

Abstract
DXP synthase catalyzes the formation of 1-deoxy-D-xylulose 5-phosphate, an essential precursor in pathogen isoprenoid biosynthesis. The selective inhibition of this ThDP-dependent transformation is a challenging goal in the development of isoprenoid biosynthesis inhibitors. Potent, selective inhibitors could lead to new anti-infective agents. Here, we demonstrate selective inhibition of E. coli DXP synthase by butylacetylphosphonate.
With the incidence of multidrug resistance on the rise, new targets need to be identified toward the development of new anti-infective agents. The methylerythritol phosphate (MEP) pathway (Fig. 1) is the sole source of essential isoprenoids in many human pathogens.1 Although numerous efforts have been made to identify inhibitors of pathogen isoprenoid biosynthesis, few potent inhibitors of the MEP pathway have been reported.2–6 Fosmidomycin potently inhibits the second enzyme in the pathway (IspC), and is the only MEP pathway inhibitor under clinical evaluation.6–8 The first, rate-limiting step in non-mammalian isoprenoid biosynthesis9,10 is the formation of 1-deoxy-D-xylulose 5-phosphate (DXP) catalyzed by DXP synthase. In addition to its potential regulatory role in isoprenoid biosynthesis, DXP synthase represents a branch point in pathogen metabolism, providing the precursor for vitamin B1 and B6 biosyntheses.11–13
Fig. 1.

The methylerythritol phosphate (MEP) pathway to isoprenoids.
Despite the importance of DXP synthase in pathogen metabolism, there are few reports describing inhibitors of this enzyme.2,3,6,14 DXP synthase catalyzes formation of DXP from pyruvate and D-glyceraldehyde 3-phosphate (D-GAP) in a thiamine diphosphate (ThDP)-dependent manner and shares weak sequence homology (20% identity) with other ThDP-dependent enzymes, including transketolase (TK) and pyruvate dehydrogenase E1 subunit (PDH), although cofactor binding sites are highly conserved.15 The similarities in cofactor binding sites suggest that achieving selectivity of inhibition against DXP synthase could be challenging. A previous study attempted to develop selective M. tuberculosis DXP synthase inhibitors using a target-based approach starting from a known, thiamin-based transketolase inhibitor, 3-(4-chloro-phenyl)-5-benzyl-4H-pyrazolo[1,5-a]pyrimidin-7-one.2 While the best enzyme inhibitor in this study showed in vivo activity against M. tuberculosis cultures (IC50 = 7.6 μM), these thiamine analogs also exhibited toxicity against mammalian cells, suggesting off-target activity against mammalian ThDP-dependent enzymes. This observation underscores the challenge in gaining selectivity of inhibition at the cofactor binding site which is highly conserved within the ThDP-dependent enzyme class.15
In principle, selective inhibition can be achieved by targeting the unique kinetic mechanism and/or conformational dynamics of DXP synthase. All other ThDP-dependent enzymes are known to follow classical ping-pong kinetics. However, Eubanks, et al.14 have provided compelling evidence for the requirement of a ternary complex in DXP synthase catalysis. In addition, we have shown that E. coli DXP synthase follows a random sequential kinetic mechanism where pyruvate and D-GAP bind independently and reversibly to DXP synthase, en route to the catalytically active ternary complex.16 We have also demonstrated flexibility in the active site of E. coli DXP synthase toward non-polar acceptor substrates, including aliphatic aldehdyes.17 Taken together, these results suggest it should be possible to selectively inhibit DXP synthase using analogs that incorporate elements of the donor substrate, pyruvate, and a non-polar acceptor substrate. Here we present the design and synthesis of alkylacetylphosphonate analogs and demonstrate selective inhibition against DXP synthase.
Methylacetylphosphonate (MAP) is a pyruvate analog that is incapable of undergoing activation by decarboxylation, and is a well-characterized inhibitor of ThDP-dependent enzymes that utilize pyruvate as substrate.18 Previously, we investigated the inhibitory activity of MAP against DXP synthase during studies to elucidate the random sequential mechanism of this enzyme.16 The observation that MAP potently inhibits DXP synthase prompted speculation about the potential utility of alkylacylphosphonates as bisubstrate analogs for selective inhibition of DXP synthase. Two compound series were envisioned that incorporate an acylphosphonate group as the pyruvate mimic (Fig. 2). Modification of either the acyl or alkyl groups of the phosphonate could mimic a non-polar acceptor substrate and test the importance of acylphosphonate orientation in bisubstrate analogs. Although DXP synthase exhibits relaxed substrate specificity for non-polar acceptor substrates, α-ketoacids modified at the acyl position are poor alternative donor substrates for this enzyme.19 On this basis, we hypothesized that phosphonates modified at the alkyl position (Fig. 2, series A) should have more potent inhibitory activity against DXP synthase compared to phosphonates modified at the acyl position (Fig. 2, series B).
Fig. 2.
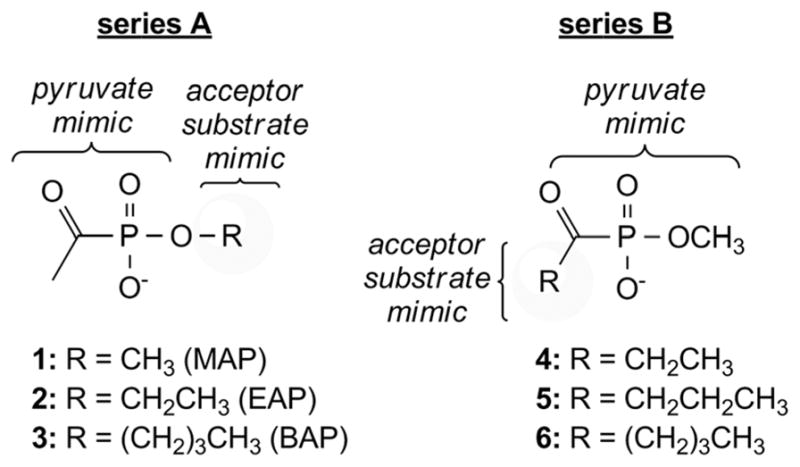
Design of acylphosphonate inhibitors of DXP synthase.
The general synthetic route used to access alkylacylphosphonates 2–6 employs the Michaelis–Arbuzov reaction between commercially available trialkyl phosphites and acyl chlorides to generate alkylacylphosphonate diesters in reasonable yields under mild conditions (Scheme 1).20 Subsequent cleavage of a single alkyl phosphonate ester is accomplished using stoichiometric LiBr to yield the corresponding lithium salt.20
Scheme 1.

Synthesis of alkylacylphosphonates 2–6.
Alkylacylphosphonates 1–6 were evaluated as inhibitors of E. coli DXP synthase using a spectrophotometric, coupled assay.6 As reported previously,16 MAP (1) is a potent competitive inhibitor against pyruvate (Ki MAP = 0.96 ± 0.3 μM, ESI Fig. S2†). As expected, increasing the length of the acyl group (compounds 4–6, Fig. 2) results in dramatic reduction of inhibitory activity against DXP synthase (Table 1), highlighting the importance of the small acetyl group in this pyruvate mimic. In contrast, compounds bearing a non-polar acceptor substrate mimic at the alkylphosphonate group (2 and 3, Fig. 2) are potent inhibitors of DXP synthase. Ethylacetylphosphonate (EAP, 2) and butylacetylphosphonate (BAP, 3) exhibit comparable inhibitory activity to MAP, with Ki values of 6.7 ± 0.03 μM (ESI Fig. S3A†) and 5.6 ± 0.8 μM (Fig. 3), respectively (Table 1). Similarly, both compounds show a competitive mode of inhibition with respect to pyruvate.
Table 1.
Inhibition of ThDP-dependent enzymes DXP synthase (E. coli), PDH (porcine) and TK (S. cerevisiae) by alkylacylphosphonates
|
E. coli DXP synthase (Kmpyruvate = 40.8 ± 4.6 μM)
| |
|---|---|
| Compound | Ki (μM) |
| 1 (MAP) | 0.96 ± 0.3 |
| 2 (EAP) | 6.7 ± 0.03 |
| 3 (BAP) | 5.6 ± 0.8 |
| 4 | 258.4 ± 67.8 |
| 5 | >1 mM |
| 6 | >1 mM |
|
| |
| Porcine pyruvate dehydrogenase E1 subunit (Kmpyruvate = 54.0 ± 5.3 μM) | |
|
| |
| Compound | Ki (μM) |
|
| |
| 1 (MAP) | 29.9 ± 12.6 |
| 2 (EAP) | 46.6 ± 1.9 |
| 3 (BAP) | 335.4 ± 7.9 |
| 4 | >1 mM |
| 5 | >1 mM |
| 6 | >1 mM |
|
| |
| S. cerevisiae transketolase | |
|
| |
| Compound | Ki |
|
| |
| 1 (MAP) | >1 mM |
| 2 (EAP) | >1 mM |
| 3 (BAP) | >1 mM |
| 4 | >1 mM |
| 5 | >1 mM |
| 6 | >1 mM |
Fig. 3. Competitive inhibition by butylacetylphosphonate (BAP).

A) DXP synthase. The concentration of pyruvate was varied with increasing concentrations of BAP: 0 (○), 10 (●), 25 (□), and 50 (■) μM BAP. B) E1 subunit of pyruvate dehydrogenase. The concentration of pyruvate was varied at with increasing concentrations of BAP: 0 (○), 0.2 (●), 0.5 (□), and 1.0 (■) mM BAP.
The goal of this work is to demonstrate selective inhibition of DXP synthase over other members of the ThDP-dependent class of enzymes. Compounds bearing an acylphosphonate mimic of pyruvate are not expected to exhibit inhibitory activity against transketolase as this enzyme does not recognize pyruvate as a substrate. As expected, none of the compounds reported here shows inhibitory activity against transketolase at concentrations up to 1 mM (Table 1). In contrast, pyruvate dehydrogenase E1 subunit (PDH) uses pyruvate as its natural substrate, and the pyruvate analogs MAP and the related phosphinates are known inhibitors of this enzyme.21,22 However, the kinetic mechanism of PDH is distinct from DXP synthase, and small hydrogen and methyl phosphinates show a marked increase in inhibitory activity against PDH compared to MAP.22 We hypothesized that EAP and BAP may exhibit high affinity for DXP synthase which is known to accommodate both substrates in its active site prior to decarboxylation of pyruvate; however, these sterically demanding analogs should be less potent inhibitors of PDH. Indeed, all three alkylacylphosphonates tested, MAP, EAP and BAP, are considerably less potent inhibitors of porcine PDH compared to DXP synthase, with Ki values against PDH of 29.9 ± 12.6 μM, 46.6 ± 1.9 μM and 335.4 ± 7.9 μM, respectively (Table 1). Even the smallest alkylacylphosphonate, MAP, exhibits selective inhibition toward DXP synthase (KiPDH/Ki DXP synthase = 31). Selectivity of inhibition increases with increasing chain length, as demonstrated by the greatly reduced inhibitory activity of BAP against PDH compared to DXP synthase (Ki PDH/Ki DXP synthase = 60). Compounds 4–6 bearing modified acyl groups are not inhibitors of porcine PDH at concentrations up to 1 mM (Table 1).
The present study shows that alkylacetylphosphonates selectively target DXP synthase. Presumably these analogs exhibit high affinity for a DXP synthase active site that uniquely accommodates both donor and acceptor substrates. Our results indicate that modification of the acetyl group abbrogates inhibitory activity of acylphosphonates, consistent with the observation that acyl-modified α-ketoacids are not substrates for DXP synthase. However, lengthening the carbon chain of the alkyl phosphonate produces inhibitors with comparable activity to MAP. These analogs are considerably less potent inhibitors of porcine PDH and do not exhibit measurable inhibitory activity against transketolase. Taken together, these results suggest a unique inhibition mechanism in ThDP-dependent enzymology, and suggest an exciting new direction for the development of inhibitors targeting early-stage isoprenoid biosynthesis.
Supplementary Material
Acknowledgments
This work was supported by funding from the National Institutes of Health (GM084998 for C.F.M.; T32GM007445-34 for J.M.S. T32GM08018901 for R.J.V.).
Footnotes
Electronic supplementary information (ESI) available: Synthesis and characterization of compounds 2–6. Enzyme inhibition assays. See DOI: 10.1039/c1md00233c
References
- 1.Rodriguez-Concepcion M, Boronat A. Plant Physiol. 2002;130:1079–1089. doi: 10.1104/pp.007138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mao J, Eoh H, He R, Wang Y, Wan B, Franzblau SG, Crick DC, Kozikowski AP. Bioorg Med Chem Lett. 2008;18:5320–5323. doi: 10.1016/j.bmcl.2008.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matsue Y, Hiroko M, Tomita T, Tadao A, Makoto N, Kuzuyama T. J Antibiot. 2010:1–6. [Google Scholar]
- 4.Wang W, Li J, Wang K, Huang C, Zhang Y, Oldfield E. Proc Natl Acad Sci U S A. 2010;107:11189–11193. doi: 10.1073/pnas.1000264107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang W, Li J, Wang K, Smirnova TI, Oldfield E. J Am Chem Soc. 2011;133:6525. doi: 10.1021/ja2008455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jomaa H, Wiesner J, Sanderbrand S, Altincicek B, Weidemeyer C, Hintz M, Türbachova I, Eberl M, Zeidler J, Lichtenthaler HK, Soldati D, Beck E. Science. 1999;285:1573–1576. doi: 10.1126/science.285.5433.1573. [DOI] [PubMed] [Google Scholar]
- 7.Missinou MA, Borrmann S, Schindler A, Issifou S, Adegnika AA, Matsiegui PB, Binder R, Lell B, Wiesner J, Baranek T, Jomaa H, Kremsner PG. Lancet. 2002;360:1941. doi: 10.1016/S0140-6736(02)11860-5. [DOI] [PubMed] [Google Scholar]
- 8.Wiesner J, Henschker D, Hutchinson D, Beck E, Jomaa H. Antimicrob Agents Chemother. 2002;46:2889. doi: 10.1128/AAC.46.9.2889-2894.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuzuyama T, Takagi M, Takahashi S, Seto H. J Bacteriol. 2000;15:891. doi: 10.1128/jb.182.4.891-897.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Estévez JM, Cantero A, Reindl A, Reichler S, León P. J Biol Chem. 2001;276:22901. doi: 10.1074/jbc.M100854200. [DOI] [PubMed] [Google Scholar]
- 11.Sprenger GA, Schörken U, Wiegert T, Grolle S, de Graaf AA, Taylor SV, Begley TP, Bringer-Meyer S, Sahm H. Proc Natl Acad Sci U S A. 1997;94:12857. doi: 10.1073/pnas.94.24.12857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lois LM, Campos N, Putra SR, Danielsen K, Rohmer M, Boronat A. Proc Natl Acad Sci U S A. 1998;95:2105. doi: 10.1073/pnas.95.5.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hill RE, Himmeldirk K, Kennedy IA, Pauloski RM, Sayer BG, Wolf E, Spenser ID. J Biol Chem. 1996;271:30426. doi: 10.1074/jbc.271.48.30426. [DOI] [PubMed] [Google Scholar]
- 14.Eubanks LM, Poulter CD. Biochemistry. :1140. doi: 10.1021/bi0205303. [DOI] [PubMed] [Google Scholar]
- 15.Xiang S, Usunow G, Lange G, Busch M, Tong L. J Biol Chem. 2007;282:2676–2682. doi: 10.1074/jbc.M610235200. [DOI] [PubMed] [Google Scholar]
- 16.Brammer LA, Smith JM, Wade H, Freel Meyers C. J Biol Chem. 2011 doi: 10.1074/jbc.M111.25974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brammer LA, Freel Meyers C. Org Lett. 2009:4748. doi: 10.1021/ol901961q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arjunan P, Sax M, Brunskill A, Chandrasekhar K, Nemeria N, Zhang S, Jordan F, Furey W. J Biol Chem. 2006;281:15296. doi: 10.1074/jbc.M600656200. [DOI] [PubMed] [Google Scholar]
- 19.Schürmann M, Schürmann M, Sprenger GA. J Mol Catal B: Enzym. 2002;19–20:247–252. [Google Scholar]
- 20.Fang M, Toogood RD, Macova A, Ho K, Franzblau SG, McNeil MR, Sanders DAR, Palmer DRJ. Biochemistry. 2010:2672. doi: 10.1021/bi901432d. [DOI] [PubMed] [Google Scholar]
- 21.Arjunan P, Sax M, Brunskill A, Chandrasekhar K, Nemeria N, Zhang S, Jordan F, Furey W. J Biol Chem. 2006;281:15296. doi: 10.1074/jbc.M600656200. [DOI] [PubMed] [Google Scholar]
- 22.Nemeria NS, Korotchkina LG, Chakraborty S, Patel MS, Jordan F. Bioorg Chem. 2006;34:362–379. doi: 10.1016/j.bioorg.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.