domain is composed of LA1-LA7, of which LA3-LA6 have been shown to be essential for binding apoE [25]. The construct used in this study represents the essential ligand binding elements of the extracellular soluble portion of the LDLr, which recapitulates the essential structural and functional features of the intact receptor and serves as a ‘mini-receptor’ [25].
Lastly, rat apoE binds heparin with comparable affinity as human apoE; previous studies show that ~400 mM NaCl is required to elute human apoE from the heparin column [27]. This is an ionic interaction wherein the Lys side chains interact with the acidic sulfated moieties of HSPG. The residues essential for heparin binding are predicted to be Lys135 and Lys138, corresponding to Lys143 and Lys146 in apoE3 [2].
In summary, we report the biochemical and biophysical characteristics of a bacterially- expressed recombinant rat apoE using an optimized-codon system. Our studies indicate that although rat apoE bears an Arg at a position corresponding to Arg112 in human apoE4, it displays the biophysical behavior of apoE3, possibly because of the presence of Thr53. Rat apoE is a highly helical, tetrameric two-domain protein that displays the ability to bind lipids, the LDLr and heparin efficiently. Future studies will allow the use of recombinant rat apoE as a structural and mechanistic model for in vivo studies involving rats or other rodents.
Highlights.
Codon-optimized recombinant rat apoE was purified by affinity chromatography
Chemical-induced denaturation indicates 2 independently folded domains in rat apoE
Rat apoE has the ability to bind lipids, LDL receptor and heparin
It resembles human apoE3, but not apoE4, from a biophysical perspective
Acknowledgments
This work was supported by partial funding from Tobacco Related Disease Research Program (TRDRP 17RT-0165), Faculty Development Grant, California State University Program to Enhance Research in Biotechnology (CSUPERB) (VN), NIH HL096365 (TNT, CG & VN), CSUPERB-Howell Foundation and NSF/CSU-LSAMP (HRD-0802628) (TNT).
LIST OF ABBREVIATIONS
- ApoE
Apolipoprotein E
- CD
Circular dichroism
- CT
C-Terminal
- DMPC
1,2-Dimyristoyl-sn-glycero-3-phosphocholine
- EDTA
Ethylenediaminetetraacetic acid
- GdnHCl
Guanidine hydrochloride
- IP
Immunoprecipitation
- LDL
Low-density lipoprotein
- LDLr
Low-density lipoprotein receptor
- MLV
Multilamellar vesicles
- NT
N-terminal
- PBS
phosphate buffered saline, 10 mM sodium phosphate, pH 7.4, 150 mM NaCl
- RP-HPLC
Reversed phase high-performance liquid chromatography
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mahley RW, Rall SC., Jr Annu Rev Genomics Hum Genet. 2000;1:507–537. doi: 10.1146/annurev.genom.1.1.507. [DOI] [PubMed] [Google Scholar]
- 2.Weisgraber KH. Adv Protein Chem. 1994;45:249–302. doi: 10.1016/s0065-3233(08)60642-7. [DOI] [PubMed] [Google Scholar]
- 3.Hatters DM, Peters-Libeu CA, Weisgraber KH. Trends Biochem Sci. 2006;31:445–454. doi: 10.1016/j.tibs.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 4.Saito H, Lund-Katz S, Phillips MC. Prog Lipid Res. 2004;43:350–380. doi: 10.1016/j.plipres.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 5.de Chaves EP, Narayanaswami V. Future Lipidol. 2008;3:505–530. doi: 10.2217/17460875.3.5.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahley RW, Huang Y. Curr Opin Lipidol. 1999;10:207–217. doi: 10.1097/00041433-199906000-00003. [DOI] [PubMed] [Google Scholar]
- 7.Narayanaswami V, Kiss RS, Weers PM. Comp Biochem Physiol A Mol Integr Physiol. 2010;155:123–133. doi: 10.1016/j.cbpa.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hauser PS, Narayanaswami V, Ryan RO. Prog Lipid Res. 2011;50:62–74. doi: 10.1016/j.plipres.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fullerton SM, Clark AG, Weiss KM, Nickerson DA, Taylor SL, Stengard JH, Salomaa V, Vartiainen E, Perola M, Boerwinkle E, Sing CF. Am J Hum Genet. 2000;67:881–900. doi: 10.1086/303070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aggerbeck LP, Wetterau JR, Weisgraber KH, Wu CS, Lindgren FT. J Biol Chem. 1988;263:6249–6258. [PubMed] [Google Scholar]
- 11.Wetterau JR, Aggerbeck LP, Rall SC, Jr, Weisgraber KH. J Biol Chem. 1988;263:6240–6248. [PubMed] [Google Scholar]
- 12.Wilson C, Wardell MR, Weisgraber KH, Mahley RW, Agard DA. Science. 1991;252:1817–1822. doi: 10.1126/science.2063194. [DOI] [PubMed] [Google Scholar]
- 13.Sivashanmugam A, Wang J. J Biol Chem. 2009;284:14657–14666. doi: 10.1074/jbc.M901012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen J, Li Q, Wang J. Proc Natl Acad Sci USA. 2011;108:14813–14818. doi: 10.1073/pnas.1106420108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dong LM, Weisgraber KH. J Biol Chem. 1996;271:19053–19057. doi: 10.1074/jbc.271.32.19053. [DOI] [PubMed] [Google Scholar]
- 16.Dong LM, Wilson C, Wardell MR, Simmons T, Mahley RW, Weisgraber KH, Agard DA. J Biol Chem. 1994;269:22358–22365. [PubMed] [Google Scholar]
- 17.Patel AB, Khumsupan P, Narayanaswami V. Biochemistry. 2010;49:1766–1775. doi: 10.1021/bi901902e. [DOI] [PubMed] [Google Scholar]
- 18.Sreerama N, Venyaminov SY, Woody RW. Anal Biochem. 2000;287:243–251. doi: 10.1006/abio.2000.4879. [DOI] [PubMed] [Google Scholar]
- 19.Morrow JA, Segall ML, Lund-Katz S, Phillips MC, Knapp M, Rupp B, Weisgraber KH. Biochemistry. 2000;39:11657–11666. doi: 10.1021/bi000099m. [DOI] [PubMed] [Google Scholar]
- 20.Dettloff M, Niere M, Ryan RO, Luty R, Kay CM, Wiesner A, Weers PM. Biochemistry. 2002;41:9688–9695. doi: 10.1021/bi0200108. [DOI] [PubMed] [Google Scholar]
- 21.Weers PM, Abdullahi WE, Cabrera JM, Hsu TC. Biochemistry. 2005;44:8810–8816. doi: 10.1021/bi050502v. [DOI] [PubMed] [Google Scholar]
- 22.Fisher CA, Narayanaswami V, Ryan RO. J Biol Chem. 2000;275:33601–33606. doi: 10.1074/jbc.M002643200. [DOI] [PubMed] [Google Scholar]
- 23.Narayanaswami V, Szeto SS, Ryan RO. J Biol Chem. 2001;276:37853–37860. doi: 10.1074/jbc.M102953200. [DOI] [PubMed] [Google Scholar]
- 24.Raussens V, Drury J, Forte TM, Choy N, Goormaghtigh E, Ruysschaert JM, Narayanaswami V. Biochem J. 2005;387:747–754. doi: 10.1042/BJ20041536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fisher C, Abdul-Aziz D, Blacklow SC. Biochemistry. 2004;43:1037–1044. doi: 10.1021/bi035529y. [DOI] [PubMed] [Google Scholar]
- 26.Kiss RS, Weers PM, Narayanaswami V, Cohen J, Kay CM, Ryan RO. J Biol Chem. 2003;278:21952–21959. doi: 10.1074/jbc.M302676200. [DOI] [PubMed] [Google Scholar]
- 27.Tamamizu-Kato S, Wong JY, Jairam V, Uchida K, Raussens V, Kato H, Ruysschaert JM, Narayanaswami V. Biochemistry. 2007;46:8392–8400. doi: 10.1021/bi700289k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perugini MA, Schuck P, Howlett GJ. J Biol Chem. 2000;275:36758–36765. doi: 10.1074/jbc.M005565200. [DOI] [PubMed] [Google Scholar]
- 29.Segall ML, Dhanasekaran P, Baldwin F, Anantharamaiah GM, Weisgraber KH, Phillips MC, Lund-Katz S. J Lipid Res. 2002;43:1688–1700. doi: 10.1194/jlr.m200157-jlr200. [DOI] [PubMed] [Google Scholar]
- 30.Pearson K, Liu M, Shen L, Tso P, Davidson WS. Protein Expr Purif. 2005;41:447–453. doi: 10.1016/j.pep.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 31.Kapust RB, Tozser J, Copeland TD, Waugh DS. Biochem Biophys Res Commun. 2002;294:949–955. doi: 10.1016/S0006-291X(02)00574-0. [DOI] [PubMed] [Google Scholar]
- 32.Narayanaswami V, Maiorano JN, Dhanasekaran P, Ryan RO, Phillips MC, Lund-Katz S, Davidson WS. J Biol Chem. 2004;279:14273–14279. doi: 10.1074/jbc.M313318200. [DOI] [PubMed] [Google Scholar]
- 33.Sakamoto T, Tanaka M, Vedhachalam C, Nickel M, Nguyen D, Dhanasekaran P, Phillips MC, Lund-Katz S, Saito H. Biochemistry. 2008;47:2968–2977. doi: 10.1021/bi701923h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dong LM, Parkin S, Trakhanov SD, Rupp B, Simmons T, Arnold KS, Newhouse YM, Innerarity TL, Weisgraber KH. Nat Struct Biol. 1996;3:718–722. doi: 10.1038/nsb0896-718. [DOI] [PubMed] [Google Scholar]
- 35.Dong LM, Wilson C, Wardell MR, Simmons T, Mahley RW, Weisgraber KH, Agard DA. J Biol Chem. 1994;269:22358–22365. [PubMed] [Google Scholar]
- 36.Wetterau JR, Aggerbeck LP, Rall SC, Jr, Weisgraber KH. J Biol Chem. 1988;263:6240–6248. [PubMed] [Google Scholar]
- 37.Choy N, Raussens V, Narayanaswami V. J Mol Biol. 2003;334:527–539. doi: 10.1016/j.jmb.2003.09.059. [DOI] [PubMed] [Google Scholar]
- 38.Westerlund JA, Weisgraber KH. J Biol Chem. 1993;268:15745–15750. [PubMed] [Google Scholar]
- 39.Hatters DM, Zhong N, Rutenber E, Weisgraber KH. J Mol Biol. 2006;361:932–944. doi: 10.1016/j.jmb.2006.06.080. [DOI] [PubMed] [Google Scholar]
- 40.Weers PM, Narayanaswami V, Choy N, Luty R, Hicks L, Kay CM, Ryan RO. Biophys Chem. 2003;100:481–492. doi: 10.1016/s0301-4622(02)00300-9. [DOI] [PubMed] [Google Scholar]
- 41.Morrow JA, Hatters DM, Lu B, Hochtl P, Oberg KA, Rupp B, Weisgraber KH. J Biol Chem. 2002;277:50380–50385. doi: 10.1074/jbc.M204898200. [DOI] [PubMed] [Google Scholar]
- 42.Raffai RL, Dong LM, Farese RV, Jr, Weisgraber KH. Proc Natl Acad Sci U S A. 2001;98:11587–11591. doi: 10.1073/pnas.201279298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davidson WS, Thompson TB. J Biol Chem. 2007;282:22249–22253. doi: 10.1074/jbc.R700014200. [DOI] [PubMed] [Google Scholar]
- 44.Jeon H, Blacklow SC. Annu Rev Biochem. 2005;74:535–562. doi: 10.1146/annurev.biochem.74.082803.133354. [DOI] [PubMed] [Google Scholar]
- 45.Huang X, Miller M. Adv Appl Math. 1991;12:337–357. [Google Scholar]