

Abstract
2-Amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) is a heterocyclic aromatic amine that is formed during the cooking of meats. PhIP is a potential human carcinogen: it undergoes metabolic activation to form electrophilic metabolites that bind to DNA and proteins, including serum albumin (SA). The structures of PhIP-SA adducts formed in vivo are unknown and require elucidation before PhIP protein adducts can be implemented as biomarkers in human studies. We previously examined the reaction of genotoxic N-oxidized metabolites of PhIP with human SA in vitro and identified covalent adducts formed at cysteine34 (Cys34); however, other adduction products were thought to occur. We have now identified adducts of PhIP formed at multiple sites of SA reacted with isotopic mixtures of electrophilic metabolites of PhIP and 2-amino-1-methyl-6-[2H5]-phenylimidazo[4,5-b]pyridine ([2H5]-PhIP). The metabolites used for study were: 2-nitro-1-methyl-6-phenylimidazo[4,5-b]pyridine (NO2-PhIP), 2-hydroxyamino-1-methyl-6-phenylimidazo[4,5-b]pyridine (HONH-PhIP), or N-acetyloxy-2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (N-acetoxy-PhIP). Following proteolytic digestion, PhIP-adducted peptides were separated by ultra performance liquid chromatography and characterized by ion trap mass spectrometry, employing isotopic data-dependent scanning. Analysis of the tryptic or tryptic/chymotryptic digests of SA modified with NO2-PhIP revealed that adduction occurred at Cys34, Lys195, Lys199, Lys351, Lys541, Tyr138, Tyr150, Tyr401, and Tyr411, whereas the only site of HONH-PhIP adduction was detected at Cys34. N-Acetoxy-PhIP, a penultimate metabolite of PhIP that reacts with DNA to form covalent adducts, did not appear to form stable adducts with SA; instead, PhIP and 2-amino-1-methyl-6-(5-hydroxy)-phenylimidazo[4,5-b]pyridine, an aqueous reaction product of the proposed nitrenium ion of PhIP, were recovered during the proteolysis of N-acetoxy-PhIP-modified SA. Some of these SA adduction products of PhIP may be implemented in molecular epidemiology studies to assess the role of well-done cooked meat, PhIP, and the risk of cancer.
Introduction
2-Amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) is a carcinogenic heterocyclic aromatic amine (HAA) that is formed, by reaction of creatinine with phenylalanine, during the cooking of meats, poultry, and fish.1 The concentrations of PhIP can reach up to 480 parts per billion in well-done cooked poultry.2 PhIP comprises about 70% of the daily mean intake of HAAs in the United States.3 Many epidemiological studies have reported a positive association between frequent consumption of well-done cooked meats containing PhIP and an increased risk of stomach, colon, pancreas, prostate, and breast cancers, although some studies have failed to find associations between well-done meat and cancer risk.4–6 A major limiting factor in most epidemiological studies is the uncertainty in quantitative estimates of chronic exposure to PhIP or other HAAs, and thus, the association of HAAs formed in cooked meat and cancer risk has been difficult to establish. In the 11th Report on Carcinogens, PhIP and several other HAAs were classified as ‘reasonably anticipated to be human carcinogens’.7 However, there is a critical need to establish long-term biomarkers of HAAs that can be implemented in molecular epidemiology studies to firmly evaluate the health risk of these genotoxicants.
Long-term biomarkers of carcinogens, such as DNA or protein adducts, are a measure of the biologically effective dose and have been used for human risk assessment of environmental and dietary genotoxicants.8,9 Electrophilic genotoxic metabolites of PhIP react with DNA and proteins.10 PhIP undergoes N-oxidation by cytochrome P450 enzymes to form 2-hydroxyamino-1-methyl-6-phenylimidazo[4,5-b]pyridine (HNOH-PhIP) which can directly react with DNA, or HONH-PhIP can undergo conjugation reactions with phase II enzymes to form highly reactive esters that covalently bind to DNA.10 PhIP-DNA adducts have been detected in human tissues and fluids, by immunohistochemistry methods,11,12 accelerator mass spectrometry,13 or by liquid chromatography/mass spectrometry.14 However, DNA adduct measurements are often precluded by the unavailability of biopsy samples in large scale human studies. Moreover, DNA adducts are generally repaired, and the adduct levels can be below the limit of detection, even when measured by sensitive tandem mass spectrometry techniques.
Hemoglobin (Hb) and serum albumin (SA) carcinogen adducts have been used as an alternative to DNA adducts for biomarkers of several different classes of carcinogens.9,15 Stable carcinogen protein adducts are expected to accumulate and follow the kinetics of the lifetime of Hb or half-life of SA, during chronic exposure, and augment the sensitivity of adduct detection.16 The N-hydroxylated metabolites of many aromatic amines, carcinogens that are structurally related to HAAs, bind to human hemoglobin (Hb) at appreciable levels.17 The arylhydroxylamines penetrate the erythrocyte and undergo a co-oxidation reaction with oxy-hemoglobin (oxy-Hb), to form the arylnitroso intermediates and methemoglobin (met-Hb).18 The arylnitroso compounds can react with the Cys93 residue of the human β-Hb chain to form a sulfinamide adduct.9,19 However, the covalent binding of HONH-PhIP and other N-hydroxylated-HAAs to Hb in rodents and humans is very low, and the HAA-Hb sulfinamide adduct does not appear to be a promising biomarker to assess human exposure.10
Many genotoxicants and toxic electrophiles also bind covalently to human SA.9,15,16,20 Among the 585 amino acids of the mature SA sequence, the thiol group of Cys34, the imidazole nitrogen atoms of histidine, the amino and guanidine groups of the side chain of lysine and arginine, the carboxyl groups of the side chain of aspartic and glutamic acid, and the phenolic group of tyrosine are most frequently involved in the formation of SA adducts.20 PhIP was reported to bind to human SA in vivo at levels that may be sufficient to establish mass spectrometry techniques for biomonitoring.13 At least one of the PhIP-SA adducts undergoes hydrolysis under acidic pH conditions to regenerate PhIP.21 However, the reactive species responsible for adduct formation and the structures of the intact PhIP-SA adduct(s) are unknown. N-Oxidation products of PhIP covalently bind to rat or human SA in vitro.22–24 2-Nitroso-1-methyl-6-phenylimidazo[4,5-b]pyridine (NO-PhIP) reacted with SA in vitro to produce the N2-[cystein-S-yl-PhIP]-S-oxide at Cys34.24 This sulfinamide adduct is labile towards acid and some portion of the acid-labile adduct(s) of PhIP formed with SA in vivo may exist as the sulfinamide. The Cys34 of SA also reacted with 2-nitro-1-methyl-6-phenylimidazo[4,5-b]pyridine (NO2-PhIP) in vitro to form a stable sulfur-carbon linked adduct with PhIP.24 However, other PhIP-SA adduction products are likely to have been formed and remain to be characterized.
Isotope pattern-dependent mass spectrometric scanning methods have been employed to analyze the formation of reactive metabolites from a number of toxicants.25 Isotopic data-dependent scanning has also been used to identify proteins in liver microsomal preparations chemically modified with unlabeled and 14C-radiolabeled furan containing drugs.26 The goal of our current study was to investigate the reactivity of human SA with electrophilic N-oxidation products of PhIP in vitro and to characterize the adduction products by mass spectrometric techniques. We have exploited the isotopic data-dependent scanning technique to map the sites of PhIP binding to human SA modified with a mixture of unlabeled and [2H5]-labeled N-oxidized metabolites of PhIP.
Materials and Methods
Caution: PhIP is a carcinogen and should be handled in a well-ventilated fume hood with the appropriate protective clothing.
Chemicals and Materials
PhIP was purchased from Toronto Research Chemicals (Toronto, ON, Canada). 2-Amino-1-methyl-6-[2H5]-phenylimidazo[4,5-b]pyridine ([2H5]-PhIP, 99% isotopic purity) was a gift from Dr. Mark Knize and Dr. Kristen Kulp, (Lawrence Livermore National Laboratory, Livermore, CA). Human SA, cysteine, tyrosine, lysine, tryptophan, β-mercaptoethanol, iodoacetamide, dithiothreitol, N-ethylmaleimide (NEM) and 4-chloromercuribenzoic acid (4-CMB) were obtained from Sigma (St. Louis, MO). The human plasma was purchased from Bioreclamation LLC (Hicksville, NY). Trypsin gold, sequencing grade trypsin, and chymotrypsin were purchased from Promega (Madison, WI). Pronase E, leucine aminopeptidase, and prolidase were purchased from Sigma (St. Louis, MO). All solvents were high-purity B & J Brand from Honeywell Burdick and Jackson (Muskegon, MI). ACS reagent-grade formic acid (88%) was purchased from J.T. Baker (Phillipsburg, NJ). Isolute C18 SPE column (25 mg) was from Biotage (Charlotte, NC). HiTrap Blue affinity and GE P10 columns were obtained from GE Healthcare (Piscataway, NJ). Spire PEP tips were a gift from Thermo (Bellefonte, PA). All other chemical reagents were ACS grade and purchased from Sigma- Aldrich.
Synthesis of N-Oxidized Metabolites of PhIP
Unlabeled PhIP and [2H5]-PhIP were mixed at a molar ratio of 1:1. NO2-PhIP and NO2-[2H5]-PhIP were synthesized by diazotization of PhIP with NaNO2, as described previously.27 Subsequently, the NO2-PhIP analogues were reduced with hydrazine, using Pd/C as a catalyst, to produce HONH-PhIP and HONH-[2H5]-PhIP.27 The method of synthesis of N-acetoxy-PhIP is a modification of procedures reported in the literature.28,29 An equimolar mixture of HONH-PhIP and HONH-[2H5]-PhIP (20 μg, 83 nmol) in C2H5OH (50 μL) at −5 °C was reacted with acetic anhydride in acetic acid (10% v/v, 2 μL) for 16 min. The solution was then diluted with chilled, deionized H2O (500 μL) and applied to the Spire PEP tip (12 mg) under a gentle vacuum. The resin was washed with chilled deionized H2O (500 μL), and the mixture of N-acetoxy-PhIP and N-acetoxy-[2H5]-PhIP was eluted with chilled C2H5OH (~100 μL). The product was characterized by UPLC-ESI/MS2 with a triple stage quadrupole mass spectrometer (vide infra). The N-acetoxy-PhIP derivatives were prepared immediately prior to their reaction with peptides or SA.
2-Hydroxy-1-methyl-6-phenylimidazo[4,5-b]pyridine (2-HO-PhIP) was prepared by incubation of NO2-PhIP (1.7 μg) in 90 mM NH4OH (500 μL) at 40 °C for 1 h. 2-Amino-1-methyl-6-(5-hydroxy)-phenylimidazo[4,5-b]pyridine (5-HO-PhIP) was obtained by the solvolysis of N-acetoxy-PhIP as described previously.22,30 The identities of 2-HO-PhIP and 5-HO-PhIP were confirmed by their characteristic product ion spectra.24,30
Amino acid adducts of NO2-PhIP were synthesized by the reaction of cysteine, tryptophan, tyrosine or lysine (0.1 μmol) with NO2-PhIP (0.1 μmol) in 50 mM ammonium bicarbonate buffer, pH 8.5 (1 mL) at 37 °C for 18 h. The adducts were isolated by enrichment with C18 SPE resins that were prewashed with CH3OH and H2O. The reaction products were applied to the resin, and the resin was washed with H2O (2 mL). The adducts were eluted with CH3OH (1 mL) and concentrated to dryness by vacuum centrifugation. The modified amino acids were purified by HPLC with Agilent 1100 HPLC system (Palo Alto, CA) and an Agilent Eclipse XDB-C18 column (4.6 × 250 mm). A linear gradient was employed, starting from 100% A solvent (0.1% HCO2H) and reaching 100% B solvent (95% CH3CN containing, 4.9% H2O, and 0.1% HCO2H) at 20 min, at a flow rate of 1 mL/min. The wavelength was monitored at 210 and 320 nm. The structures of the PhIP-modified amino acids were determined by LC-ESI/MSn with a linear quadrupole ion trap MS (LTQ MS, Thermo Fisher, San Jose, CA). The approximate yields of the NO2-PhIP-modified cysteine and tyrosine adducts were ~50%, and the yield of the NO2-PhIP-modified lysine adduct was ~5% on the basis of UV measurements. NO2-PhIP did not form an adduct with tryptophan, under these reaction conditions.
Modification of Human SA and Plasma with N-Oxidized Metabolites of PhIP
Commerical human SA was pretreated with β-mercaptoethanol to reduce mixed disulfides formed at Cys34.31 For some studies, the Cys34 of SA was blocked by titration of the thiol residue to its end point with 4-CMB,31,32 or by reaction with a 5-fold mol excess of NEM for 5 h at room temperature, followed by gel filtration. The sulfhydryl content of SA was determined using Ellman’s reagent.33 The SA reduced with β-mercaptoethanol contained a sulfhydryl content of 0.95 mol -SH/mol SA, whereas the sulfhydryl content of 4-CMB or NEM modified SA was 0.02 mol -SH/mol SA.
A solution of NO2-PhIP and NO2-[2H5]-PhIP (90 nmol in 6.6 μL DMSO), or HONH-PhIP and HONH-[2H5]-PhIP (90 nmol in 13 μL C2H5OH), or N-acetoxy-PhIP and N-acetoxy-[2H5]-PhIP mixture (90 nmol in 40 μL C2H5OH) was reacted with reduced SA (2 mg, 30 nmol) in 1 mL 10 mM potassium phosphate buffer (pH 7.4). The reactions of NO2-PhIP and HONH-PhIP with SA were performed at 37°C for 18 h, whereas the reaction of N-acetoxy-PhIP with SA was performed at 37 °C for 2 h. Unbound PhIP derivatives were removed from SA by solvent extraction with ethyl acetate (3 mL, 3 times), followed by gel-filtration chromatography of the SA with the PD-10 column containing 10 mM potassium phosphate buffer (pH 7.4).
Human plasma was diluted with PBS by 5-fold. A 1 mL solution of diluted plasma was then treated with a 1:1 isotopic mixture of NO2-PhIP and NO2-[2H5]-PhIP (450 nmol in 33 μL DMSO) at 37 °C for 18 h. The purification of SA from plasma matrix was done with a HiTrap Blue affinity column.24 In brief, the treated plasma was diluted with buffer A (50 mM KH2PO4, pH 7.0) and centrifuged to remove particulates, before it was applied to a HiTrap Blue affinity column. Buffer B (50 mM KH2PO4 buffer (pH 7.0) containing, 1.5 M KCl (3 mL) was used to elute SA from the affinity column. Thereafter, any remaining unbound PhIP derivatives were removed from SA by solvent extraction, followed by gel-filtration chromatography of the SA in 10 mM potassium phosphate buffer as described above.
Trypsin Digestion
Enzymes were prepared as described previously.24 Three different proteolytic conditions were employed to digest SA adducts modified by N-oxidized-PhIP derivatives.
Trypsin digestion: PhIP-modified SA (0.5 mg) was concentrated to dryness by vacuum centrifugation and dissolved in 0.25 M Tris buffer containing 6 M guanidine-HCl (pH 7.4, 0.5 mL). Dithiothreitol (6.5 mg, 20 mM) was added to the solution, and the mixture was incubated at 55 °C for 1 h. After the solution was cooled to room temperature, excess iodoacetamide (6 mg, 65 mM) was added, and the mixture was incubated in the dark for 30 min. The denatured and alkylated SA was then subjected to gel-filtration chromatography with the PD-10 column in 50 mM ammonium bicarbonate buffer (pH 8.5), to remove excess iodoacetamide. The SA solution (6 μg in 35 μL) was mixed with CaCl2 (1 μL of 50 mM stock solution), followed by addition of trypsin at a protease:SA ratio of 1:50 (w/w). The digestion was performed for 20 h at 37 °C. Thereafter, the protein digest was diluted with H2O (1 mL) and applied to a C18 SPE. The CH3OH eluents were concentrated to dryness by vacuum centrifugation and resuspended in 1:1 H2O: DMSO for UPLC-ESI/MSn analysis.
Trypsin/chymotrypsin digestion: The denatured and alkylated PhIP-modified SA (5 μg, 75 pmol, in 35 μL 50 mM ammonium bicarbonate buffer (pH 8.5) containing CaCl2 (1 mM) was digested with trypsin at a protease:protein ratio of 1:50 (w/w) and chymotrypsin at a protease:SA ratio of 1:12.5 (w/w). The digestion was performed at 37° C for 20 h, followed by the same SPE purification procedure described above.
Pronase E/leucine aminopeptidase/prolidase digestion: This enzymatic digestion was performed as described previously.24 In brief, the PhIP-modified SA (5 μg, 75 pmol) in 50 mM ammonium bicarbonate buffer (pH 8.5) containing 1 mM MnCl2 was treated with Pronase E at a protease:protein ratio of 1:2 (w/w), leucine aminopeptidase at a protease:protein ratio of 1:30 (w/w), and prolidase at a protease:SA ratio of 1:8 (w/w). The digested was conducted at 37° C for 20 h, followed by the same SPE purification procedure described above.
Mass Spectrometry Methods
Product ion spectra of N-oxidized PhIP derivatives were acquired by infusion with a Finnigan Quantum Ultra triple stage quadrupole mass spectrometer (TSQ MS, Thermo Fisher, San Jose, CA) interfaced with a CaptiveSpray™ source from Michrom Bioresource Inc. (Auburn, CA). Typical instrument tuning parameters were: capillary temperature 200 °C, source spray voltage 1.4 kV, tube lens offset 95 V, capillary offset 35 V, and in-source fragmentation 5 V. Argon, set at 1.5 mTorr, was used as the collision gas, and the collision energy was variable and set between 15 to 35 eV. In some instances, metabolites were characterized online with a Waters NanoAcquity UPLC (New Milford, MA) interfaced with the TSQ MS. LC separation of peptides was performed with a Magic C18AQ column (0.3 × 150 mm) (Michrom Bioresource Inc.). Metabolites were resolved with a linear gradient starting from 90% A solvent (0.01% HCO2H) to 100% B solvent (95% CH3CN containing, 4.99% H2O, and 0.01% HCO2H) over 20 min, at a flow rate of 5 μL/min.
Accurate mass measurements of the synthesized amino acid and peptide adducts of NO2-PhIP were acquired on the LTQ Orbitrap XL (Thermo Fisher Scientific, Bremen, Germany) at the Proteomics Core Facility in the Center for Biotechnology and Interdisciplinary Studies, Rensselaer Polytechnic Institute, Troy, NY. The MS was interfaced with the Michrom CaptiveSpray™ ion source. Full scan mass spectra were acquired from a scan range of m/z 100 to 700 at a resolution of R = 60000 at m/z 400. The injection time was 500 ms and data were acquired with a μscan count of 1. The detected ions were recalibrated on the fly using phthalate as the lock mass at m/z 149.02332. The spray voltage was 2.2 kV, capillary temperature was 200 °C, and the tube lens was 100 V. The isolation width was 2 amu, and the collision-induced dissociation (CID) in the linear ion trap was set at a normalized collision energy of 35%.
UPLC-ESI/MSn
The separation of peptides was performed with a NanoAcquity UPLC system (Waters Corp., Milford, MA) equipped with a Magic C18AQ column (0.3 × 150 mm) from Michrom Bioresource Inc. The Digests (3 μL) were injected and peptides were resolved with a linear gradient starting from 90% A solvent (0.01% HCO2H) to 100% B solvent (95% CH3CN containing, 4.99% H2O, and 0.01% HCO2H) over 60 min, at a flow rate of 5 μL/min. MS spectra were acquired with a linear quadrupole ion trap mass spectrometer (LTQ, Thermo Fisher, San Jose, CA). Xcalibur version 2.07 software was used for data manipulations. All analyses were conducted in the positive ionization mode and employed an Advance CaptiveSpray™ source from Michrom Bioresource Inc. The temperature of capillary tube was set at 200 °C; the spray voltage was 1.5 kV; and the in-source fragmentation was 10 V. There was no sheath or auxiliary gas. Helium was used as the collision and damping gas in the ion trap and was set at a pressure of 1 mTorr. One μscan was used for data acquisition. The automatic gain control (AGC) settings were full MS target 30,000 and MSn target 10,000, and the maximum injection time was 10 ms.
Data-Dependent MS/MS
A full scan was obtained for eluting peptides in the range of 300–2000 amu, followed by three data-dependent MS/MS scans. The MS/MS spectra were recorded using dynamic exclusion of previously analyzed precursors for 180 s with a repeat of 3 and a repeat duration of 60 s. MS/MS spectra were generated by CID of the peptide ions at a normalized collision energy of 35% to generate a series of b- and y-ions as major fragments. For isotopically labeled experiments, the mass spectrometer was programmed to switch from the full survey scan MS to the MS/MS scan mode, which was triggered by the characteristic isotopic pattern of unlabelled PhIP/[2H5]-PhIP at a partner intensity ratio of 65 – 100%, employing the mass tags scanning option. The mass to charge differences were set at m/z 5.00, 2.50, or 1.67, respectively, for singly, doubly, or triply charged peptide species. The mass tag of singly charged species (m/z 5) was employed to scan for amino acid adducts recovered from SA digested with Pronase E/leucine aminopeptidase/prolidase, whereas the mass tags of doubly and triply charged ions (m/z 2.50 and 1.67) were employed to scan for SA peptide adducts recovered from tryptic or tryptic/chymotryptic digests. The four most abundant ions above 1000 counts that displayed the isotopic pattern were selected for CID with a normalized collision energy of 35%, employing an isolation width of 2 amu.
Data Analysis
The mass spectral data PhIP- and [2H5]-PhIP-modified amino acid and peptide adducts were acquired by mass tags with the Xcalibur software. Scan filters were employed to extract the protonated ions and MS/MS spectral data of the isotopic pairs. The tandem mass spectra of peptides were interpreted manually and facilitated by online software (Protein Prospector, Univ. of California, San Francisco, http://us.expasy.org/proteomics). Fully automated data analysis on peptide adducts was conducted by converting RAW data to mz5 format in the ProteoWizard msConvert tool.34 Spectra were identified by the MyriMatch algorithm,35 version 2.1.111, using a 31 protein subset database containing SA from an initial search against the RefSeq human protein database, version 37.3. Searching for modifications in a protein database containing only a handful of sequences can inappropriately force spectra to match to modified peptides. Therefore, we generated a database of 31 proteins by a fully tryptic search of the LC-MS/MS data generated here in combination with a separate LC-MS/MS experiment from unrelated major proteins frequently observed in pull-downs with immunoprecipitation to add protein distractors to the database. The sequence database was reversed so that each protein sequence appeared in both normal and reversed orientations (totaling 62 sequences), enabling false discovery rate estimation. MyriMatch was configured to have cysteines to contain carbamidomethyl (+57.021 Da) as a dynamic modification and to allow for the possibility of oxidation (+15.996) on methionines, and deamidation (−17.03 Da) of N-terminal glutamines. Peptides modified with NO2-PhIP and NO2-[2H5]-PhIP were searched with [C,K,Y] allowed to have dynamic modifications of 207.1 and 212.1 amu; peptides modified with HONH-PhIP and HONH-[2H5]-PhIP or their N-acetoxy derivatives were allowed to have dynamic modifications at [C,K,Y,S,T,W,H] of 222.1 and 227.1 amu, for adduction with PhIP, or dynamic modifications at [C] of 238.1 and 243.1 amu for adduction with NO-PhIP (sulfinamide adducts); and 254.1 and 259.1 amu (sulfonamide adducts). Candidate peptides were allowed to have trypsin cleavages or protein termini at one or both termini (semi-tryptic search), and up to 2 missed cleavages were permitted. The precursor error was set at 1.25 m/z, but fragment ions were required to match within 0.5 m/z. Analyses of modified SA were also performed with trypsin/chymotrypsin digests, employing the same configurations. The IDPicker algorithm36 v3.0.420 filtered the identifications for each spectrum with a 5% identification false discovery rate at the peptide-spectrum match level.
Results
Mapping of Human SA Modified with NO2-PhIP and NO2-[2H5]-PhIP
Data-Dependent Analysis of Human SA Modified with NO2-PhIP Followed by Digestion with Pronase E, Leucine aminopeptidase and Prolidase
This three enzyme mixture efficiently digests many proteins to amino acids.37,38 The full scan mode was monitored from the mass range of 300 to 600 Da, a range that encompasses the molecular masses [M+H]+ of all possible amino acid adducts formed with NO2-PhIP. The mass chromatograms of the enzymatic digests of unmodified and NO2-PhIP-modified SA acquired by the data-dependent MS/MS with mass tags enabled are presented in Supporting Information, Figure S-1.
Three pairs of adducts were detected (tR = 6.7, 13.9, and 17.8 min). The first adduct was characterized as desamino-PhIP-K. The product ion spectra of the adduct ([M+H]+ at m/z 354.2) and its isotopic labeled species ([M+H]+ at m/z 359.2) are shown in Figure 1. The masses are consistent with the molecular weight of a reaction product formed between lysine (146.2 Da) and NO2-PhIP or NO2-[2H5]-PhIP (254.1 or 259.1 Da), where the amine group of the side chain of lysine displaced the nitro moiety of PhIP. The product ion spectra of desamino-PhIP-K ([M+H]+ at m/z 354.2) and [2H5]-desamino-PhIP-K ([M+H]+ at m/z 359.2) exhibited a ‘twin’ pattern of ions that differ by 5 Da. The product ion spectrum of desamino-PhIP-K contained 3 major fragment ions at m/z 337.2, 309.2 and 291.2, corresponding to the losses of NH3, NH3+CO, and NH3+HCO2H, respectively. The peaks observed at m/z 225.1 and 230.1, respectively, in the spectra of desamino-PhIP-K and desamino-[2H5]-PhIP-K are attributed to protonated PhIP and [2H5]-PhIP, which were produced by cleavage of the bond between the ε-carbon and the side chain amine group of Lys. The product ion spectra acquired at the MS3 scan stage on m/z 225.1 and 230.1 are identical to the spectra of PhIP and [2H5]-PhIP (data not shown). The synthetic desamino-PhIP-K adducts exhibit the identical tR and product ion spectra (data not shown). The assignments of these fragment ions were supported by exact-mass measurements (Table 1).
Figure 1.

Product ion mass spectra of desamino-PhIP-K ([M+H]+, m/z 354.2) recovered from NO2-PhIP-modified SA (upper panel) and desamino-[2H5]-PhIP-K ([M+H]+, m/z 359.2) recovered from NO2-[2H5]-PhIP modified SA (bottom panel), following digestion of with Pronase E, leucine aminopeptidase, and prolidase.
Table 1.
Accurate mass measurements of PhIP-SA adducts formed with Lys, Tyr, and Tyr*-Thr-Lys
| PhIP adducts | Assignment | Observed m/z | Calcd m/z | Error (ppm) | Formula |
|---|---|---|---|---|---|
desamino-PhIP-K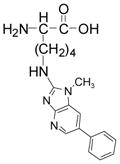
|
[M+H]+ | 354.1926 | 354.1925 | 0.28 | C19H24N5O2 |
| [M+H-NH3]+ | 337.1657 | 337.1659 | −0.59 | C18H21N4O | |
| [M+H-NH3-CO]+ | 309.1709 | 309.1709 | −0.11 | C18H21N4O | |
| [M+H-NH3-HCO2H]+ | 291.1605 | 291.1604 | 0.34 | C18H19N4 | |
| [M+H-C6H11NO2]+ | 225.1135 | 225.1135 | 0.18 | C13H13N4 | |
|
| |||||
desamino-PhIP-Y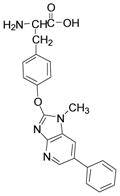
|
[M+H]+ | 389.1609 | 389.1608 | 0.26 | C22H21N4O3 |
| [M+H-NH3-CO]+ | 344.1390 | 344.1394 | −1.16 | C21H18N3O2 | |
| [M+H-HCO2H]+ | 343.1556 | 343.1553 | 0.87 | C21H19N4O | |
| [M+H-NH3-CO2]+ | 328.1444 | 328.1444 | 0.02 | C21H18N3O | |
| [M+H-C2H4NO2•]+ | 315.1365 | 315.1366 | −0.32 | C20H17N3O | |
| [M+H-C9H9NO2]+a | 226.0975 | 226.0975 | 0.16 | C13H12N3O | |
| [M+H-C9H10NO3•]+b | 209.0948 | 209.0947 | 0.11 | C13H11N3 | |
|
| |||||
desamino-PhIP-Y*TK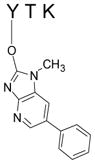
|
[M+2H]2+ | 309.6554 | 309.6554 | −0.12 | C32H41N7O6 |
| b2* | 472.1978 | 472.1979 | −0.21 | C26H26N5O4 | |
| a2* | 444.2030 | 444.2029 | 0.22 | C25N26N5O3 | |
| Y* | 343.1553 | 343.153 | −0.17 | C21H19N4O | |
| b2*+H2O | 490.2084 | 490.2085 | −0.20 | C26H28N5O5 | |
| b2*-H2O | 454.1873 | 454.1874 | −0.22 | C26H24N5O3 | |
| b2*-CO2 | 428.2080 | 428.2081 | −0.23 | C25H26N5O2 | |
Product ion at the MS3 scan stage (389.16 > 343.16 → 226.10)
Product ion at the MS3 scan stage (389.16 > 315.14 → 209.09)
The product ion spectra of adducts ([M+H]+ at m/z 329.2 and 334.2) of the second pair of adducts (tR = 13.9 min are identical to the spectra that we reported previously for desamino-PhIP-C adduct, where the thiol group of Cys34 displaced the nitro moiety of PhIP.24
The most abundant amino acid adducts (tR = 17.8 min) have molecular weights of 388.2 and 393.2 Da; these masses are consistent with masses of adducts formed between tyrosine (181.1 Da) and 209.1 (desamino-PhIP) or 214.1 Da (desamino-[2H5]-PhIP), less two protons. Major fragment ions are observed in the product ion spectrum of desamino-PhIP-modified tyrosine ([M+H]+ at m/z 389.3) (Figure 2A) at m/z 344.2 [M+H-NH3-CO]+, 343.2 [M+H-HCO2H]+ (tyrosine immonium ion plus desamino-PhIP), 328.2 [M+H–NH3-CO2]+, 315.2 [M+H-C2H4NO2•]+, and at m/z 209.2 [M+H-C9H10NO3•]+, which is attributed to the radical cation of desamino-PhIP. The product ion spectrum of the desamino-[2H5]-PhIP-Y ([M+H]+ at m/z 394.1) displays the corresponding fragment ions at m/z 349.2, 348.2, 333.2, 320.2, and 214.2 (Figure 2B). Consecutive reaction monitoring was performed at the MS3 and MS4 scan stages for several fragment ions of desamino-PhIP-Y to determine the site of bond formation between NO2-PhIP and tyrosine (Figures 2C – 2E). The radical cation at m/z 315.2 [M+H-C2H4NO2•]+ (m/z 320.2 for desamino-[2H5]-PhIP-Y) is proposed to arise by homolytic cleavage of the bond between the α and β-carbon atoms of tyrosine; this is an uncommon mechanism of fragmentation of amino acids by ESI-MS under low-energy CID conditions (Scheme 1).39 The second generation product ion spectrum (MS3) of the ion at m/z 315.2 (m/z 320.2 for desamino-[2H5]-PhIP-Y) contains the ion at m/z 209.2 (m/z 214.2 for desamino-[2H5]-PhIP-Y) as the base peak (Figure 2C). This ion is proposed to occur by cleavage of the bond of the C-2 atom of the imidazole moiety of PhIP and the phenolic oxygen of tyrosine, to produce the radical cation of desamino-PhIP and the quinone methide (106.1 Da) as a neutral species (Scheme 1). The second generation product ion spectrum (MS3) of the m/z 343.2 ([M+H-HCO2H]+ (tyrosine immonium ion containing desamino-PhIP) (Figure 3D) contains a major fragment at m/z 226.2, which is the m/z of the protonated 2-HO-PhIP. The third generation product ion spectrum (MS4) of m/z 226.2 produced ions at m/z 211.2, 198.2 and 183.2; these ions correspond to, respectively, the loss of the CH3•, CO, and the losses of CH3• and CO from 2-HO-PhIP (Figure 2E). The mass spectrum is in excellent agreement to the spectrum of synthetic 2-HO-PhIP (data not shown). The mass spectral data demonstrate that bond formation adduct occurred between the 4-HO group of tyrosine and the C-2 imidazole atom of PhIP. The assignments of these fragment ions were supported by exact-mass measurements (Table 1).
Figure 2.

Product ion mass spectra of A) desamino-PhIP-Y ([M+H]+ at m/z 389.2) recovered from NO2-PhIP-modified SA; B) desamino-[2H5]-PhIP-Y ([M+H]+ at m/z 394.2) recovered from NO2-[2H5]-PhIP-modified SA, following digestion with Pronase E, leucine aminopeptidase, and prolidase; C) second generation product ion spectrum of the ion at m/z 315.2; D) second generation product ion spectrum of ion at m/z 343.2; and E) third generation product ion spectrum of m/z 226.2 (2-HO-PhIP) from desamino-PhIP-Y.
Scheme 1.

Proposed mechanisms of fragmentation of desamino-PhIP-Y
Figure 3.

Data-dependent MS/MS scanning with mass tags of NO2-PhIP and NO2-[2H5]-PhIP-modified SA following trypsin digestion, either unmodified (upper panel) or SA modified with a 3 mol excess of an equimolar mixture of NO2-PhIP and NO2-[2H5]-PhIP modified (bottom panel). The chromatograms of the MS/MS data were acquired on the ions exhibiting a difference of m/z 2.5 in doubly charged form and a difference of m/z 1.67 in triply charged form, employing the mass tags scanning option with a partner intensity ratio of 65 to 100% for PhIP/[2H5]-PhIP. The ion intensities are normalized to the same scale for untreated and NO2-PhIP-modified SA.
Data-Dependent Analysis of Human SA Modified with NO2-PhIP Following Protein Digestion with Trypsin
Data mining of the peptide chemical features by MyriMatch revealed that the sequence coverage of unmodified SA exceeded 80% in the data dependent MS/MS mode (data not shown). Cys34 is the only unpaired cysteine residue in SA that is available for reaction with carcinogenic metabolites and toxic electrophiles.16,20 However, human SA contains 59 lysine and 18 tyrosine residues40 that can potentially react with NO2-PhIP to form adducts. The specific lysine and tyrosine residues within SA that reacted with NO2-PhIP were identified by tandem MS sequencing of the proteolytic digests of the modified SA.41
The mass chromatograms of unmodified SA peptides, and NO2-PhIP- and NO2-[2H5]-PhIP adducted peptides of SA are shown in Figure 3. A deuterium isotope effect caused the [2H5]-PhIP-peptide adducts to elute about 2 s earlier than the unlabeled adducts. These slight differences in tR values skewed the 1/1 ratio of the unlabelled PhIP/[2H5]-PhIP-adduct signals. Therefore, the partner intensity ratio for PhIP/[2H5]-PhIP was expanded to 65 – 100%, to efficiently trigger the acquisition of MS/MS spectra on both unlabeled and labeled peptide adducts. This “relaxed” ratio resulted in elevated background signals of some peptides in non-modified SA. Ten NO2-PhIP-modified peptides were identified (Table 2), and several product ion spectra of representative peptide adducts are presented Figure 4.
Table 2.
Assignment of NO2-PhIP modified amino acid residues from SA that were identified by data-dependent MS/MS analysis
| Enzyme(s) | Fraction | Position | Peptide sequence | Modified site |
|---|---|---|---|---|
| Pronase E/leucine aminopediase/prolidase | 1 | N/A | N/A | Lysine |
| 2 | N/A | N/A | Cysteine | |
| 3 | N/A | N/A | Tyrosine | |
|
| ||||
| Trypsin | 1 | 191–197 | ASSAK*QR | Lys195 |
| 2 | 539–545 | ATK*EQLK | Lys541 | |
| 3 | 198–205 | LK*CASLQK | Lys199 | |
| 4 | 349–359 | LAK*TYETTLEK | Lys351 | |
| 5 | 411–413 | Y*TK | Tyr411 | |
| 6 | 30–41 | YLQQC*PFEDHVK | Cys34 | |
| 7 | 28–41 | AQYLQQC*PFEDHVK | Cys34 | |
| 8 | 138–144 | Y*LYEIAR | Tyr138 | |
| 9 | 390–402 | QNCELFEQLGEY*K | Tyr401 | |
| 10 | 149–159 | FY*APELLFFAK | Tyr150 | |
|
| ||||
| Trypsin/chymotrypsin | 1 | 191–197 | ASSAK*QR | Lys195 |
| 2 | 539–545 | ATK*EQLK | Lys541 | |
| 3 | 411–413 | Y*TK | Tyr411 | |
| 4 | 29–41 | LQQC*PFEDHVK | Cys34 | |
| 5 | 31–36 | LQQC*PF | Cys34 | |
| 6 | 149–155 | FY*APELL | Tyr150 | |
| 7 | 149–156 | FY*APELLF | Tyr150 | |
| 8 | 149–157 | FY*APELLFF | Tyr150 | |
Figure 4.

Product ion mass spectra of A) Fraction 1, ASSAK*QR (desamino-PhIP-K) adduct ([M+2H]2+ at m/z 477.8) recovered from NO2-PhIP-modified SA; B) ASSAK*QR (desamino-[2H5]-PhIP-K) adduct ([M+2H]2+ at m/z 480.3) recovered from NO2-[2H5]-PhIP modified SA; C) Fraction 5, Y*TK (desamino-PhIP-Y) adduct ([M+2H]2+ at m/z 309.9) recovered from NO2-PhIP-modified; D) Y*TK (desamino-[2H5]-PhIP-Y) adduct ([M+2H]2+ at m/z 312.4); E) Fraction 8, Y*LYEIAR (desamino-PhIP-Y) adduct ([M+2H]2+ at m/z 568.1) recovered from NO2-PhIP-modified SA; and F) Y*LYEIAR (desamino-[2H5]-PhIP-Y) adduct ([M+2H]2+ at m/z 570.6) recovered from NO2-[2H5]-PhIP modified SA by trypsin digestion.
The first pair of modified peptides (tR = 4.1 min) were present as doubly charged ions [M+2H]2+ at m/z 477.8 and 480.3. This molecular mass corresponds to the mass of the amino acid sequence 191–197 of SA (ASSAKQR, [M+H]+ at m/z 747.4) plus desamino-PhIP or desamino-[2H5]-PhIP, respectively (addition of 207 or 212 Da). The product ion spectra of the proposed ASSAK*QR adducts ([M+2H]2+ at m/z 477.8 and 480.3) are shown in Figure 4A and 4B. The ions observed at m/z 291.1 and m/z 296.1, respectively, in the spectrum of NO2-PhIP- and NO2-[2H5]-PhIP-modified ASSAKQR are at the same m/z observed in the spectra of desamino-PhIP-K ([K*-NH3-HCO2H]+ (Figure 1), suggesting that NO2-PhIP had bound to lysine. The peptide fragment ions (y2, y2-NH3, b3 and b4) were detected, respectively, at m/z 303.1, 286.2, 246.2 and 317.2 for both NO2-PhIP- and NO2-[2H5]-PhIP modified peptides, as would be expected for fragment ions in the unmodified peptide ASSAKQR. The peptide fragment ions (y3*, y4*, y5*, y6*, b5* and b6*) were shifted by 207 and 212 Da, respectively for the NO2-PhIP- and the NO2-[2H5]-PhIP-modified peptides.
Fraction 2 was found to contain another adducted peptide of NO2-PhIP at a lysine residue (tR = 5.7 min). The sequence was identified as ATK*EQLK, which covers the amino acid sequence 539–545 of SA. The product ion spectra of the NO2-PhIP- and NO2-[2H5]-PhIP-adducted ATK*EQLK are presented in Supporting Information, Figure S-2.
Two more adducted peptides containing NO2-PhIP modified lysine residues were detected in Fraction 3 (tR = 5.9 min) and Fraction 4 (tR = 8.3 min) at relatively low abundance, which encompasses the amino acid sequences 198–205 (LK*CASLQK) and 349–359 (LAK*TYETTLEK) of SA, respectively. (Table 1)
The fifth NO2-PhIP-modified peptide displayed a strong signal (tR = 9.7 min), and both singly charged [M+H]+ (m/z 618.4 and 623.4) and doubly charged species [M+2H]2+ (m/z 309.9 and 312.4) were observed. This modified peptide was assigned to the amino acid sequence 411–413 of SA (YTK ([M+H]+ at m/z 411.2), and contained a mass increment of 207 and 212 Da, consistent with adduction by desamino-PhIP and desamino-[2H5]-PhIP. Either Tyr411 or Lys413 of SA could be the site of modification. The product ion spectra of the modified YTK peptide contained several prominent fragment ions (Figures 4C and 4D). The y1 ion observed at m/z 147.1 corresponds to lysine, and the fragment ions observed at m/z 230.1 and 248.2 (y2 and y2-H2O) in the spectra for both NO2-PhIP-and NO2-[2H5]-PhIP modified peptides are also seen in the spectrum of YTK (data not shown). The fragment ions (a2*, b2* and b2*-H2O) contain mass increments of desamino-PhIP (207 Da) and desamino-[2H5]-PhIP (212 Da), respectively. The b2*-CO2 ion (C25H26N5O2) at m/z 428.3 and 433.3 for unlabeled and desamino-[2H5]-PhIP adducts is proposed to occur by a rearrangement involving the HO-group of the threonine side chain to the electrophilic carbonyl moiety, followed by loss of CO2.42 The ions at m/z 343.2 and 348.2 (y*) for desamino-PhIP- and desamino-[2H5]-PhIP adducts are assigned to the desamino-PhIP-Y immonium ions. These spectral data prove that adduction of NO2-PhIP occurred at tyrosine and not lysine. The second generation product ion spectrum of the ion at m/z 343.2 for desamino-PhIP-Y*TK and at m/z 348.2 for desamino[2H5]-PhIP-Y*TK ([M+H-HCO2H]+) contained the prominent fragment ion at m/z 226.1, attributed to 2-HO-PhIP (m/z 231.1 for 2-HO-[2H5]-PhIP), as was observed in the spectra of desamino-PhIP-Y (Figure 1). Accurate mass measurement of the monoisotopic elemental mass of synthetic Y*TK and its product ion spectra confirmed the assignment (Table 1). Thus, the structure of the modified peptide in Fraction 5 is assigned Y*TK with the site of adduction occurring between the 4-HO-tyrosine group and the C-2 imidazole atom of NO2-PhIP.
Three more fractions containing peptides modified with NO2-PhIP at tyrosine residues were observed in Fraction 8 (tR =14.9 min), Fraction 9 (tR =16.8 min), and Fraction 10 (tR =21.6 min) (Table 1). The modified peptides in Fraction 8 were doubly charged species ([M+2H]2+ at m/z 568.1 and 570.6), corresponding to the peptide containing amino acid residues 138–144 of SA (Y*LYEIAR [M+H]+ at m/z 927.5) plus the desamino-PhIP and desamino-[2H5]-PhIP moieties, respectively. The product ion spectra of this pair of modified peptides contained almost the complete series of b- and y-ions and the immonium ions for the desamino-PhIP modified tyrosine (Figures 4E and 4F), which confirmed that Tyr138 was the amino acid modified in this sequence. The peptide adduct observed in Fraction 9 corresponded to the amino acid residues 390 – 412 of SA (QNCELFEQLGEY*K, [M+2H]2+at m/z 933.2) with NO2-PhIP adducted to Y, based on the interpretation of the product ion spectra (Table 1, and Supporting Information, Figure S-3).
The pair of peptide adducts detected in Fraction 10 were consistent with the amino acid residues 149–159 of SA (FY*APELLFFAK, [M+2H]2+at m/z 777.1 and 779.6) having adductions with desamino-PhIP and desamino-[2H5]-PhIP moieties at Y, respectively.
NO2-PhIP-modified peptides of the Cys34 residue were detected in fractions 6 and 7 (tR = 11.9 and 12.2 min). Fraction 6 contained triply charged desamino-PhIP- and desamino-[2H5]-PhIP-labeled peptide adducts [M+3H]3+ at m/z 572.4 and 574.0 (Supporting Information, Figures S-4A and S-4B). The peptide sequence is ascribed to amino acid residues 30–41 of SA, with adduct formation occurring at Cys34 (YLQQC*PFEDHVK, 1505.7 Da). The ions observed at m/z 242.1 in the mass spectrum of NO2-PhIP-modified peptide and at m/z 247.1 in the spectrum of NO2-[2H5]-PhIP-modified peptide were identified as the protonated 2-thioimdazole derivatives of PhIP and [2H5]-PhIP, by their spectra at the MS3 scan stage as reported previously.24 The y and b ion series supported the proposed assignment of the adduct structure. The b2 (m/z 277.1), b3 (m/z 404.6), b4 (m/z 533.2), b5 (m/z 843.1), b6 (m/z 940.4), y2 (m/z 246.2), y3 (m/z 383.3), y4 (m/z 498.3), y5 (m/z 627.3), y6 (m/z 774.6) and y7 (m/z 871.3) observed in both product ion spectra are consistent with predicted sequence for unmodified YLQQCPFEDHVK. A number of ions display the characteristic isotopic pattern with a mass difference of 5 Da and encompass the modified amino acid residue, b6*-NH3, y9*-NH3, y9*-NH3–HS-PhIP, y10*-NH3, y10*-NH3–HS-PhIP and y11*.
A pair of PhIP and [2H5]-PhIP labeled peptide adducts are observed as triply charged species [M+3H]3+ at m/z at 638.5 and 640.2 in Fraction 7. The product ion spectrum displayed many of the peptide sequence series of y and b ions in common to that observed for YLQQC*PFEDHVK, but also contained ions attributed to 3 additional amino acid residues. The peptide was identified as AQYLQQC*PFEDHVK (1704.8 Da) modified by desamino-PhIP (207 Da) or desamino-[2H5]-PhIP (212 Da); this pair of peptide adducts occurred at amino acid residues 28 – 41 of SA (Supporting Information, Figure S-4C and S-4D).
Data-Dependent Analysis Human SA Modified with NO2-PhIP Following Protein Digestion with Trypsin/Chymotrypsin
Data-dependent scanning of modified SA digested with trypsin/chymotrypsin resulted in the discovery of six pairs of modified peptide adducts (Supporting Information, Figure S-5 and Table 2). Consistent with the trypsin digestion, Y*TK adducts derived from trypsin/chymotrypsin of modified SA were identified as doubly charged ion [M+2H]2+ at m/z 309.9 and 312.4. The NO2-PhIP-modified LQQC*PF adducts (amino acid residues 31–36 of SA) were observed as doubly charged species [M+2H]2+ (m/z 472.0 and 474.4) (tR =17.5 min). The product ion spectra were consistent with results from our previous study that showed the formation of LQQC*PF (C-desamino-PhIP or C- desamino-[2H5]-PhIP) adducts.24
Peptide adducts observed at 11.4 min displayed triply charged ions [M+3H]3+ at m/z 517.8 and 519.7, which are an addition mass of 207 and 212 Da to the protonated peptide LQQCPFEDHVK (m/z 1343.6, residues 31–41 of SA) with a missed cleavage at C-terminal of phenylalanine. The appearance of b2 (m/z 242.1), b2 – NH3 (m/z 225.1), y3 (m/z 383.2), b4*(m/z 340.8 or 343.1, doubly charged), y7 (m/z 871.3), y8*(m/z 591.4 or 594.1, doubly charged), y9*(m/z 655.5 or 657.9, doubly charged), y10*-NH3 (m/z 710.7 or 713.2, doubly charged), as well as protonated HS-PhIP and HS-[2H5]-PhIP ions (m/z 242.1 and 247.1) provided the evidence for PhIP adduction at LQQC*PFEDHVK. (Supporting Information, Figure S-6).
Another site of NO2-PhP modification of human SA was identified at Tyr150. Three peptides containing the FY*AP, due to missed cleavages with chymotrypsin, were detected at tR 22.8, 25.6 and 28.0 min (Table 2, Supporting Information, Figure S-7).
Data Dependent Analyses of SA in Human Plasma Reacted with a Mixture of NO2-PhIP and NO2-[2H5]-PhIP
The formation of PhIP-SA adduction products was also examined in human plasma treated with NO2-PhIP and NO2-[2H5]-PhIP, because plasma proteins, fatty acids, drugs, and other organic compounds that are associated with SA can induce structural changes in the conformation of SA and affect the reactivity of the amino acid residues.40,43 Data-dependent scanning of the proteolytic digest with trypsin or trypsin/chymotrypsin revealed that NO2-PhIP-modified peptides at Cys34, Lys195, Lys199, Lys351, Lys541, Tyr401, Tyr411 and Tyr150 of SA, results consistent with the adduction products of NO2-PhIP formed with commercial human SA (Supporting Information, Figure S-8).
Targeted UPLC-ESI/MS2 Analysis of Human SA Modified with NO2-PhIP Following Protein Digestion with Trypsin
The sites of NO2-PhIP binding to human SA reported above were characterized with SA that had been modified with a 3 mol excess of NO2-PhIP. However, when the reaction of NO2-PhIP with SA was performed with 10-fold less carcinogen (0.3 mol NO2-PhIP: 1 mol SA), targeted UPLC-ESI/MS2 analysis revealed that the desamino-PhIP adducts were hardly formed at Lys195, Lys199, Lys541, Cys34, Tyr138, and Tyr401, and desamino-PhIP adduct formation at Tyr411 accounted for about 99% of the total ion counts of SA adducts (Figure 5). Thus, Tyr411 is a primary binding site of NO2-PhIP.
Figure 5.

UPLC-ESI/MS2 mass chromatograms of adducts of NO2-PhIP-modified SA formed by reaction of the carcinogen with human SA at a molar ratio of 3:1 (left panel) and 0.3:1 (right panel); the SA was digested with trypsin. Targeted ESI/MS2 was done on the following modified peptides: Fraction 1, ASSAK*QR as doubly charged species ([M+2H]2+ at m/z 477.8); Fraction 2, ATK*EQLK ([M+2H]2+ at m/z 512.9); Fraction 3, LK*CASLQK ([M+2H]2+ at m/z 577.8); Fraction 4, LAK*TYETTLEK ([M+3H]3+ at m/z 501.9, extracted ions: m/z 378.8, 660.6 and 720.4); Fraction 5, Y*TK ([M+2H]2+ at m/z 309.2); Fraction 7, AQYLQQC*PFEDHVK ([M+3H]3+ at m/z 638.5); Fraction 8, Y*LYEIAR ([M+2H]2+ at m/z 568.1); and Fraction 9, QNCELFEQLGEY*K ([M+2H]2+ at m/z 933.2).
Mass Spectrometric Characterization of Human SA Modified with HNOH-PhIP
Data-Dependent Analysis Human SA Modified with HONH-PhIP Following Protein Digestion with Pronase E, leucine aminopeptidase, and prolidase
The reaction of human SA with HONH-PhIP in vitro produced the N2-cysteinesulfinyl-PhIP; this sulfinamide linkage accounted for greater than 95% of the HONH-PhIP bound to SA.24 The enzymatic digestion of HONH-PhIP- and HONH-[2H5]-PhIP-modified SA with Pronase E, leucine aminopeptidase, and prolidase resulted in extensive hydrolysis of the sulfinamide linkage, and targeted MS/MS analyses of the protein digest showed that PhIP was recovered in high yields.24 The amino acid adduct N2-cysteinesulfinyl-PhIP was not detected in the 3-enzyme digest; however, data-dependent scanning identified two pairs of HONH-PhIP-tripeptide adducts (tR =17.8 and 19.3 min) (Supporting Information, Figure S-9, Table 3). The first pair of adducts was detected as singly charged species [M+H]+ at m/z 604.2 (m/z = 609.2 for [2H5]-PhIP-peptide). The product ion spectra (Figures 6A and 6B) were in excellent agreement with the mass spectra previously reported for the N2-cysteinesulfinyl-PhIP tripeptide C*PF (C-[S=O]-PhIP) and (C-[S=O]-[2H5]-PhIP) adducts.24 The second set of adducts (tR = 19.3 min) was detected as singly charged species [M+H]+ at m/z 620.2 and 625.2, respectively, for the HONH-PhIP- and HONH-[2H5]-PhIP-modified peptides. The mass of this adduct pair is 16 Da greater than the molecular mass of the C*PF (C-[S=O]-PhIP and C-[S=O]-[2H5]-PhIP). The structure of this adduct pair is proposed to be the sulfonamide C*PF (C-[SO2]-PhIP) and (C-[SO2]-[2H5]-PhIP). The assignment of the structure is supported by the product ion spectra (Figures 6C and 6D). The fragment ion observed at m/z 396.2 in the spectra of both modified peptides is attributed to the loss of PhIP (224.1 Da) and [2H5]-PhIP (229.1 Da). Thus, the 2 oxygen atoms are associated with the C*PF peptide and not the PhIP moiety. The mass difference of 30 Da observed between the fragment ion at m/z 396.2 and the protonated CPF ([M+H]+ at m/z 366.1) is consistent with the presence two sulfur-oxygen double bonds on Cys34. The ions at m/z 263.2, 313.2, 330.1, 358.1, 427.1 and 455.1 correspond to, respectively, y2, the C* immonium ion-NH3, the C* immonium ion, and the b1*, a2* and b2* ions.
Table 3.
Assignment of HONH-PhIP modified amino acid residues from SA that were identified by data-dependent MS/MS analyses
| Enzyme(s) | Fraction | Position | Peptide sequence | Modified site |
|---|---|---|---|---|
| Pronase E/leucine aminopediase/prolidase | 1 | 34–36 | C*PF (sulfinamide) | Cys34 |
| 2 | 34–36 | C*PF (sulfonamide) | Cys34 | |
|
| ||||
| Trypsin/chymotrypsin | 1 | 31–41 | LQQC*PFEDHVK (sulfinamide) | Cys34 |
| 2 | 31–41 | LQQC*PFEDHVK (sulfonamide) | Cys34 | |
| 3 | 31–36 | LQQC*PF sulfinamide | Cys34 | |
Figure 6.

Product ion mass spectra of A) C*PF (C-[S=O]-PhIP) sulfinamide adduct ([M+H]+ at m/z 604.2) recovered from HONH-PhIP-modified SA; B) C*PF (C-[S=O]-[2H5]-PhIP) sulfinamide adduct ([M+H]+ at m/z 609.2) recovered from HONH-[2H5]-PhIP modified SA; C) C*PF (C-[SO2]-PhIP) sulfonamide adduct ([M+H]+ at m/z 620.2) recovered from HONH-PhIP-modified SA, and C*PF (C- [SO2]-[2H5]-PhIP) sulfonamide adduct ([M+H]+ at m/z 625.2) recovered from HONH-[2H5]-PhIP modified SA digested with Pronase E, leucine aminopeptidase, and prolidase.
Data-Dependent Analysis Human SA Modified with HONH-PhIP Following Protein Digestion with Trypsin/chymotrypsin
The PhIP sulfinamide linkage at Cys34 of SA undergoes hydrolysis to produce PhIP, by heat and treatment with DTT during the denaturation of SA (Peng, L., unpublished observations). However, the hexapeptide N2-cysteinesulfinyl-PhIP LQQC*PF (C-[S=O]-PhIP, tR = 16.8 min, [M+2H]2+ m/z 487.4 and 489.7 for PhIP and [2H5]-PhIP-modified peptides) was recovered from HONH-PhIP-modified SA, following digestion with trypsin/chymotrypsin without heat denaturation and DTT treatment. The missed cleavage adducts LQQC*PFEDHVK (C-[S=O]-PhIP) and LQQC*PFEDHVK (C-[SO2]-PhIP) were also observed, respectively at tR = 11.3 min and 13.4 min, occurring as triply protonated species [M+3H]3+ (Supporting Information, Figure S-10 and Figure S-11).
Mass Spectrometric Characterization of N-Acetoxy-PhIP and its Adduction Products with Human SA
N-Acetoxy-PhIP is a penultimate metabolite of PhIP that reacts with DNA to form stable covalent adducts.28,29,44 The conditions of synthesis of N-acetoxy-PhIP were optimized to produce this reactive intermediate in high yield with minimal side-reaction products (Figure 7A). The amount of N-acetoxy-PhIP was about 100-fold greater than the amount of its inactive isomer, the hydroxamic acid, N-hydroxy-N-(1-methyl-6-phenylimidazo[4,5-b]pyridin-2-yl)-acetamide (N-hydroxy-N-acetyl-PhIP), assuming comparable ionization efficiencies of the compounds by ESI/MS. The structures of these acetylated isomers of HONH-PhIP ([M+H]+ at m/z 283.1) are readily distinguished by their product ion spectra (Figures 7B and 7C). The product ion spectrum of N-acetoxy-PhIP contains major fragment ions at m/z 223.0 and 224.0, corresponding, respectively, to losses of CH3CO2• and CH3CO2H, whereas the product ion spectrum of N-hydroxy-N-acetyl-PhIP contains a base peak ion at m/z 241.1, which is attributed to the loss of ketene (CH2CO).
Figure 7.

(A) UPLC-ESI/MS analysis of reaction products formed by reaction of HONH-PhIP with acetic anhydride, following SPE. The peaks in the chromatogram are PhIP (tR = 10.0), 5-HO-PhIP (tR = 10.2), N-hydroxy-N-acetyl-PhIP (tR = 10.9), and N-acetoxy-PhIP (tR = 13.7). Product ion spectra were acquired on acetylated isomers of HONH-PhIP ([M+H]+ at m/z 283.1): (B) product ion spectrum of N-acetoxy-PhIP and (C) product ion spectrum N-hydroxy-N-acetyl-PhIP.
Data-Dependent Analysis Human SA Modified with N-Acetoxy-PhIP Following Protein Digestion with Pronase E, Leucine Aminopeptidase, and Prolidase
The UV spectrum of human SA modified with N-acetoxy-PhIP displays a maximum at 320 – 325 nm, whereas non-modified SA does not possess a chromophore around these wavelengths. The level of chemical modification of SA by N-acetoxy-PhIP was estimated at ~170 pmol PhIP/nmol SA, based on the assumption that N-acetoxy-PhIP-SA adducts possess molar absorption coefficients that are comparable to that of PhIP (315 nm; ε (M−1 cm−1) at 22,220). However, the isotopic data-dependent scanning was not successful in detecting SA adducts of N-acetoxy-PhIP, irrespective of the enzymatic digestion conditions. Nucleophilic amino acids of SA were expected to react with N-acetoxy-PhIP or the proposed nitrenium ion45 to form adducts with amino acids or peptides with mass increments of 223 or 228 Da, attributed respectively, to reactive PhIP or [2H5]-PhIP species, less one proton from the amino acid residue. Although data-dependent scanning analyses failed to detect stable covalent adducts, targeted UPLC-ESI/MS2 analysis of the N-acetoxy-PhIP-modified SA digested with the Pronase E, amino leucine peptidase, and prolidase revealed the presence of high levels of PhIP and 5-HO-PhIP. The identities of PhIP (data not shown), and 5-HO-PhIP and 5-HO-[2H5]-PhIP were corroborated by their product ion spectra (Figure 8). Pretreatment of SA with the selective thiol reagents 4-CMB or NEM prior to reaction with N-acetoxy-PhIP, decreased the amounts of PhIP and 5-HO-PhIP recovered from the proteolytic digest by 3-fold. This finding suggests that a substantial portion of the N-acetoxy-PhIP had bound to Cys34 as unstable adducts that underwent hydrolysis to form PhIP and 5-HO-PhIP, during proteolytic digestion.
Figure 8.

Product ion mass spectra of A) 5-HO-PhIP ([M+H]+ at m/z 241.2) recovered from N-acetoxy-PhIP-modified SA (upper panel) and 5-HO-[2H5]-PhIP (([M+H]+ at m/z 246.2) recovered from N-acetoxy-[2H5]-PhIP-modified SA (bottom panel) digested with Pronase E, leucine aminopeptidase, and prolidase. The ions observed at m/z 227.2 and m/z 226.2 in the product ion spectrum of 5-HO-[2H5]-PhIP are attributed to losses of HDO and D2O.
Discussion
NO2-PhIP, HONH-PhIP, and N-acetoxy-PhIP are reactive species that contribute to the deleterious biological effects of PhIP through DNA and protein adduct formation.10,46 Metabolites of PhIP are known to bind to human SA in vivo;13 however, the structures of the adducts remain to be elucidated. Human SA is comprised of three homologous domains and each domain contains two subdomains.47 Studies have shown that micro environments of pH within the different domains of SA can lower the pKa values of some nucleophilic side chain groups of amino acids and enhance their reactivity with electrophiles43,48 Moreover, the tertiary structure of SA can exert an influence on noncovalent protein-electrophile interactions and direct ensuing covalent adduct formation between specific amino acids and electrophiles.49 Thus, we had anticipated that nucleophilic amino acids of SA would display different degrees of reactivity towards these distinct electrophilic metabolites of PhIP, resulting in an array of covalent adducts of PhIP within different sequence locations of SA.
We employed a two tier approach to map the sites of human SA modification with NO2-PhIP, HONH-PhIP and N-acetoxy-PhIP. In the first approach, SA samples modified with metabolites of PhIP were enzymatically digested with a mixture of Pronase E, leucine aminopeptidase, and prolidase, to produce amino acid containing adducts.37,38 Data-dependent scanning of NO2-PhIP-modified SA revealed that adducts were formed with Cys, Lys, and Tyr. This 3-enzyme mixture did not generate amino acid adducts of SA modified with HONH-PhIP, but produced tripeptide adducts of PhIP formed at Cys34. The Cys34 residue of human SA is known to react with many genotoxicants and toxic electrophiles to form adducts with acrylamide,50 nitrogen mustard,51 α,β-unsaturated aldehydes,52 the neurotoxin brevetoxin B,53 acetaminophen,54 benzene,55 2-amino-3-methylimidazo[4,5-f]quinoline (IQ),31 as well as PhIP.22–24 Many of these adducts do not undergo digestion beyond the tripeptide stage with Pronase. Thus, the efficacy of proteoloytic digestion of chemically-modified SA is highly dependent upon structure of the toxicant and the nature of the bond formed between the toxicant and the sulfhydryl group of Cys34.
With knowledge about the primary amino acids that had formed adducts with PhIP metabolites, the sequence locations of PhIP adducts were mapped in tryptic or tryptic/chymotrypic digests obtained from SA modified with a 3-fold excess of an equimolar mixture of unlabeled and [2H5]-labeled PhIP metabolites. The dual isotope label provided a characteristic fingerprint for the unambiguous identification of PhIP-modified peptides. Data-dependent scanning of NO2-PhIP-modified SA enabled us to identify adducts at nine sequence locations: Lys195, Lys199, Lys351, Lys541, Cys34, Tyr138, Tyr150, Tyr401 and Tyr411. The identification of SA modifications by N-oxidized derivatives of PhIP was greatly accelerated, by the MyriMatch algorithm over manual interpretation. However, the major adduct formed at Tyr411, Y*TK, escaped detection by MyriMatch, probably because there was an insufficient number of y and b ions to identify the modified peptide by the algorithm (Figure 3C). Similarly, the C*PF adducts formed with NO-PhIP that were recovered from the 3-enzyme digest also escaped detection by MyriMatch. The employment of the isotopic mixture of the N-oxidized PhIP metabolites was critical for the successful identification of these tripeptide adducts by manual inspection.
The major site of binding of NO2-PhIP to SA occurred at Tyr411, this amino acid is present in the center of a binding pocket with Arg410 in subdomain IIIA of human SA.48 The phenolic HO group of tyrosine has a pKa of ~10, whereas the pKa of phenolic HO group of Tyr411 is lowered to 7.9 due to the electrostatic interaction with guanidinium ω-NH of Arg410.56 Tyr411 is a major site for covalent adduct with nitrophenyl acetate,48 organophosphorous pesticides,57 and nerve agents (tabun, sarin, cyclosarin, VX); secondary adduction sites with many of these toxicants occurs at Tyr148, Tyr150 and Tyr161 of SA.57–60
NO2-PhIP also formed adducts with SA at Lys195, Lys199, and Lys541. The Lys195 and Lys199 residues occur at the interface of subdomains IB and IIA, and Lys541 resides within subdomain III B.47 These lysine residues are known to form acyl-linked adducts with glucuronide conjugates of tolmetin and permethrin.61,62 Lys195 or Lys199 was also reported to form an adduct with 2,4-dinitro-1-chlorobenzene,63 and both lysine residues are reactive sites in SA and form adducts with 4-hydroxy-trans-2-nonenal.52
Cys34 is another reactive site in human SA that forms adducts with NO2-PhIP. The Cys34 resides in a shallow crevice of SA and predominantly exists in the thiolate form due to its interaction with three ionizable residues, Asp38, His39 and Tyr84, which are within close vicinity because of the tertiary structure of SA.43,48,64 As a result, the thiol of Cys34 has an unusually low pKa value of 6.5 compared to about 8.0 – 8.5 in many other proteins or peptides at physiological pH.43,52 These molecular and structural features of SA can help to explain the high reactivity of Cys34 towards many low molecular weight electrophiles of diverse structure.20,65
The sites of NO2-PhIP binding to human SA were characterized with SA that had been modified with a 3 mol excess of NO2-PhIP. However, when the reaction of NO2-PhIP with SA was performed with a limiting amount of carcinogen, desamino-PhIP-Y*TK adduct formation at Tyr411 accounted for about 99% of the total ion counts of SA adducts. These binding data indicate that Tyr411 is a primary binding site of NO2-PhIP and a likely adduction site of NO2-PhIP in vivo.
The adduction of HONH-PhIP to SA was found to occur only with Cys34; this data is in agreement with our previous finding.24 The adduct exists as the sulfur-nitrogen linked N2-[cystein-S-yl-PhIP]-S-oxide. Therefore, the reactivity of HONH-PhIP with SA was poor and protein adduct formation only occurred following the oxidation of HONH-PhIP to NO-PhIP. The sulfhydryl SH of Cys34 reacted with the N=O bond of NO-PhIP to form a semimercaptal, which underwent rearrangement to the more stable sulfinamide structure.66 We also identified the sulfonamide adduct of PhIP formed at Cys34; this adduct was not previously reported and its mechanism of formation is uncertain.24 The reaction of HONH-PhIP with oxygen results in the production of NO-PhIP and superoxide anion.67 The superoxide anion may have oxidized a portion of the Cys34 to the sulfinic acid prior to its reaction with N-oxidized-PhIP, resulting in formation of the sulfonamide adduct.68 Alternatively, the superoxide anion generated over the time, by HONH-PhIP, may have oxidized a portion of the N2-[cystein-S-yl-PhIP]-S-oxide linkage to the sulfonamide structure; the oxidation of N2-[cystein-S-yl-PhIP]-S-oxide to the sulfonamide adduct during enzymatic digestion also cannot be excluded.69
N-Acetoxy-PhIP, a penultimate metabolite of PhIP that forms stable covalent adducts at appreciable levels with DNA,29,44 was the least efficient of the N-oxidized metabolites of PhIP to react with human SA and form stable covalent adducts. Both HONH-PhIP and N-acetoxy-PhIP bind to DNA, almost exclusively at dG.29,44,70 DNA adduct formation with N-acetoxy-PhIP is believed to occur through the proposed nitrenium ion45 via an SN1 or an intermediate SN2 mechanism.71 N-Acetoxy-PhIP undergoes solvolysis more facilely than HONH-PhIP, to produce the nitrenium ion, and explains the superior reactivity of N-acetoxy-PhIP towards DNA.45 Adduct formation of N-acetoxy-PhIP with SA was expected to occur by a similar mechanism. Although stable adducts of N-acetoxy-PhIP were not detected, appreciable levels of PhIP and 5-HO-PhIP were recovered from the N-acetoxy-PhIP-treated SA digested with Pronase E, leucine aminopeptidase, and prolidase. The pretreatment of SA with the selective thiol reagents 4-CMB or NEM decreased the amounts of PhIP and 5-HO-PhIP recovered from the digests of N-acetoxy-PhIP-modified SA by about 3-fold. These findings point to the formation of unstable adducts of PhIP at Cys34. A previous study reported that certain adducts formed between N-acetoxy-PhIP and rat SA were unstable and underwent hydrolysis to produce 5-HO-PhIP.22 The reaction of N-acetoxy-PhIP with cysteine or glutathione also produced labile adducts, tentatively assigned as sulfenamide conjugates, which underwent hydrolysis to form 5-HO-PhIP.22 Thus, a large portion of the PhIP and 5-HO-PhIP recovered from N-acetoxy-PhIP bound to rat or human SA may be attributed to an unstable sulfenamide linked adduct formed at Cys34 (Scheme 2).
Scheme 2.

Proposed mechanisms of hydrolysis of Cys34 PhIP-sulfenamide of SA to produce 5-HO-PhIP and PhIP.
The Tyr411 adduct formed with NO2-PhIP and the Cys34 sulfinamide adduct formed with NO-PhIP are distinct structures derived from different N-oxidized metabolites of PhIP; however, both adducts are biomarkers of the genotoxic metabolite, HONH-PhIP. The N-Hydroxy metabolites of aromatic amines and HAAs are well known to undergo oxidation, by enzymatic and non-enzymatic chemistry, to form their corresponding nitroso derivatives.67,72,73 We have also observed that NO2-PhIP is formed from HONH-PhIP, through the NO-PhIP intermediate, under aerobic conditions in phosphate buffered saline (pH 7.4) (Turesky R, unpublished observations). Moreover, NO2-PhIP is formed during the metabolism of PhIP in hepatocytes of rats pretreated with polychlorinated biphenyls, and the desamino-PhIP-gluathione conjugate, glutathionyl-1-methyl-6-phenylimidazo[4,5-b]pyridine has been detected in bile of rats exposed to PhIP.74 The pathway by which NO2-PhIP is formed in hepatocytes or in liver of rodents is not clear, but the findings indicate that NO2-PhIP occurs during the N-oxidation of PhIP.
In summary, our mapping study of N-oxidized metabolites of PhIP adducted to human SA point to the Tyr411 and Cys34 residues as major sites of binding, respectively, for NO2-PhIP and NO-PhIP. Sensitive tandem MS methods are under development to determine if these SA adducts of PhIP are present in humans. Our goal is to implement stable covalent SA protein adducts of PhIP as biomarkers in molecular epidemiology studies to measure exposure and assess the role of this HAA in diet-related cancers.
Supplementary Material
Acknowledgments
Comments and discussion of the data with Dr. Paul Vouros, Northeastern University; Dr. Paul Skipper, MIT; and Dr. Dan Liebler, Vanderbilt University, are greatly appreciated.
Funding Source: This research was supported by grant R01 CA122320 (L.P. and R.J.T.), and R01 CA126479 (D.T. and S.D.) from the National Cancer Institute.
Abbreviations
- PhIP
2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine
- 5-HO-PhIP
2-amino-1-methyl-6-(5-hydroxy)-phenylimidazo[4,5-b]pyridine
- HONH-PhIP
2-hydroxyamino-1-methyl-6-phenylimidazo[4,5-b]pyridine
- 2-HO-PhIP
2-hydroxy-1-methyl-6-phenylimidazo[4,5-b]pyridine
- NO2-PhIP
2-nitro-1-methyl-6-phenylimidazo[4,5-b]pyridine
- NO-PhIP
2-nitroso-1-methyl-6-phenylimidazo[4,5-b]pyridine
- N-acetoxy-PhIP
N-(acetyloxy)-1-methyl-6-phenylimidazo[4,5-b]pyridine-2-amine
- N-hydroxy-N-acetyl-PhIP
N-hydroxy-N-(1-methyl-6-phenylimidazo[4,5-b]pyridin-2-yl)-acetamide
- IQ
2-amino-3-methylimidazo[4,5-f]quinoline
- 4-CMB
4-chloromercuribenzoic acid
- Hb
hemoglobin
- HAA
heterocyclic aromatic amine
- LTQ MS
linear quadrupole ion trap MS
- LC-ESI/MS/MS
liquid chromatography-electrospray ionization/tandem mass spectrometry
- NEM
N-ethylmaleimide
- SA
serum albumin
- SPE
solid phase extraction
- TSQ MS
triple stage quadrupole mass spectrometer
- UPLC
ultra performance liquid chromatography
Footnotes
Supporting Information Available: Additional information as noted in text (Figure S-1 - Figure S-11). This material is available free of charge via the Internet at http://pubs.acs.org.
Reference List
- 1.Felton JS, Jagerstad M, Knize MG, Skog K, Wakabayashi K. Contents in foods, beverages and tobacco. In: Nagao M, Sugimura T, editors. Food Borne Carcinogens Heterocyclic Amines. John Wiley & Sons Ltd; Chichester, England: 2000. pp. 31–71. [Google Scholar]
- 2.Sinha R, Rothman N, Brown ED, Salmon CP, Knize MG, Swanson CS, Rossi SC, Mark SD, Levander OA, Felton JS. High concentrations of the carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) occur in chicken but are dependent on the cooking method. Cancer Res. 1995;55:4516–4519. [PubMed] [Google Scholar]
- 3.Keating GA, Bogen KT. Estimates of heterocyclic amine intake in the US population. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;802:127–133. doi: 10.1016/j.jchromb.2003.10.047. [DOI] [PubMed] [Google Scholar]
- 4.Sinha R. An epidemiologic approach to studying heterocyclic amines. Mutat Res. 2002;506–507:197–204. doi: 10.1016/s0027-5107(02)00166-5. [DOI] [PubMed] [Google Scholar]
- 5.Knize MG, Felton JS. Formation and human risk of carcinogenic heterocyclic amines formed from natural precursors in meat. Nutr Rev. 2005;63:158–165. doi: 10.1111/j.1753-4887.2005.tb00133.x. [DOI] [PubMed] [Google Scholar]
- 6.Zheng W, Lee SA. Well-done meat intake, heterocyclic amine exposure, and cancer risk. Nutr Cancer. 2009;61:437–446. doi: 10.1080/01635580802710741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.National Toxicology Program. Report on Carcinogenesis. 11. U.S. Department of Health and Human Services, Public Health Service; Research Triangle Park, N.C: 2005. [Google Scholar]
- 8.Jarabek AM, Pottenger LH, Andrews LS, Casciano D, Embry MR, Kim JH, Preston RJ, Reddy MV, Schoeny R, Shuker D, Skare J, Swenberg J, Williams GM, Zeiger E. Creating context for the use of DNA adduct data in cancer risk assessment: I. Data organization. Crit Rev Toxicol. 2009;39:659–678. doi: 10.1080/10408440903164155. [DOI] [PubMed] [Google Scholar]
- 9.Skipper PL, Tannenbaum SR. Protein adducts in the molecular dosimetry of chemical carcinogens. Carcinogenesis. 1990;11:507–518. doi: 10.1093/carcin/11.4.507. [DOI] [PubMed] [Google Scholar]
- 10.Turesky RJ, Le Marchand L. Metabolism and biomarkers of heterocyclic aromatic amines in molecular epidemiology studies: lessons learned from aromatic amines. Chem Res Toxicol. 2011;24:1169–1214. doi: 10.1021/tx200135s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu J, Chang P, Bondy ML, Sahin AA, Singletary SE, Takahashi S, Shirai T, Li D. Detection of 2-amino-1-methyl-6-phenylimidazo[4,5-b]-pyridine-DNA adducts in normal breast tissues and risk of breast cancer. Cancer Epidemiol Biomarkers Prev. 2003;12:830–837. [PubMed] [Google Scholar]
- 12.Zhu J, Rashid A, Cleary K, Abbruzzese JL, Friess H, Takahashi S, Shirai T, Li D. Detection of 2-amino-1-methyl-6-phenylimidazo [4,5-b]-pyridine (PhIP)-DNA adducts in human pancreatic tissues. Biomarkers. 2006;11:319–328. doi: 10.1080/13547500600667911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dingley KH, Curtis KD, Nowell S, Felton JS, Lang NP, Turteltaub KW. DNA and protein adduct formation in the colon and blood of humans after exposure to a dietary-relevant dose of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine. Cancer Epidemiol Biomarkers Prev. 1999;8:507–512. [PubMed] [Google Scholar]
- 14.Bessette EE, Spivack SD, Goodenough AK, Wang T, Pinto S, Kadlubar FF, Turesky RJ. Identification of carcinogen DNA adducts in human saliva by linear quadrupole ion trap/multistage tandem mass spectrometry. Chem Res Toxicol. 2010;23:1234–1244. doi: 10.1021/tx100098f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liebler DC. Proteomic approaches to characterize protein modifications: new tools to study the effects of environmental exposures. Environ Health Perspect. 2002;110(Suppl 1):3–9. doi: 10.1289/ehp.02110s113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tornqvist M, Fred C, Haglund J, Helleberg H, Paulsson B, Rydberg P. Protein adducts: quantitative and qualitative aspects of their formation, analysis and applications. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;778:279–308. doi: 10.1016/s1570-0232(02)00172-1. [DOI] [PubMed] [Google Scholar]
- 17.Bryant MS, Vineis P, Skipper PL, Tannenbaum SR. Hemoglobin adducts of aromatic amines: associations with smoking status and type of tobacco. Proc Natl Acad Sci U S A. 1988;85:9788–9791. doi: 10.1073/pnas.85.24.9788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kiese M, Taeger K. The fate of phenylhydroxylamine in human red cells. Naunyn Schmiedebergs Arch Pharmacol. 1976;292:59–66. doi: 10.1007/BF00506490. [DOI] [PubMed] [Google Scholar]
- 19.Ringe D, Turesky RJ, Skipper PL, Tannenbaum SR. Structure of the single stable hemoglobin adduct formed by 4-aminobiphenyl in vivo. Chem Res Toxicol. 1988;1:22–24. doi: 10.1021/tx00001a003. [DOI] [PubMed] [Google Scholar]
- 20.Rubino FM, Pitton M, Di FD, Colombi A. Toward an “omic” physiopathology of reactive chemicals: thirty years of mass spectrometric study of the protein adducts with endogenous and xenobiotic compounds. Mass Spectrom Rev. 2009;28:725–784. doi: 10.1002/mas.20207. [DOI] [PubMed] [Google Scholar]
- 21.Magagnotti C, Orsi F, Bagnati R, Celli N, Rotilio D, Fanelli R, Airoldi L. Effect of diet on serum albumin and hemoglobin adducts of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) in humans. Int J Cancer. 2000;88:1–6. doi: 10.1002/1097-0215(20001001)88:1<1::aid-ijc1>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 22.Reistad R, Frandsen H, Grivas S, Alexander J. In vitro formation and degradation of 2-amino-1-methyl-6- phenylimidazo[4,5-b]pyridine (PhIP) protein adducts. Carcinogenesis. 1994;15:2547–2552. doi: 10.1093/carcin/15.11.2547. [DOI] [PubMed] [Google Scholar]
- 23.Chepanoske CL, Brown K, Turteltaub KW, Dingley KH. Characterization of a peptide adduct formed by N-acetoxy-2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP), a reactive intermediate of the food carcinogen PhIP. Food Chem Toxicol. 2004;42:1367–1372. doi: 10.1016/j.fct.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 24.Peng L, Turesky RJ. Mass spectrometric characterization of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine N-oxidized metabolites bound at Cys34 of human serum albumin. Chem Res Toxicol. 2011;24:2004–2017. doi: 10.1021/tx2003504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma S, Subramanian R. Detecting and characterizing reactive metabolites by liquid chromatography/tandem mass spectrometry. J Mass Spectrom. 2006;41:1121–1139. doi: 10.1002/jms.1098. [DOI] [PubMed] [Google Scholar]
- 26.Tzouros M, Pahler A. A targeted proteomics approach to the identification of peptides modified by reactive metabolites. Chem Res Toxicol. 2009;22:853–862. doi: 10.1021/tx800426x. [DOI] [PubMed] [Google Scholar]
- 27.Turesky RJ, Lang NP, Butler MA, Teitel CH, Kadlubar FF. Metabolic activation of carcinogenic heterocyclic aromatic amines by human liver and colon. Carcinogenesis. 1991;12:1839–1845. doi: 10.1093/carcin/12.10.1839. [DOI] [PubMed] [Google Scholar]
- 28.Goodenough AK, Schut HA, Turesky RJ. Novel LC-ESI/MS/MSn method for the characterization and quantification of 2′-deoxyguanosine adducts of the dietary carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine by 2-D linear quadrupole ion trap mass spectrometry. Chem Res Toxicol. 2007;20:263–276. doi: 10.1021/tx0601713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin D, Kaderlik KR, Turesky RJ, Miller DW, Lay JO, Jr, Kadlubar FF. Identification of N-(deoxyguanosin-8-yl)-2-amino-1-methyl-6-phenylimidazo [4,5-b]pyridine as the major adduct formed by the food-borne carcinogen, 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine, with DNA. Chem Res Toxicol. 1992;5:691–697. doi: 10.1021/tx00029a016. [DOI] [PubMed] [Google Scholar]
- 30.Langouët S, Paehler A, Welti DH, Kerriguy N, Guillouzo A, Turesky RJ. Differential metabolism of 2-amino-1-methyl-6-phenylimidazo[4,5- b]pyridine in rat and human hepatocytes. Carcinogenesis. 2002;23:115–122. doi: 10.1093/carcin/23.1.115. [DOI] [PubMed] [Google Scholar]
- 31.Turesky RJ, Skipper PL, Tannenbaum SR. Binding of 2-amino-3-methylimidazo[4,5-f]quinoline to hemoglobin and albumin in vivo in the rat. Identification of an adduct suitable for dosimetry. Carcinogenesis. 1987;8:1537–1542. doi: 10.1093/carcin/8.10.1537. [DOI] [PubMed] [Google Scholar]
- 32.Boyer PD. Spectrophotometric Study of the Reaction of Protein Sulfhydryl Groups with Organic Mercurials. J Am Chem Soc. 1954;76:4331–4337. [Google Scholar]
- 33.Riener CK, Kada G, Gruber HJ. Quick measurement of protein sulfhydryls with Ellman’s reagent and with 4,4′-dithiodipyridine. Anal Bioanal Chem. 2002;373:266–276. doi: 10.1007/s00216-002-1347-2. [DOI] [PubMed] [Google Scholar]
- 34.Kessner D, Chambers M, Burke R, Agus D, Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 2008;24:2534–2536. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tabb DL, Fernando CG, Chambers MC. MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J Proteome Res. 2007;6:654–661. doi: 10.1021/pr0604054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma ZQ, Dasari S, Chambers MC, Litton MD, Sobecki SM, Zimmerman LJ, Halvey PJ, Schilling B, Drake PM, Gibson BW, Tabb DL. IDPicker 2.0: Improved protein assembly with high discrimination peptide identification filtering. J Proteome Res. 2009;8:3872–3881. doi: 10.1021/pr900360j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsao M, Otter DE. Quantification of glutamine in proteins and peptides using enzymatic hydrolysis and reverse-phase high-performance liquid chromatography. Anal Biochem. 1999;269:143–148. doi: 10.1006/abio.1998.3091. [DOI] [PubMed] [Google Scholar]
- 38.Baxter JH, Lai CS, Phillips RR, Dowlati L, Chio JJ, Luebbers ST, Dimler SR, Johns PW. Direct determination of methionine sulfoxide in milk proteins by enzyme hydrolysis/high-performance liquid chromatography. J Chromatogr A. 2007;1157:10–16. doi: 10.1016/j.chroma.2007.04.035. [DOI] [PubMed] [Google Scholar]
- 39.Papayannopoulos IA. The interpretation of collision-induced dissociation tandem mass spectra of peptides. Mass Spectrom Rev. 1995;14:49–73. [Google Scholar]
- 40.Peters T., Jr Serum albumin. Adv Protein Chem. 1985;37:161–245. doi: 10.1016/s0065-3233(08)60065-0. [DOI] [PubMed] [Google Scholar]
- 41.Steen H, Mann M. The ABC’s (and XYZ’s) of peptide sequencing. Nat Rev Mol Cell Biol. 2004;5:699–711. doi: 10.1038/nrm1468. [DOI] [PubMed] [Google Scholar]
- 42.Delatour T, Richoz J, Vouros P, Turesky RJ. Simultaneous determination of 3-nitrotyrosine and tyrosine in plasma proteins of rats and assessment of artifactual tyrosine nitration. J Chromatogr B. 2002;779:189–199. doi: 10.1016/s1570-0232(02)00370-7. [DOI] [PubMed] [Google Scholar]
- 43.Stewart AJ, Blindauer CA, Berezenko S, Sleep D, Tooth D, Sadler PJ. Role of Tyr84 in controlling the reactivity of Cys34 of human albumin. FEBS J. 2005;272:353–362. doi: 10.1111/j.1742-4658.2004.04474.x. [DOI] [PubMed] [Google Scholar]
- 44.Frandsen H, Grivas S, Andersson R, Dragsted L, Larsen JC. Reaction of the N2-acetoxy derivative of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) with 2′-deoxyguanosine and DNA. Synthesis and identification of N2-(2′-deoxyguanosin-8-yl)-PhIP. Carcinogenesis. 1992;13:629–635. doi: 10.1093/carcin/13.4.629. [DOI] [PubMed] [Google Scholar]
- 45.Nguyen TM, Novak M. Synthesis and decomposition of an ester derivative of the procarcinogen and promutagen, PhIP, 2-amino-1-methyl-6-phenyl-1H-imidazo[4,5-b]pyridine: unusual nitrenium ion chemistry. J Org Chem. 2007;72:4698–4706. doi: 10.1021/jo070306p. [DOI] [PubMed] [Google Scholar]
- 46.Glatt H. Metabolic factors affecting the mutagenicity of heteroyclic amines. In: Skog K, Alexander J, editors. Acrylamide and Other Hazardous Compounds in Heat-Treated Foods. Woodhead Publishing Ltd; Cambridge, England: 2006. pp. 358–404. [Google Scholar]
- 47.Sugio S, Kashima A, Mochizuki S, Noda M, Kobayashi K. Crystal structure of human serum albumin at 2.5 A resolution. Protein Eng. 1999;12:439–446. doi: 10.1093/protein/12.6.439. [DOI] [PubMed] [Google Scholar]
- 48.Carter DC, Ho JX. Structure of serum albumin. Adv Protein Chem. 1994;45:153–203. doi: 10.1016/s0065-3233(08)60640-3. [DOI] [PubMed] [Google Scholar]
- 49.Skipper PL. Influence of tertiary structure on nucleophilic substitution reactions of proteins. Chem Res Toxicol. 1996;9:918–923. doi: 10.1021/tx960028h. [DOI] [PubMed] [Google Scholar]
- 50.Noort D, Fidder A, Hulst AG. Modification of human serum albumin by acrylamide at cysteine-34: a basis for a rapid biomonitoring procedure. Arch Toxicol. 2003;77:543–545. doi: 10.1007/s00204-003-0484-5. [DOI] [PubMed] [Google Scholar]
- 51.Noort D, Hulst AG, Jansen R. Covalent binding of nitrogen mustards to the cysteine-34 residue in human serum albumin. Arch Toxicol. 2002;76:83–88. doi: 10.1007/s00204-001-0318-2. [DOI] [PubMed] [Google Scholar]
- 52.Aldini G, Regazzoni L, Orioli M, Rimoldi I, Facino RM, Carini M. A tandem MS precursor-ion scan approach to identify variable covalent modification of albumin Cys34: a new tool for studying vascular carbonylation. J Mass Spectrom. 2008;43:1470–1481. doi: 10.1002/jms.1419. [DOI] [PubMed] [Google Scholar]
- 53.Wang Z, Ramsdell JS. Analysis of interactions of brevetoxin-B and human serum albumin by liquid chromatography/mass spectrometry. Chem Res Toxicol. 2011;24:54–64. doi: 10.1021/tx1002854. [DOI] [PubMed] [Google Scholar]
- 54.Damsten MC, Commandeur JN, Fidder A, Hulst AG, Touw D, Noort D, Vermeulen NP. Liquid chromatography/tandem mass spectrometry detection of covalent binding of acetaminophen to human serum albumin. Drug Metab Dispos. 2007;35:1408–1417. doi: 10.1124/dmd.106.014233. [DOI] [PubMed] [Google Scholar]
- 55.Bechtold WE, Willis JK, Sun JD, Griffith WC, Reddy TV. Biological markers of exposure to benzene: S-phenylcysteine in albumin. Carcinogenesis. 1992;13:1217–1220. doi: 10.1093/carcin/13.7.1217. [DOI] [PubMed] [Google Scholar]
- 56.Ahmed N, Dobler D, Dean M, Thornalley PJ. Peptide mapping identifies hotspot site of modification in human serum albumin by methylglyoxal involved in ligand binding and esterase activity. J Biol Chem. 2005;280:5724–5732. doi: 10.1074/jbc.M410973200. [DOI] [PubMed] [Google Scholar]
- 57.John H, Breyer F, Thumfart JO, Hochstetter H, Thiermann H. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) for detection and identification of albumin phosphylation by organophosphorus pesticides and G- and V-type nerve agents. Anal Bioanal Chem. 2010;398:2677–2691. doi: 10.1007/s00216-010-4076-y. [DOI] [PubMed] [Google Scholar]
- 58.Li B, Schopfer LM, Hinrichs SH, Masson P, Lockridge O. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry assay for organophosphorus toxicants bound to human albumin at Tyr411. Anal Biochem. 2007;361:263–272. doi: 10.1016/j.ab.2006.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Noort D, Hulst AG, van ZA, van RE, van der Schans MJ. Covalent binding of organophosphorothioates to albumin: a new perspective for OP-pesticide biomonitoring? Arch Toxicol. 2009;83:1031–1036. doi: 10.1007/s00204-009-0456-5. [DOI] [PubMed] [Google Scholar]
- 60.Li B, Nachon F, Froment MT, Verdier L, Debouzy JC, Brasme B, Gillon E, Schopfer LM, Lockridge O, Masson P. Binding and hydrolysis of soman by human serum albumin. Chem Res Toxicol. 2008;21:421–431. doi: 10.1021/tx700339m. [DOI] [PubMed] [Google Scholar]
- 61.Ding A, Zia-Amirhosseini P, McDonagh AF, Burlingame AL, Benet LZ. Reactivity of tolmetin glucuronide with human serum albumin. Identification of binding sites and mechanisms of reaction by tandem mass spectrometry. Drug Metab Dispos. 1995;23:369–376. [PubMed] [Google Scholar]
- 62.Noort D, van ZA, Fidder A, van OB, Hulst AG. Protein adduct formation by glucuronide metabolites of permethrin. Chem Res Toxicol. 2008;21:1396–1406. doi: 10.1021/tx8000362. [DOI] [PubMed] [Google Scholar]
- 63.Aleksic M, Pease CK, Basketter DA, Panico M, Morris HR, Dell A. Investigating protein haptenation mechanisms of skin sensitisers using human serum albumin as a model protein. Toxicol In Vitro. 2007;21:723–733. doi: 10.1016/j.tiv.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 64.Christodoulou J, Sadler PJ. 1H NMR of albumin in human blood plasma: drug binding and redox reactions at Cys34. FEBS Lett. 1995;376:1–5. doi: 10.1016/0014-5793(95)01231-2. [DOI] [PubMed] [Google Scholar]
- 65.Rappaport SM, Li H, Grigoryan H, Funk WE, Williams ER. Adductomics: Characterizing exposures to reactive electrophiles. Toxicol Lett. doi: 10.1016/j.toxlet.2011.04.002. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dolle B, Topner W, Neumann HG. Reaction of arylnitroso compounds with mercaptans. Xenobiotica. 1980;10:527–536. doi: 10.3109/00498258009033787. [DOI] [PubMed] [Google Scholar]
- 67.Hiramoto K, Negishi K, Namba T, Katsu T, Hayatsu H. Superoxide dismutase-mediated reversible conversion of 3-hydroxyamino-1-methyl-5H-pyrido[4,3-b]indole, the N-hydroxy derivative of Trp-P-2, into its nitroso derivative. Carcinogenesis. 1988;9:2003–2008. doi: 10.1093/carcin/9.11.2003. [DOI] [PubMed] [Google Scholar]
- 68.Umemoto A, Grivas S, Yamaizumi Z, Sato S, Sugimura T. Non-enzymatic glutathione conjugation of 2-nitroso-6-methyldipyrido [1,2-a: 3′,2′-d] imidazole (NO-Glu-P-1) in vitro: N-hydroxy-sulfonamide, a new binding form of arylnitroso compounds and thiols. Chem -Biol Interact. 1988;68:57–69. doi: 10.1016/0009-2797(88)90006-3. [DOI] [PubMed] [Google Scholar]
- 69.Finch JW, Crouch RK, Knapp DR, Schey KL. Mass spectrometric identification of modifications to human serum albumin treated with hydrogen peroxide. Arch Biochem Biophys. 1993;305:595–599. doi: 10.1006/abbi.1993.1466. [DOI] [PubMed] [Google Scholar]
- 70.Tang Y, LeMaster DM, Nauwelaers G, Gu D, Langouet S, Turesky RJ. UDP-Glucuronosyltransferase-mediated metabolic activation of the tobacco carcinogen 2-amino-9H-pyrido[2,3-b]indole. J Biol Chem. 2012;287:14960–14972. doi: 10.1074/jbc.M111.320093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bentley TW, Schleyer PR. Role of nucleophilic solvent assistance and nucleophilically solvated ion pair intermediates in solvolysea of primary and secondary arenesulfonates. J Am Chem Soc. 1976;98:7658–7666. [Google Scholar]
- 72.Kim D, Kadlubar FF, Teitel CH, Guengerich FP. Formation and reduction of aryl and heterocyclic nitroso compounds and significance in the flux of hydroxylamines. Chem Res Toxicol. 2004;17:529–536. doi: 10.1021/tx034267y. [DOI] [PubMed] [Google Scholar]
- 73.Lindeke B. The Non- and postenzymatic chemistry of N-oxygenated molecules. Drug Metab Rev. 1982;13:71–121. doi: 10.3109/03602538209002232. [DOI] [PubMed] [Google Scholar]
- 74.Alexander J, Wallin H, Rossland OJ, Solberg KE, Holme JA, Becher G, Andersson R, Grivas S. Formation of a glutathione conjugate and a semistable transportable glucuronide conjugate of N2-oxidized species of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) in rat liver. Carcinogenesis. 1991;12:2239–2245. doi: 10.1093/carcin/12.12.2239. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.