

Abstract
Background:
Nitric oxide (NO) is an important molecule in maintaining endothelial survival and normal function. It is a unique mediator, which may promote or suppress both inflammation and apoptosis. Endothelial cell (EC) injury, dysfunction, and death in response to cytokines, especially interferon gamma (IFN-γ), represent the critical event for the initiation of several inflammatory diseases.
Objective(s):
EC injury or death result in endothelial dysfunction that precedes the development of atherosclerosis and its subsequent vascular events. We examine the effect of different concentrations of IFN-γ on human umbilical vein ECs (HUVECs) NO production and apoptosis.
Materials and Methods:
HUVECs were cultured at 37°C for 24 h in the absence (control) or presence of 10, 100, and 1000 μg IFN-γ, respectively. The apoptotic cells were determined as annexin V-positive propidium iodide (PI)-negative cells by flow cytometry. Total nitrite concentration was measured in cell cultures supernatant by Griess method.
Results:
A comparison of the effect of IFN-γ on EC NO production with untreated cells showed that pretreatment of HUVEC with IFN-γ failed to have a significant effect on NO production by these cells at 10 and 100 U/mL, whereas it led to a significant decreased NO production at 1000 U/mL (P < 0.05). The cells stimulated with IFN-γ showed significantly higher apoptotic cells (PI negative and annexin V-positive cells) after 24 h, compared with cells with no stimulations (P < 0.05).
Conclusion:
IFN-γ has detrimental effects on ECs in high doses. This might be due to inducible NO synthase activation.
Keywords: Apoptosis, endothelial cells, IFN-γ, nitric oxide
INTRODUCTION
The endothelial cells (ECs) have a key role in the regulation of local immune and inflammatory reactions, and processes of vascular injury.[1,2] Vascular ECs can interact with complement, chemokines, or humoral components;[1,2] express receptors for blood leukocytes or modify immune reactions;[2] generate and respond to cytokines;[3] and modulate local vascular reactions at inflammatory sites.[2,3] Endothelial activation, as an early step in vascular dysfunction, can be induced by several cytokines.[3] Because of the EC strategic location of the lining of the vasculature, they are major targets of immune-mediated injury.
EC injury or death results in endothelial dysfunction that is associated with some situations, such as chronic inflammatory diseases. Endothelial dysfunction precedes the development of atherosclerosis and its subsequent vascular events as well as several systemic autoimmune diseases, such as vacuities, inflammatory bowel disease, and multiple sclerosis.[3]
Atherosclerosis is a chronic inflammatory state characterized by lesions that contain abundant immune cells, particularly monocytes and T lymphocytes.[4] The interaction between macrophages and lymphocytes within the atherosclerotic lesion microenvironment exemplifies a site where both innate and adaptive immunity contribute toward disease progression.[4] Interferon gamma (IFN- γ) that is the classic macrophage-activating factor, has been localized at atherosclerotic lesions. IFN- γ acting on all the major cell types of the plaque, has a complex effect on atherosclerosis progression.[4] The introduction of leukocytes into the blood vessel intima at the site of plaque formation requires cell adhesion molecules displayed on the luminal surface of the endothelium.[5] These events make endothelium as a major player in atherosclerosis process.[6]
It has been shown that IFN- γ increases T helper-1 cells (Th1) immune responses by upregulation of class I and II antigens in endothelial, vascular smooth muscle cells and monocyte–macrophages as well as foam cells and increase monocyte and lymphocyte infiltration into the vascular lesion by increases in chemokines and surface adhesion molecules.[2,7] However, the direct effect of IFN- γ on ECs (not through leukocyte-mediated mechanisms) has been overlooked.
Healthy ECs respond to a number of stimuli by releasing nitric oxide (NO). NO plays a strategic role in the protection exerted by the endothelium against risk factors and stressors.[7] NO not only prevents abnormal constriction (vasospasm) of the vessels, but also inhibits the aggregation of platelets, the expression of adhesion molecules at the surface of the ECs, and hence the adhesion and penetration of leukocytes. When this protective role of NO is flawed, the inflammatory response that leads to atherosclerosis is initiated.[8]
IFN-γ, being the hallmark of the Th1 response, has been extensively studied with respect to its expression and regulation of immune function.[9] Although IFN- γ increases Th1 immune responses,[10,11] the direct effect of IFN- γ on ECs (not through leukocyte-mediated mechanisms) has been less mentioned.
In this study, we aimed to examine the effect of different concentrations of IFN- γ on human umbilical vein ECs (HUVECs) NO production and apoptosis.
MATERIALS AND METHODS
Cell culture and treatment
The HUVECs (National Cell bank of Iran affiliated to Pasteur Institute, Tehran, Iran) were cultured in endothelial basal medium supplemented with EC growth factor, gentamicin, amphotericin B, and 10% fetal calf serum until the third passage before experiments were performed. All the cell culture material were from Gibco, Oklahoma USA. Before stimulation HUVECs were plated in 6-well plates and divided into 4 groups. HUVECs in the control group and IFNγ groups were further cultured at 37°C for 24 h, in the absence (control) or presence of 10, 100, and 1000 μg IFN-γ. The IFN-γ was kindly gifted by Exir Pharmaceutical Company (Tehran, Iran). All the experiments were performed at least in triplicate and repeated at least twice.
Apoptosis detection
In the early stages of apoptosis one of the important plasma membrane modifications is the translocation of phosphatidylserine (PS) from the inner side of the plasma membrane to the outer layer, by which PS becomes exposed at the external surface of the cell. Annexin V is a Ca2+-dependent phospholipid-binding protein with high affinity for PS. Hence this protein can be used as a sensitive probe for PS exposure upon the cell membrane. Translocation of PS to the external cell surface is not unique to apoptosis, but occurs also during cell necrosis. The difference between these 2 forms of cell death is that during the initial stages of apoptosis the cell membrane remains intact, while at the very moment that necrosis occurs the cell membrane loses its integrity and becomes leaky. Therefore, the measurement of annexin V binding to the cell surface as indicative for apoptosis has to be performed in conjunction with a dye exclusion test to establish integrity of the cell membrane, such as propidium iodide (PI).
For each staining, a total number of 105 cells per sample were washed once with ice-cold PBS and the cells were stained by annexin–PI (R&D Systems, Minneapolis, USA) as follows: Cells (105/mL) were incubated with 1 μL annexin V–fluorescein isothiocyanate and 0.5 μL PI (10 mg/mL) in the binding buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2).
Subsequently, the cells were analyzed by fluorescence-activated cell sorting (FACScan, Becton–Dickinson, Miami, USA). Apoptotic cells were distinguished as annexin V–PI– cells. Data analysis was performed with Cell Quest software.
NO metabolite (nitrite) measurement
The levels of NO in the culture supernatants were determined using the Griess reaction by parameter TM, total NO Assay kit catalog number KGE001 (R&D Systems Company, USA). This assay determines nitrite concentrations as NO metabolite. Nitrite is detected calorimetrically as an Azo dye product of the Griess reaction. The Griess reaction is based on the 2-step diazotization reaction in which acidified NO2 produces a nitrosating agent, which reacts with sulfanilic acid to produce the diazonium ion. This ion is then coupled to N-(1-naphthyl) Ethylenediamine to form the chromophoric azo-derivative, which absorbs light at 540–570 nm. The results were calculated using the standard curve equation.
Statistical analysis
Data were tested for normal distribution by the Kolmogorov–Smirnov test. Significance of variation between groups was evaluated using Kruskal–Wallis test followed by Mann–Whitney test. The correlation relationships will be evaluated by Spearman test. P < 0.05 will be considered significant.
RESULTS
As it has been illustrated in Figure 1, comparison of the effect of 3 different doses of IFN-γ (10, 100, and 1000 U/mL) on EC NO production with untreated cells showed that pretreatment of HUVECs with IFN-γ failed to have a significant effect on NO production by these cells at 10 and 100 U/mL, whereas it led to significant decreased NO production at 1000 U/mL (P < 0.05).
Figure 1.
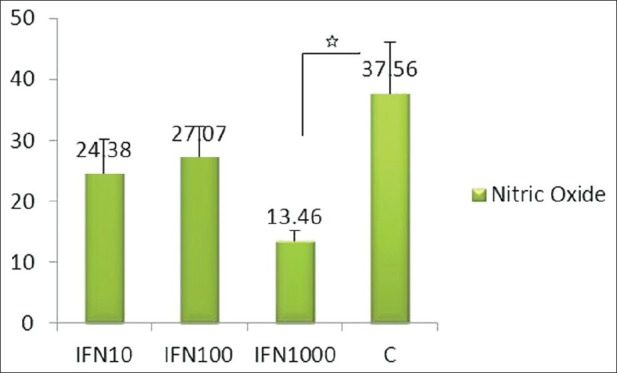
The effect of interferon γ (10, 100, and 1000 U) on endothelial cell nitric oxide production. Experiments were performed in triplicate and repeated at least twice
As shown in Figure 2, cells stimulated with IFN-γ showed significantly higher apoptotic cells (annexin V–FITC-positive cells and PI negative) after 24 h, compared with cells with no stimulations (P < 0.05).
Figure 2.
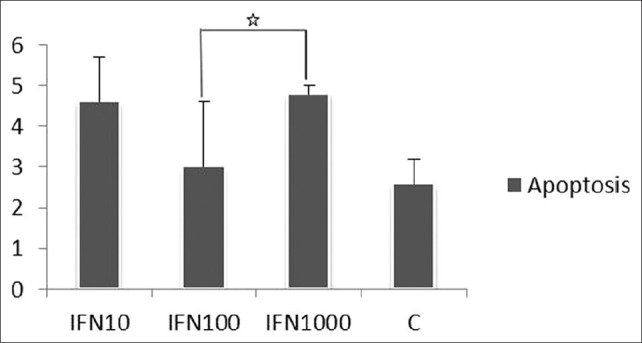
The effect of interferon γ (10, 100, and 1000 U) on endothelial cell apoptosis. Experiments were performed in triplicate and repeated at least twice
Results were as follows: Cells without stimulations, 2.58 ± 0.6%; cells stimulated with IFN-γ (10 U/mL): 4.61 ± 1.08, IFN-γ (100 U/mL): 3 ± 1.6 and IFN-γ (1000 U/mL): 4.8 ± 0.19.
DISCUSSION
ECs are routinely exposed to circulating immune cells and effector molecules of immunity and consequently are main targets of their adverse effects. Injury to vascular ECs is a critical event in tissue damage seen in acute and chronic inflammation. However, the exact mechanism has not yet been precisely elucidated.
The results of our study showed that IFN-γ induced both apoptosis and reduced NO production in 1000 U/mL.
In addition to the key role of the IFN-γ in host defense, its excessive release has been associated with the pathogenesis of chronic inflammatory and autoimmune diseases.[12] In fact, knockout models reveal that IFN-γ plays a key role in mediating a number of pathologic processes related to chronic immune activation.[12–15]
However, IFN-γ has various effects on vascular cells through orchestration of a spectrum of hundreds of genes and different cellular programs.[16] Microarray analysis of human cell lines and primary vascular cells reveal a wide range of transcripts that are regulated by IFN-γ, including some with known immune modulatory functions and a variety of genes with unknown functional significance.[17,18]
Especially in some of the human diseases with prominent endothelial injury, such as atherosclerosis, T lymphocytes are the main components of all stages of the disease.[4,10] This cell type has also been identified in atherosclerotic lesions from rabbits[19] and mice.[20,21] Both protein and mRNA for this cytokine have been detected in atherosclerotic lesions from humans[23,24] and mice.[25] IFN-γ has a range of biological properties in cultured cells that could influence the development of atherosclerotic lesions. It has been shown that cultured ECs express low levels of Fas, but IFN-γ increases Fas and procaspase-8 expression, sensitizing ECs to Fas-mediated apoptosis.[25] In addition, IFN-γ stimulation of ECs isolated from aortas of male Wistar rats was associated with increased expression of cathepsin B, caspase-3, and FasL and triggers the apoptotic process.[9] Furthermore, it has been demonstrated that IFN-γ inhibits EC proliferation by a mechanism, which involves growth factors.[9]
A product of ECs that is a potent anti-inflammatory agent is NO, and therefore induction or suppression of NO by cytokines has the potential to enhance or inhibit the inflammatory response.[26]
Strong evidence suggests that endothelial NO synthase (eNOS) plays an important role in the protection of the ECs. NO has been shown to inhibit apoptosis both in vitro and in vivo in certain cell types.[26,27] NO inhibits apoptosis induced by many different stimuli, including growth factor withdrawal, tumor necrosis factor (TNF), or Fas.[28] It has become evident that decreased bioavailability of endothelial NO produced from eNOS, referred to as ED, plays a crucial role in the development and progression of several vascular diseases, such as atherosclerosis.[29] In our study, the decreased levels of NO production in 1000 U/mL IFN-γ may be attributed to decreased eNOS as a protective molecule, which subsequently leads to ECs apoptosis. Conversely, the decreased NO may be the result of the increased apoptosis, so, decreased NO may be both the cause and effect of EC apoptosis.
It has been reported that IFNγ, in cooperation with lipopolysaccharide (LPS) or other cytokines, such as TNF-α, interleukin (IL) IL-2,and IL-4 results in upregulation of inducible NO synthase (iNOS) gene expression in several cell types, including ECs and macrophages.[30]
Accordingly, blockade of the proinflammatory cytokine IFN in human allografts is sufficient to prevent EC dysfunction and loss of endothelial NO expression.[30] Thus, cytokine stimulation of ECs can both positively and negatively modulate the expression of endothelial gene products that control vascular tone and the ability of the vessel to respond to vasodilatory signals.
In our study, IFN-γ has no significant effect on NO production in doses of 10 and 100 U/mL. In a similar report to our study, Weiss et al. have shown that treatment of HUVEC with IFN-γ (200 U) for 24 h had no effect on NOS activity in cell lysates, whereas Lamas et al. have reported that IFN-γ (1000 U) failed to exert a significant effect on basal release of NO or metabolic conversion of L-[“C]arginine to L-[‘4C citrulline in bovine aortic ECs.[31]
In another study, Morikawa et al. have reported that IFN-γ exhibited contrary effects on NO production, depending on its concentration; 1 ng/mL of IFN-γ significantly reduced NO production of END-D cells, whereas IFN-γ at the intermediate concentration (10 ng/mL) did not significantly alter NO production. They also showed that a low concentration of IFN-γ, LPS, and TNF-a reduced NO production through downregulating eNOS.[32,33]
Moreover, Yamaoka et al. have shown that cytotoxicity of IFN-γ on vascular ECs is mediated by NO.[34] It has been reported that ONO-1714, a potent iNOS-specific inhibitor, completely blocked both cytokine-induced cytotoxicity and NO production in murine vascular ECs F-2, whereas NO scavengers, such as carboxy-PTIO and hemoglobin, blocked cytotoxicity. In the same experiment, exogenous NO from NO donor caused cytotoxicity as well.[33]
CONCLUSION
In conclusion, it seems that despite the consistent regulation of inflammatory factors with a relatively predictable cellular phenotype, the effects of IFN-γ on EC survival are variable and condition dependent. IFN-γ has detrimental effects on endothelium only in very high concentrations. In addition, it is possible that the unfavorable effects may be enhanced in the presence of the other inflammatory factors, such as TNFα. The current study suggested the presence of a complicated regulation of NOS expression in vascular ECs by proinflammatory cytokines, such as IFN-γ. However, it was unclear whether the in vitro findings in the present study could be applied to the in vivo phenomenon or not. The exact role of NO produced in vivo by different isoenzymes of NOS in vascular ECs stimulated with IFN-γ is still a matter for speculation.
ACKNOWLEDGMENTS
This study was supported by Isfahan University of Medical sciences (Grant No. 287240). Special thanks to Maede Samadi, Vida Homayouni, and Tayyebeh Karimi for their valuable help.
Footnotes
Source of Support: Isfahan University of Medical sciences (Grant No. 287240).
Conflict of Interest: None declared
REFERENCES
- 1.Cotran RS, Pober JS. Endothelial activation and inflammation. ProgImmunol. 1989;8:747. [Google Scholar]
- 2.Pober JS, Cotran RS. The role of endothelial cells in inflammation. Transplantation. 1990;50:537–44. doi: 10.1097/00007890-199010000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Pober JS, Min W, Bradley JR. Mechanisms of endothelial dysfunction, injury, and death. Annu Rev Pathol. 2009;4:71–95. doi: 10.1146/annurev.pathol.4.110807.092155. [DOI] [PubMed] [Google Scholar]
- 4.Hansson GK, Libby P, Schönbeck U, Yan ZQ. Innate and Adaptive Immunity in the Pathogenesis of Atherosclerosis. Circ Res. 2002;91:281–91. doi: 10.1161/01.res.0000029784.15893.10. [DOI] [PubMed] [Google Scholar]
- 5.Steinberg D. Atherogenesis in perspective: Hypercholesterolemia and inflammation as partners in crime. Nat Med. 2002;8:1211–7. doi: 10.1038/nm1102-1211. [DOI] [PubMed] [Google Scholar]
- 6.Aird WC. Endothelial cell heterogeneity and Atherosclerosis. Curr Atheroscler Rep. 2006;8:69–75. doi: 10.1007/s11883-006-0067-z. [DOI] [PubMed] [Google Scholar]
- 7.Flammer AJ, Lüscher TF. Human endothelial dysfunction: EDRFs. Pflugers Arch. 2010;459:1005–13. doi: 10.1007/s00424-010-0822-4. [DOI] [PubMed] [Google Scholar]
- 8.Cooke JP. The pivotal role of nitric oxide for vascular health. Can J Cardiol. 2004;20(Suppl B):7B–15B. [PubMed] [Google Scholar]
- 9.Tellides G, Pober JS. Interferon-gamma axis in graft arteriosclerosis. Circ Res. 2007;100:622–32. doi: 10.1161/01.RES.0000258861.72279.29. [DOI] [PubMed] [Google Scholar]
- 10.Hansson GK, Jonasson L, Holm J, Clowes MM, Clowes AW. Gamma-interferon regulates vascular smooth muscle proliferation and Ia antigen expression in vivo and in vitro. Circ Res. 1988;63:712–9. doi: 10.1161/01.res.63.4.712. [DOI] [PubMed] [Google Scholar]
- 11.Mach B, Steimle V, Martinez-Soria E, Reith W. Regulation of MHC class II genes: Lessons from a disease. Annu Rev Immunol. 1996;14:301–31. doi: 10.1146/annurev.immunol.14.1.301. [DOI] [PubMed] [Google Scholar]
- 12.Miller CH, Maher SG, Young HA. Clinical Use of Interferon-gamma. Ann N Y Acad Sci. 2009;1182:69–79. doi: 10.1111/j.1749-6632.2009.05069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Billiau A, Matthys P. Interferon-gamma: A historical perspective. Cytokine Growth Factor Rev. 2009;20:97–113. doi: 10.1016/j.cytogfr.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 14.McLaren JE, Ramji DP. Interferon gamma: A master regulator of atherosclerosis. Cytokine Growth Factor Rev. 2009;20:125–35. doi: 10.1016/j.cytogfr.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 15.Schoenborn JR, Wilson CB. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol. 2007;96:41–101. doi: 10.1016/S0065-2776(07)96002-2. [DOI] [PubMed] [Google Scholar]
- 16.Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annu Rev Immunol. 1997;15:749–95. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- 17.Zohlnhofer D, Richte T, Neumann F, Nuhrenberg T, Wessely R, Brandl R, et al. Transcriptome analysis reveals a role of interferon-gamma in human neointima formation. Mol Cell. 2001;7:1059–69. doi: 10.1016/s1097-2765(01)00239-8. [DOI] [PubMed] [Google Scholar]
- 18.Der SD, Zhou A, Williams BR, Silverman RH. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci U S A. 1998;95:15623–8. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roselaar SE, Schonfeld G, Daugherty A. Enhanced development of atherosclerosis in cholesterol-fed rabbits by suppression of cell-mediated immunity. J Clin Invest. 1995;96:1389–94. doi: 10.1172/JCI118174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roselaar SE, Kakkanathu PX, Daugherty A. Lymphocyte populations in atherosclerotic lesions of apoE-/- and LDL receptor-/- mice: Decreasing density with disease progression. Arterioscler Thromb Vasc Biol. 1996;16:1013–8. doi: 10.1161/01.atv.16.8.1013. [DOI] [PubMed] [Google Scholar]
- 21.Zhou XH, Stemme S, Hansson GK. Evidence for a local immune response in atherosclerosis: CD4(+) T cells infiltrate lesions of apolipoprotein-E-deficient mice. Am J Pathol. 1996;149:359–66. [PMC free article] [PubMed] [Google Scholar]
- 22.Emeson EE, Shen ML, Bell CG, Qureshi A. Inhibition of atherosclerosis in CD4 T-cell-ablated and nude (nu/nu) C57BL/6 hyperlipidemic mice. Am J Pathol. 1996;149:675–85. [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou XH, Paulsson G, Stemme S, Hansson GK. Hypercholesterolemia is associated with a T helper (Th) 1/Th2 switch of the autoimmune response in atherosclerotic apo E-knockout mice. J Clin Invest. 1998;101:1717–25. doi: 10.1172/JCI1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fong LG, Fong TA, Cooper AD. Inhibition of mouse macrophage degradation of acetyl low density lipoprotein by interferon-γ. J Biol Chem. 1990;265:11751–60. [PubMed] [Google Scholar]
- 25.Li JH, Kluger MS, Madge LA, Zheng L, Bothwell AL, Pober JS. Interferon-γ augments CD95(APO-1/Fas) and procaspase-8 expression and sensitizes human vascular endothelial cells to CD95-mediated apoptosis. Am J Pathol. 2002;161:1485–95. doi: 10.1016/s0002-9440(10)64424-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laroux FS, Pavlick KP, Hines IN, Kawachi S, Harada H, Bharwani S, et al. Role of nitric oxide in inflammation. Acta Physiol Scand. 2001;173:113–8. doi: 10.1046/j.1365-201X.2001.00891.x. [DOI] [PubMed] [Google Scholar]
- 27.Kim YM, Talanian RV, Billiar TR. Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J Biol Chem. 1997;272:31138–48. doi: 10.1074/jbc.272.49.31138. [DOI] [PubMed] [Google Scholar]
- 28.Javanmard SH, Nematbakhsh M, Mahmoodi F, Mohajeri MR. l-Arginine supplementation enhances eNOS expression in experimental model of hypercholesterolemic rabbits aorta. Pathophysiology. 2009;16:9–13. doi: 10.1016/j.pathophys.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 29.Marumo T, Nakaki T, Adachi H, Esumi H, Suzuki H, Saruta T, et al. Nitric oxide synthase mRNA in endothelial cells: Synergistic induction by interferon-gamma, tumor necrosis factor-alpha and lipopolysaccharide and inhibition by dexamethasone. Jpn J Pharmacol. 1993;63:327–34. doi: 10.1254/jjp.63.327. [DOI] [PubMed] [Google Scholar]
- 30.Cornicelli JA, Butteiger D, Rateri DL, Welch K, Daugherty A. Interleukin-4 augments acetylated LDL induced cholesterol esterification in macrophages. J Lipid Res. 2000;41:376–83. [PubMed] [Google Scholar]
- 31.Lamas S, Michel T, Collins T, Brenner BM, Marsden PA. Effects of interferon-gamma on nitric oxide synthase activity and endothelin-1 production by vascular endothelial cells. J Clin Invest. 1992;90:879–87. doi: 10.1172/JCI115963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morikawa A, Koide N, Kato Y, Sugiyama T, Chakravortty D, Yoshida T, et al. Augmentation of nitric oxide production by gamma interferon in a mouse vascular endothelial cell line and its modulation by tumor necrosis factor alpha and lipopolysaccharide. Infect Immun. 2000;68:6209–14. doi: 10.1128/iai.68.11.6209-6214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kubes P, Suzuki M, Granger DN. Nitric oxide: An endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci USA. 1991;88:4651–5. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamaoka J, Kabashima K, Kawanishi M, Toda K, Miyachi Y. Cytotoxicity of IFN-gamma and TNF-alpha for vascular endothelial cell is mediated by nitric oxide. Biochem Biophys Res Commun. 2002;291:780–6. doi: 10.1006/bbrc.2002.6487. [DOI] [PubMed] [Google Scholar]