

Abstract
A developmental disruption of prefrontal cortical inhibitory circuits is thought to contribute to the adolescent onset of cognitive deficits observed in schizophrenia. However, the developmental mechanisms underlying such a disruption remain elusive. The goal of this study is to examine how repeated exposure to the NMDA receptor antagonist dizocilpine maleate (MK-801) during periadolescence [from postnatal day 35 (P35) to P40] impacts the normative development of local prefrontal network response in rats. In vivo electrophysiological analyses revealed that MK-801 administration during periadolescence elicits an enduring disinhibited prefrontal local field potential (LFP) response to ventral hippocampal stimulation at 20 Hz (beta) and 40 Hz (gamma) in adulthood (P65–P85). Such a disinhibition was not observed when MK-801 was given during adulthood, indicating that the periadolescent transition is indeed a sensitive period for the functional maturation of prefrontal inhibitory control. Accordingly, the pattern of prefrontal LFP disinhibition induced by periadolescent MK-801 treatment resembles that observed in the normal P30–P40 prefrontal cortex (PFC). Additional pharmacological manipulations revealed that these developmentally immature prefrontal responses can be mimicked by single microinfusion of the GABAA receptor antagonist picrotoxin into the normal adult PFC. Importantly, acute administration of the GABAA-positive allosteric modulator Indiplon into the PFC reversed the prefrontal disinhibitory state induced by periadolescent MK-801 to normal levels. Together, these results indicate a critical role of NMDA receptors in regulating the periadolescent maturation of GABAergic networks in the PFC and that pharmacologically induced augmentation of local GABAA-receptor-mediated transmission is sufficient to overcome the disinhibitory prefrontal state associated with the periadolescent MK-801 exposure.
Introduction
It is widely accepted that a strong developmental component underlies the pathophysiology of several neuropsychiatric conditions, including schizophrenia and depression (National Institute of Mental Health–National Advisory Mental Health Council, 2008). Of particular interest is the transition from adolescence to adulthood because the onset of major psychiatric symptoms, such as those observed in schizophrenia, often occurs during this developmental period (Kessler et al., 2007; Paus et al., 2008; Gogtay et al., 2011). In schizophrenia, a disruption of cortical interneurons is thought to contribute to the development of cognitive deficits associated with prefrontal cortex (PFC) functioning (Uhlhaas and Singer, 2006; Gonzalez-Burgos et al., 2011). In fact, PFC impairment in schizophrenia is typically accompanied by reduced beta and gamma oscillations (Uhlhaas and Singer, 2006; Uhlhaas et al., 2006), an effect believed to be attributable to reduced GABAergic transmission in cortical circuits (Wang and Buzsáki, 1996; Sohal et al., 2009). Interestingly, the acquisition of prefrontal-dependent cognitive functions in primates (Crone et al., 2006; Dumontheil et al., 2008; Ernst et al., 2009) and rodents (Andrzejewski et al., 2011; Koss et al., 2011) and the maturation of PFC GABAergic function (Tseng and O'Donnell, 2007a, 2007b) are also refined during adolescence. Thus, a developmental dysregulation of GABAergic transmission in the PFC could contribute to the late adolescent onset of cognitive deficits in schizophrenia (O'Donnell, 2011; Uhlhaas and Singer, 2011). However, the mechanism underlying the development of impaired GABAergic function is not fully understood.
Studies from animal models indicate that the functional maturation of the PFC network activity is dependent on the remodeling of local inhibitory circuits during adolescence by the influence of glutamatergic inputs, in particular those from the ventral hippocampus (Tseng et al., 2009). Accordingly, an impairment of NMDA-receptor-mediated transmission has been implicated in the development of PFC-dependent cognitive deficits (Krystal et al., 1994; Jentsch and Roth, 1999; Krystal et al., 2002; Stefani and Moghaddam, 2005; Rujescu et al., 2006). At the cellular level, acute administration of NMDA receptor antagonists, such as dizocilpine maleate (MK-801) or ketamine, typically elicits a transient augmentation of PFC output activity concurrent with an inhibition of local fast-spiking interneurons (Jackson et al., 2004; Homayoun and Moghaddam, 2007; Volman et al., 2011). A reduction of prefrontal GABAergic tone was also found after 2 d exposure to ketamine (Zhang et al., 2008), suggesting a disinhibitory mechanism mediating the enhanced PFC output after acute NMDA receptor antagonism.
NMDA receptors play a critical role in maintaining the phenotype of fast-spiking interneurons in cortical circuits (Behrens et al., 2007). Thus, a dysregulation of NMDA receptor function during sensitive periods of cortical development may interfere with the maturation of GABAergic transmission in the PFC (Powell et al., 2012). Here, we assessed whether periadolescent NMDA receptor blockade is sufficient to induce a state of prefrontal disinhibition that endures through adulthood. We conducted in vivo electrophysiological recordings and examined the impact of repeated periadolescent MK-801 exposure on PFC network activity in adulthood. More specifically, changes in prefrontal response to ventral hippocampal stimulation-induced frequency-dependent facilitation and depression of local field potentials (LFPs) were compared across age and treatment groups.
Materials and Methods
All experimental procedures met the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Rosalind Franklin University Institutional Animal Care and Use Committee. In the present study, male Sprague Dawley rats (Harlan) from different age groups were used. All animals were group housed (two to three rats per cage), maintained under conditions of constant temperature (21–23°C) and kept in a 12 h light/dark cycle with food and water available ad libitum. All chemicals were obtained from Sigma-Aldrich, except for Indiplon, which was obtained from Tocris Bioscience.
Experimental groups.
All animals were allowed to habituate for at least 5 d before being subjected to any experimental manipulation. Periadolescent [postnatal day 35 (P35)] and adult (P75) rats were randomly assigned to receive daily noncontingent (home cage) intraperitoneal injections of saline or MK-801 (0.1 mg/kg in saline) for 5 consecutive days. MK-801 was chosen because it is the highest-affinity noncompetitive NMDA receptor antagonist available known to induce behavioral deficits relevant to schizophrenia (Wong et al., 1986). The dose of MK-801 used in the present study (0.1 mg/kg) was chosen based on previous electrophysiological and behavioral studies showing a significant effect on PFC-dependent functions in adult rats (Aultman and Moghaddam, 2001; Jackson et al., 2004; Homayoun and Moghaddam, 2007; Wood et al., 2012). All recordings from the periadolescent-treated group were conducted within the P65–P85 age period, which corresponds to adulthood. Recordings from the adult-treated group were also performed within the 3–5 week window (P105–P125 age range) from the last saline or MK-801 injection. Additional recordings from three distinct age groups (P30–P40, P45–P55, and P60–P80) of naive rats were conducted to determine whether the pattern of PFC response to ventral hippocampal stimulation is developmentally regulated.
LFP recordings of PFC responses to ventral hippocampal stimulation in vivo. Animals were deeply anesthetized with 8% chloral hydrate (400 mg/kg, i.p.), placed in a stereotaxic apparatus (ASI Instruments), and maintained at 37–38°C using a Physitemp TCAT-2LV Controller (Physitemp Instruments). Lidocaine (2% lidocaine hydrochloride with 1:100,000 epinephrine; Cooke-Waite) was applied subcutaneously ∼5 min before any skin incision was made. Fifteen minutes after the rat was placed in the stereotaxic frame, a steady supplementary anesthesia level (8% chloral hydrate, 400 μl/h) was delivered through an intraperitoneal cannula (26 gauge butterfly needle) attached to a syringe minipump (BASi Baby Bee Syringe Drives). The level of general anesthesia was determined by monitoring the global cortical EEG activity as described previously (Tseng et al., 2001). Burr holes were then drilled in the skull for electrode placement in the medial PFC (3.2–2.7 mm anterior from bregma, 0.8 mm lateral from the midline, 4.2 mm below the brain surface) and ventral hippocampus (5.8 mm posterior from bregma, 5.2 mm lateral from the midline, 4.5 mm below the brain surface) (Paxinos and Watson, 1998). LFPs in the medial PFC were recorded using a concentric bipolar electrode (SNE-100× 50 mm; Rhodes Medical Instruments), amplified (Cygnus Technology), filtered (1–100 Hz bandwidth), and digitized (Digidata 1440A; Molecular Devices) at a sampling rate of 10 kHz. A second concentric bipolar electrode (SNE-100× 50 mm) was placed in the ventral hippocampus for stimulation. Hippocampal-evoked prefrontal LFPs were elicited by a computer-controlled pulse generator Master 8 Stimulator (A.M.P.I.). The intensity of stimulation was chosen from the 0.25–1.0 mA range. Typically, single square pulses of 300 μs duration delivered at 0.75 mA intensity was needed to evoke a reliable prefrontal response with <15% variability in amplitude and slope. Both single and train stimulation-evoked responses were delivered every 15 s. Each set of train comprised 10 pulses delivered at 10, 20, and 40 Hz, and changes from the onset to the peak amplitude of the evoked responses were measured.
Local prefrontal microinfusions experiments.
All microinfusion procedures were conducted following the same experimental design as described previously (Tseng et al., 2011). Briefly, a 28 gauge infusion cannula (11 mm length; Plastics One) was secured to the prefrontal recording electrode. The tip of the cannula was offset dorsally by ∼0.7 mm from the electrode tip. Before lowering the recording electrode, the cannula was filled with aCSF-containing vehicle, picrotoxin (50 μm, 0.1% DMSO) or Indiplon (10 μm, 0.04% DMSO). All microinfusions (1.0 μl for picrotoxin and 0.6 μl for Indiplon) were performed at 0.1 μl/min, and changes in hippocampal train stimulation-induced LFPs in the PFC were determined within the 10–40 min postinfusion period.
Tissue processing and histological confirmation of the electrode placements.
After completion of the recordings, the rat was transcardially perfused with cold saline (150 ml), followed by 4% paraformaldehyde (PFA; 200 ml) in phosphate buffer (0.1 m). The brain was removed, incubated overnight in 4% PFA, and stored in PBS containing 30% sucrose for 3 d. Serial coronal sections, 50 μm thick, were obtained from the PFC and ventral hippocampus. For verification of electrode placement, sections were mounted on Superfrost Plus slides (VWR) and exposed to formol ethanol, before undergoing dehydration, treatment with xylene, rehydration, and staining with cresyl violet. After staining, slides were washed, dehydrated again, and coverslipped with Permount (Thermo Fisher Scientific).
Statistical analyses.
All measures are expressed as mean ± SEM. Differences among experimental conditions were considered statistically significant when p < 0.05. The effects along two or more variables were determined by one- and two-way ANOVA using Statistica (StatSoft). More specifically, one-way ANOVA was used to determine the effects of a given train stimulation frequency-induced field potential response obtained in each experimental group (age or treatment). Changes in the pattern of the evoked field potentials at a given frequency across treatment conditions or age groups were assessed by a two-way (treatment or age × pulse number of the evoked field potential) ANOVA.
Results
We first determined whether repeated exposure to MK-801 during the periadolescent transition period (0.1 mg · kg−1 · d−1 for 5 d from P35 to P40; for details, see Methods and Materials) is associated with a hyperactive PFC state in adulthood, by means of LFP recordings in vivo. All recordings were conducted from the medial PFC (infralimbic and prelimbic regions), and changes in prefrontal LFPs to ventral hippocampal stimulation were assessed in adulthood within the P65–P85 age period (Fig. 1). We observed that saline- and MK-801-treated animals exhibited similar prefrontal response curves to increasing current intensities of hippocampal stimulation (Fig. 1), indicating that the basal network state in the PFC was not affected by periadolescent MK-801 exposure.
Figure 1.

a, Diagram depicting the recording arrangement used to study the impact of MK-801 exposure on ventral hippocampal-induced synaptic responses in the medial PFC (infralimbic and prelimbic regions) in vivo. Traces are examples of hippocampal stimulation-induced LFP responses recorded in the medial PFC. Calibration: 2 mV, 50 ms. Bottom inset shows the timeline of the experimental design. All electrophysiological recordings were conducted in adulthood (i.e., P65–P85) after noncontingent periadolescent saline or MK-801 (0.1 mg · kg−1 · d−1 for 5 d) injections. b, Diagram summarizing the location of the recording and stimulation sites as determined by histological analyses from Nissl-stained sections. c, Ventral hippocampal-induced LFP responses in the PFC at different current intensities recorded from adult rats with a history of saline (n = 12) or MK-801 (n = 10) treatment during periadolescence. Data are mean ± SEM. PD, Postnatal day.
We next assessed the impact of periadolescent MK-801 treatment on hippocampal train stimulation-induced facilitation and depression of LFPs in the PFC. Hippocampal train stimulation at 10 Hz (10 pulses at 100 ms interval/15 s) elicited a distinct pattern of sustained facilitation of the evoked field potential response in the PFC that was undistinguishable among saline- and MK-801-treated animals (Fig. 2a). This was not the case when the hippocampal train stimulation was delivered at 20 Hz (10 pulses at 50 ms interval/15s). Although a transient attenuation of the prefrontal response was observed in the saline group, a shift to sustained facilitation of the field potential response emerges in the PFC of MK-801-treated animals (Fig. 2b). Similarly, MK-801 exposure during periadolescence also disrupted the prefrontal response to ventral hippocampal drive at 40 Hz. A pattern of profound suppression of the evoked field potential response was observed in the PFC of saline- and MK-801-treated animals (Fig. 2c). However, the magnitude of this inhibition was markedly reduced in the PFC of animals with a history of periadolescent MK-801 treatment (Fig. 2c). Together, these results indicate that periadolescent MK-801 exposure selectively diminished the normal inhibitory control of prefrontal processing of ventral hippocampal inputs in a frequency-dependent manner.
Figure 2.
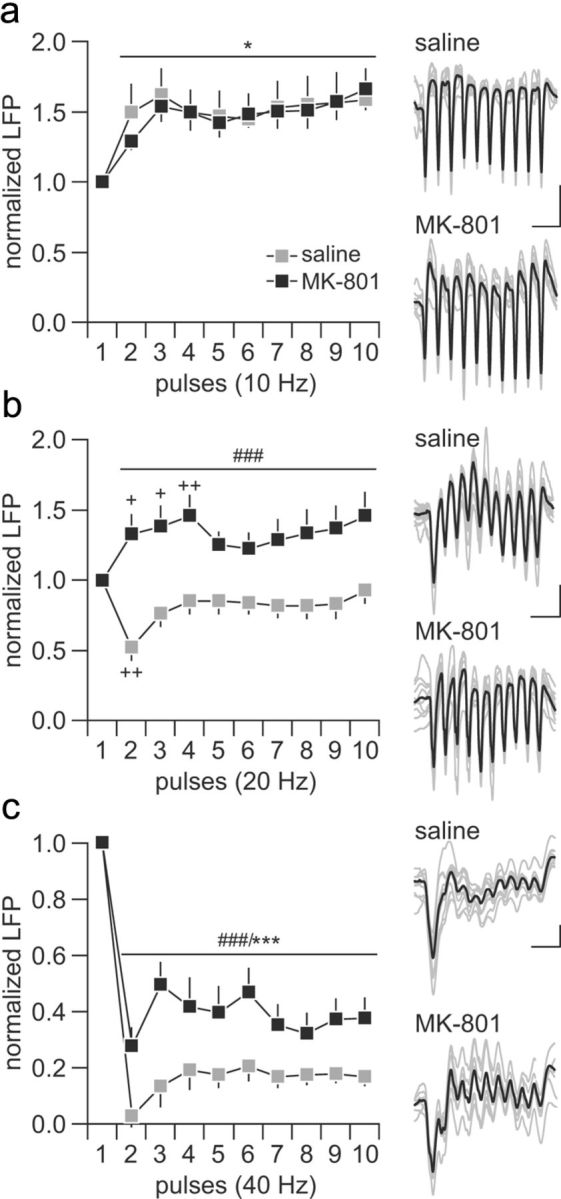
Disruption of ventral hippocampal-induced frequency-dependent prefrontal LFP inhibition by periadolescent MK-801 treatment. a, Saline-treated (n = 12) and MK-801-treated (n = 10) rats exhibited similar degrees of LFP facilitation in the PFC in response to hippocampal train stimulation at 10 Hz (main effect of pulse number, F(9,200) = 2.47, *p < 0.05). b, At 20 Hz, a transient LFP inhibition (++p < 0.005 vs first pulse, least significant difference post hoc test after significant one-way ANOVA) was observed in the PFC of saline-treated rats, whereas recordings from the MK-801-treated group revealed a facilitation (+p < 0.05, ++p < 0.005 vs first pulse, least significant difference post hoc test after significant one-way ANOVA) of the prefrontal LFP response (main effect of treatment, F(1,200) = 12.51, ###p < 0.0005, two-way ANOVA). c, Both saline- and MK-801-treated rats exhibited marked prefrontal LFP depression in response to hippocampal stimulation at 40 Hz. However, a significant attenuation of the magnitude of the 40 Hz-induced LFP inhibition was observed in the MK-801-treated group (main effect of pulse number, F(9,200) = 30.57, ***p < 0.0005; main effect of treatment, F(1,200) = 57.7984, ###p < 0.0005, two-way ANOVA). Insets are example traces of ventral hippocampal-induced LFP facilitation and suppression recorded in the PFC illustrating the effects shown in a–c. Calibration: a, 5 mV, 200 ms; b, 5 mV, 100 ms; c, 3 mV, 50 ms.
To determine whether the frequency-dependent disruption of prefrontal inhibition observed in the periadolescent-treated group is age dependent, the impact of MK-801 exposure during adulthood (i.e., P75–P80) was assessed following the same experimental design described above. As for the periadolescent cohort, all recordings were conducted within 3–5 weeks (i.e., P105–P125 range) from the last saline or MK-801 injection (Fig. 3a). Similar to the periadolescent-treated group, the characteristic hippocampal 10 Hz-induced facilitation of the prefrontal response was indistinguishable among saline- and MK-801-treated animals (Fig. 3b). Interestingly, MK-801 treatment in adulthood failed to alter the pattern of 20 and 40 Hz-induced field potential inhibition in the PFC (Fig. 3c). Together, these results indicate that MK-801-induced disruption of the ventral hippocampal-induced frequency-dependent prefrontal inhibition is age dependent. Notably, both adolescent- and adult-exposed saline groups exhibited similar pattern of prefrontal responses at 20 and 40 Hz despite the differences in testing ages (P65–P85 vs P105–P125; main effect of group, p = 0.9, two-way ANOVA).
Figure 3.

Summary of the effects of MK-801 treatment administered during adulthood. a, Diagram illustrating the experimental design. Five noncontingent injections of saline (n = 8) or MK-801 (n = 8) were performed in adult rats, and changes in prefrontal LFP response to ventral hippocampal train stimulation were assessed within the P105–P125 age period. Bottom shows the location of all recording and stimulation sites determined by means of histological analyses from Nissl-stained sections. b, Similar to the adolescent-treated groups, vehicle and MK-801 treatment in adulthood did not alter the characteristic prefrontal LFP facilitation induced by hippocampal stimulation at 10 Hz (main effect of pulse number, F(9,140) = 5.12, ***p < 0.0005, two-way ANOVA). c, At 20 Hz, a transient LFP inhibition was observed in the PFC of both saline- and MK-801-treated rats (main effect of pulse number, F(9,150) = 2.70, *p < 0.05, two-way ANOVA). d, Similarly, the magnitude of prefrontal LFP inhibition observed after ventral hippocampal train stimulation at 40 Hz remained intact when MK-801 exposure occurred in adulthood (main effect of pulse number, F(9,150) = 120.74, ***p < 0.0005, two-way ANOVA). PD, Postnatal day.
We next asked the question of whether the prefrontal disruptions observed after periadolescent MK-801 treatment could be attributed to a developmental impairment of the normal frequency-dependent regulation of synaptic transmission in the PFC by the ventral hippocampus. Toward this goal, the pattern of prefrontal response to ventral hippocampal stimulation at 10, 20, and 40 Hz was compared across three developmentally distinct age groups of naive rats: P30–P40, P45–P55, and P60–P80. We observed no differences in the 10 Hz-induced prefrontal facilitation among the three age groups (Fig. 4a). However, the characteristic 20 Hz-induced transient attenuation of the evoked field potential observed in adult animals became apparent only in the P45–P55 age group (Fig. 4b). In the P30–P40 PFC, a 20 Hz-induced sustained facilitation of the evoked response was found instead (Fig. 4b), resembling that observed in the adult PFC of periadolescent MK-801-exposed rats (for comparison, see Fig. 2d). Similarly, the degree of prefrontal field potential suppression observed at 40 Hz in the P30–P40 animals was significantly less pronounced when compared with the response observed in the P45–P55 and P60–P80 age groups (Fig. 4c). Once again, such attenuated 40 Hz-induced prefrontal inhibition in the P30–P40 group resembles that recorded in the PFC of adult rats with a periadolescent history of MK-801 treatment. Together, these results indicate that the periadolescent MK-801-induced prefrontal disinhibition could be attributable to a developmental disruption of the normal acquisition of frequency-dependent inhibitory mechanism that occurs after P40 (Fig. 4d).
Figure 4.

Developmental regulation of ventral hippocampal-induced facilitation and suppression of LFP transmission in the PFC. a, The pattern of prefrontal LFP facilitation to ventral hippocampal stimulation at 10 Hz were indistinguishable among rats from P30–P40 (n = 10), P45–P55 (n = 8), and P60–P80 (n = 6) (main effect of pulse number, F(9,210) = 2.55, *p < 0.05, two-way ANOVA). b, At 20 Hz, a facilitation of LFP response was observed only in the P30–P40 age group PFC. Note that such facilitation resembles that observed in the adult PFC from adolescent MK-801-exposed rats (see Fig. 2b). In contrast, a characteristic 20 Hz-induced transient LFP attenuation was observed in the PFC of P45–P55 and P60–P80 age groups (main effect of age, F(2,210) = 23.49, ###p < 0.0005, two-way ANOVA). c, Similarly, the P30–P40 PFC exhibited a marked attenuation of the 40 Hz-induced LFP suppression, whereas the magnitude of prefrontal inhibition obtained in the P45–P55 age group was identical to that from the adult PFC (main effect of pulse number, F(9,210) = 20.88, ***p < 0.0005; main effect of age, F(2,210) = 25.61, ###p < 0.0005, two-way ANOVA). d, Bar graph summarizing the magnitude of hippocampal-induced LFP facilitation and inhibition in the PFC across age groups. The average values were calculated from pulses 2 to 10 as shown in a–c (i.e., marked in gray). Data from adolescent saline- and MK-801-treated animals (from Fig. 2) were included for comparison. Both the degrees of LFP facilitation at 20 Hz and LFP attenuation at 40 Hz observed in the periadolescent MK-801-treated group resemble those from the P30–P40 age group (#p < 0.02 vs P45–P55, vs P60–P80, vs saline, least significant difference post hoc test after significant one-way ANOVA). e, Location of all recording and stimulation sites determined by means of histological analyses from Nissl-stained sections. PD, Postnatal day.
It is well known that GABAergic interneurons play a crucial role in mediating the enhanced prefrontal inhibitory response observed during the normal periadolescent transition period (Tseng and O'Donnell, 2007a, 2007b). To test this hypothesis, we first examined whether the effects of periadolescent MK-801 treatment could be acutely reproduced through a disruption of local prefrontal GABAergic transmission via infusion of the GABAA receptor antagonist picrotoxin into the PFC of adult naive rats (P65–P85). We found that, although the pattern of hippocampal 10 Hz-induced prefrontal facilitation remained unaffected (Fig. 5a), a shift from the distinct 20 Hz-induced transient attenuation to a sustained facilitation of the evoked field potential response was observed in the PFC after local infusion of picrotoxin (Fig. 5b). Similarly, the characteristic hippocampal 40 Hz-induced suppression in the prefrontal field potential response was markedly attenuated by picrotoxin (Fig. 5c). In summary, these results indicate that the distinctive hippocampal-induced frequency-dependent field potential inhibition observed in the normal adult PFC is mediated by local prefrontal GABAergic transmission. Furthermore, a functional impairment of this latter is sufficient to elicit a PFC state resembling that induced by periadolescent MK-801 exposure (Fig. 5d).
Figure 5.

Prefrontal disinhibition induced by local administration of the GABAA antagonist picrotoxin. a, The magnitude of ventral hippocampal-induced prefrontal LFP facilitation at 10 Hz was not affected by local microinjection of picrotoxin (50 μm) or aCSF (1 μl/10 min; n = 8 per group; main effect of pulse number, F(9,120) = 9.99, ***p < 0.0005, two-way ANOVA). b, In contrast, picrotoxin shifted the characteristic 20 Hz-induced transient LFP attenuation to a sustained facilitation (main effect of treatment, F(1,140) = 59.98, ###p < 0.0005, two-way ANOVA). c, Similarly, the profound suppression of LFP responses observed in the PFC during the 40 Hz train stimulation of the ventral hippocampus was markedly attenuated after local prefrontal administration of picrotoxin (main effect of pulse number, F(9,140) = 32.39, ***p < 0.0005; main effect of treatment, F(1,140) = 96.58, ###p < 0.0005, two-way ANOVA). d, Summary of the results obtained with picrotoxin (average values calculated from pulses 2 to 10 shown in a–c) and those recorded in the PFC of adult animals exposed to saline or MK-801 during periadolescence. Note that the magnitude of LFP facilitation at 20 Hz and the level of LFP attenuation at 40 Hz observed in the periadolescent MK-801-treated group resembles those induced by local picrotoxin in naive animals (##p < 0.005 vs aCSF; ###p < 0.0005 vs saline-treated animals, least significant difference post hoc test after significant one-way ANOVA). e, Diagram showing the location of the recording and stimulating electrodes for this cohort of experiments.
The above findings led us to further hypothesize that periadolescent MK-801 treatment could exert a developmental dysregulation of local prefrontal GABAergic transmission. To test this, we examined the impact of the GABAAα1-receptor-positive allosteric modulator Indiplon in reversing the abnormal disinhibited state observed in the PFC of animals treated with MK-801 during periadolescence. We found that local prefrontal infusion of Indiplon did not alter the pattern of prefrontal facilitation induced by hippocampal 10 Hz stimulation (Fig. 6a). However, the abnormal 20 Hz-induced facilitation of the evoked field potential response observed in the adult PFC of periadolescent MK-801-treated rats was no longer apparent after local administration of Indiplon (Fig. 6b). Instead, a shift to the typical 20 Hz-induced transient attenuation of the evoked response was obtained, which resembles the pattern observed in saline controls (for comparison, see Fig. 2b). Similarly, local prefrontal application of Indiplon rescued the abnormally attenuated 40 Hz-induced suppression of the evoked field potential response observed in MK-801-exposed rats to saline control levels (Fig. 6c). Together, these results indicate that an upregulation of local prefrontal GABAA receptor-mediated transmission is sufficient to normalize the enduring PFC disinhibitory state induced by periadolescent MK-801 exposure (Fig. 6d).
Figure 6.

Reversal actions of the GABAAα1-positive allosteric modulator Indiplon on MK-801 (MK)-induced prefrontal LFP disinhibition. a, At 10 Hz, local prefrontal infusion of Indiplon did not change the pattern of ventral hippocampal stimulation-induced facilitation of LFP transmission in the PFC of adolescent MK-801-treated rats (main effect of pulse number, F(9,120) = 2.11, *p < 0.05, two-way ANOVA; n = 7 per treatment group). b, In contrast, the abnormal prefrontal LFP facilitation observed in the periadolescent MK-801-treated group in response to 20 Hz stimulation was normalized after local administration of Indiplon (main effect of treatment, F(1,120) = 81.87, ###p < 0.0005, two-way ANOVA; aCSF, n = 6; Indiplon, n = 8). c, Indiplon also normalized the magnitude of 40 Hz-induced suppression of prefrontal LFP transmission in the periadolescent MK-801-treated group (main effect of pulse number, F(9,120) = 35.59, ***p < 0.0005; main effect of treatment, F(1,120) = 64.95, ###p < 0.0005, two-way ANOVA; aCSF, n = 6; Indiplon, n = 8). d, Summary of the reversal effects of Indiplon recorded in the PFC of adult animals exposed to MK-801 during periadolescence. Note that the abnormal facilitation at 20 Hz and the reduced LFP inhibition at 40 Hz were no longer observed after local prefrontal infusion of Indiplon (##p < 0.005 vs aCSF or periadolescent MK-801-treated animals, least significant difference post hoc test after significant one-way ANOVA). e, Diagram showing the location of the recording and stimulating electrodes for this cohort of experiments.
Discussion
Transient exposure to the NMDA receptor antagonist MK-801 during the periadolescent transition period resulted in a long-lasting frequency-dependent disinhibition of prefrontal processing of ventral hippocampal inputs in adulthood. Such susceptibility to MK-801-induced enduring PFC disruption was not observed in adult-exposed animals, further supporting the idea that periadolescence is a sensitive period for the functional maturation of prefrontal inhibitory control. Our data also indicate that the prefrontal disinhibitory state induced by periadolescent MK-801 treatment could be associated with a developmental impairment in the gain of local GABAergic function in response to 20 and 40 Hz inputs that typically emerges during the normal periadolescent transition period (Fig. 7). Accordingly, the pattern of prefrontal disinhibition observed in the PFC of P30–P40 animals is comparable with that recorded in the adult PFC after local application of the GABAA antagonist picrotoxin. Notably, acute local prefrontal administration of the GABAAα1-positive allosteric modulator Indiplon normalized the disinhibitory state observed in the adult PFC of periadolescent MK-801-treated rats. In summary, these results are suggestive of a downregulation of prefrontal GABAA-mediated transmission contributing to the enduring PFC disinhibition associated with periadolescent MK-801 exposure.
Figure 7.

Summary of the prefrontal field potential response to ventral hippocampal train stimulation at 10, 20, and 40 Hz across the different experimental groups. The percentage change of prefrontal LFP response was calculated from the mean amplitude value of pulses 2 to 10 relative to pulse 1 as shown in Figures 4–6. The control group comprised saline-treated and adult naive animals. The MK-801 group includes all periadolescent MK-801-treated rats from Figures 2 and 6 (aCSF group). Note that the characteristic facilitation of prefrontal response to 10 Hz stimulation is not developmental regulated and remained unchanged after adolescent MK-801 exposure or single acute administration of picrotoxin into the PFC (p = 0.998, one-way ANOVA). In contrast, PFC responses to 20 and 40 Hz train stimulation are developmentally regulated and sensitive to periadolescent MK-801 treatment and local prefrontal GABAA receptor blockade. In fact, MK-801-treated animals exhibited the same pattern of prefrontal responses undistinguishable from the developmentally immature P30–P40 PFC and those recorded in the presence of local GABAA receptor blockade. Importantly, single microinfusion of the GABAAα1-receptor-positive allosteric modulator Indiplon into the PFC was sufficient to acutely restore the 20 and 40 Hz-induced inhibitory prefrontal field potential response to control levels (#p < 0.02, ##p < 0.005 vs P30–P40, periadolescent, MK-801, or picrotoxin, least significant difference post hoc test after significant one-way ANOVA: F(4,55) = 8.94, p < 0.00005 for the 20 Hz response; F(4,55) = 5.96, p < 0.0005 for the 40 Hz response). PD, Postnatal day.
It is well documented that acute administration of noncompetitive NMDA receptor antagonists, such as MK-801, ketamine, or phencyclidine, is sufficient to produce a potent psychotomimetic state that is clinically indistinguishable from schizophrenia (Javitt and Zukin, 1991; Krystal et al., 1994). The neurobiology underlying such an effect appears to be mediated by an enhancement of PFC metabolic activity, as revealed by neuroimaging studies conducted in healthy volunteers exhibiting psychotic symptoms and cognitive abnormalities in response to subanesthetic doses of ketamine (Breier et al., 1997; Holcomb et al., 2001, 2005). Studies conducted in animal models further indicate that the augmented PFC output induced by acute psychotomimetic doses of NMDA antagonists (Moghaddam and Adams, 1998; Jackson et al., 2004; Labonte et al., 2009; Kiss et al., 2011; Wood et al., 2012) can be triggered by a disinhibitory mechanism mediated by a functional impairment of GABAergic interneuronal activity in cortical circuits (Grunze et al., 1996; Jackson et al., 2004; Zhang et al., 2008; Wang and Gao, 2012). Our present study expands on these findings by showing that an enduring state of prefrontal disinhibition can emerge if repeated MK-801 exposure occurs during adolescence. Importantly, such MK-801-induced disruption is age dependent because no apparent changes in PFC responses were observed in adult-treated animals.
The enduring prefrontal disinhibition resulting from periadolescent MK-801 treatment is characterized by a selective attenuation of the distinctive ventral hippocampal-induced 20 Hz (beta)- and 40 Hz (gamma)-dependent field potential inhibition observed in the normal adult PFC. A similar pattern of frequency-dependent response was found in the PFC of P30–P40 naive animals. Interestingly, acute pharmacological blockade of local prefrontal GABAA receptors was sufficient to produce a state of prefrontal disinhibition in adult naive animals mirroring that observed in the P30–P40 age group and in rats with a periadolescent history of MK-801. These results reveal that a developmental recruitment of local prefrontal GABAergic transmission is required for sustaining high-frequency-dependent inhibition. This gain of prefrontal inhibitory function that emerges after P40 is lacking in periadolescent MK-801-treated animals. Furthermore, the periadolescent MK-801-induced prefrontal deficiency can be pharmacologically rescued by increasing the gain of local GABAergic transmission with the GABAAα1-receptor-positive allosteric modulator Indiplon (Fig. 7). Thus, despite that prefrontal GABAergic transmission appears to be impaired after periadolescent MK-801 treatment, it remains functionally capable of producing prefrontal inhibition at beta and gamma frequencies upon increased levels of GABAA receptor activation.
A downregulation of GABAergic interneuron markers in the PFC has been repeatedly observed after prenatal and neonatal (younger than P7) exposure to NMDA receptor antagonists (Abekawa et al., 2007; Wang et al., 2008; Coleman et al., 2009; Turner et al., 2010; Abekawa et al., 2011). This is not surprising because NMDA-receptor-mediated signaling is required for neuronal migration (Hirai et al., 1999) and the formation of corticolimbic GABAergic circuits (Belforte et al., 2010), which are yet to be formed at these early stages of development (Erickson and Lewis, 2002). Among the different populations of GABAergic interneurons in the PFC, those containing the calcium-binding protein parvalbumin are highly susceptible to NMDA receptor antagonists (Xi et al., 2009; Abekawa et al., 2011), which reduce parvalbumin levels through an oxidative stress-mediated mechanism (Kinney et al., 2006; Behrens et al., 2007). More importantly, a functional impairment of parvalbumin-positive interneurons has been associated with the onset of prefrontal disinhibition in animal models of psychiatric disorders (Tseng et al., 2009; O'Donnell, 2011; Nakazawa et al., 2012). Thus, it remains to be determined whether a developmental impairment of parvalbumin-positive interneuron function underlies the functional attenuation of GABAergic transmission observed in the PFC when MK-801 is given during periadolescence.
In addition to the ventral hippocampus (Swanson, 1981; Jay and Witter, 1991; Cenquizca and Swanson, 2007; Hoover and Vertes, 2007), the PFC also receives strong glutamatergic afferents from the amygdala (Condé et al., 1995; Mcdonald, 1996). Although the ventral hippocampal–PFC pathway is implicated in working memory and attention as well as in suppressing impulsive behavior (Floresco et al., 1997; Chudasama et al., 2012), interactions between the PFC and the basolateral amygdala are crucial for integrating emotionally salient information (Ishikawa and Nakamura, 2003; Phillips et al., 2003). Thus, impaired maturation of local prefrontal network processing of inputs originated from these regions is expected to compromise a variety of PFC-dependent cognitive abilities and reduced impulse control. Future studies will determine the behavioral consequences of periadolescent MK-801 exposure and whether the abnormal PFC response to high-frequency glutamatergic afferent stimulation observed in MK-801-treated animals is input specific.
In summary, the results of the present study indicate that the functional maturation of GABAergic circuits in the PFC is highly susceptible to disruptions of NMDA receptor signaling during adolescence. Here, we have shown for the first time that NMDA receptor blockade during the periadolescent transition period is sufficient to produce a developmental impairment of the normative gain of local prefrontal GABAA-mediated inhibition that endures through adulthood. Such a mechanistic link is not restricted to the use of NMDA receptor antagonists because a variety of genetic and environmental factors known to play a critical role in brain development are also capable of causing NMDA receptor dysfunction and subsequent interneuronal deficits within the frontal cortical circuits (Do et al., 2009; Kantrowitz and Javitt, 2010). As in other cortical regions, proper functioning of GABAergic interneurons is critical for fine-tuning prefrontal output activity (Rao et al., 2000; Szabadics et al., 2001; Markram et al., 2004) for supporting cognitive functions, including working memory, decision making, and impulse control (Goldman-Rakic, 1995, 1999). Thus, impaired maturation of prefrontal GABAergic transmission during the periadolescent transition could result in long-term impairments in PFC cognitive functions resembling those observed after acute intra-PFC injection of GABAA receptor antagonists in animal models (Sawaguchi et al., 1988, 1989; Enomoto et al., 2011). Finally, the reversal effect of Indiplon suggests that targeting GABAAα1 receptor with positive allosteric modulators could be a novel therapeutic avenue for reversing the cognitive deficits associated with PFC disinhibition in schizophrenia and related psychiatric disorders.
Footnotes
This research was supported by Rosalind Franklin University and National Institutes of Health Grant R01-MH086507 (K.Y.T.). We thank Dr. Adriana Caballero for helpful comments and experimental insights.
References
- Abekawa T, Ito K, Nakagawa S, Koyama T. Prenatal exposure to an NMDA receptor antagonist, MK-801 reduces density of parvalbumin-immunoreactive GABAergic neurons in the medial prefrontal cortex and enhances phencyclidine-induced hyperlocomotion but not behavioral sensitization to methamphetamine in postpubertal rats. Psychopharmacology (Berl) 2007;192:303–316. doi: 10.1007/s00213-007-0729-8. [DOI] [PubMed] [Google Scholar]
- Abekawa T, Ito K, Nakato Y, Koyama T. Developmental GABAergic deficit enhances methamphetamine-induced apoptosis. Psychopharmacology (Berl) 2011;215:413–427. doi: 10.1007/s00213-011-2269-5. [DOI] [PubMed] [Google Scholar]
- Andrzejewski ME, Schochet TL, Feit EC, Harris R, McKee BL, Kelley AE. A comparison of adult and adolescent rat behavior in operant learning, extinction, and behavioral inhibition paradigms. Behav Neurosci. 2011;125:93–105. doi: 10.1037/a0022038. [DOI] [PubMed] [Google Scholar]
- Aultman JM, Moghaddam B. Distinct contributions of glutamate and dopamine receptors to temporal aspects of rodent working memory using a clinically relevant task. Psychopharmacology (Berl) 2001;153:353–364. doi: 10.1007/s002130000590. [DOI] [PubMed] [Google Scholar]
- Behrens MM, Ali SS, Dao DN, Lucero J, Shekhtman G, Quick KL, Dugan LL. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science. 2007;318:1645–1647. doi: 10.1126/science.1148045. [DOI] [PubMed] [Google Scholar]
- Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, Quinlan EM, Nakazawa K. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci. 2010;13:76–83. doi: 10.1038/nn.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breier A, Malhotra AK, Pinals DA, Weisenfeld NI, Pickar D. Association of ketamine-induced psychosis with focal activation of the prefrontal cortex in healthy volunteers. Am J Psychiatry. 1997;154:805–811. doi: 10.1176/ajp.154.6.805. [DOI] [PubMed] [Google Scholar]
- Cenquizca LA, Swanson LW. Spatial organization of direct hippocampal field CA1 axonal projections to the rest of the cerebral cortex. Brain Res Rev. 2007;56:1–26. doi: 10.1016/j.brainresrev.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudasama Y, Doobay VM, Liu Y. Hippocampal-prefrontal cortical circuit mediates inhibitory response control in the rat. J Neurosci. 2012;32:10915–10924. doi: 10.1523/JNEUROSCI.1463-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman LG, Jr, Jarskog LF, Moy SS, Crews FT. Deficits in adult prefrontal cortex neurons and behavior following early post-natal NMDA antagonist treatment. Pharmacol Biochem Behav. 2009;93:322–330. doi: 10.1016/j.pbb.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condé F, Maire-Lepoivre E, Audinat E, Crepel F. Afferent connections of the medial frontal cortex of the rat. II. Cortical and subcortical afferents. J Comp Neurol. 1995;352:567–593. doi: 10.1002/cne.903520407. [DOI] [PubMed] [Google Scholar]
- Crone EA, Wendelken C, Donohue S, van Leijenhorst L, Bunge SA. Neurocognitive development of the ability to manipulate information in working memory. Proc Natl Acad Sci U S A. 2006;103:9315–9320. doi: 10.1073/pnas.0510088103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do KQ, Cabungcal JH, Frank A, Steullet P, Cuenod M. Redox dysregulation, neurodevelopment, and schizophrenia. Curr Opin Neurobiol. 2009;19:220–230. doi: 10.1016/j.conb.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Dumontheil I, Burgess PW, Blakemore SJ. Development of rostral prefrontal cortex and cognitive and behavioural disorders. Dev Med Child Neurol. 2008;50:168–181. doi: 10.1111/j.1469-8749.2008.02026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto T, Tse MT, Floresco SB. Reducing prefrontal gamma-aminobutyric acid activity induces cognitive, behavioral, and dopaminergic abnormalities that resemble schizophrenia. Biol Psychiatry. 2011;69:432–441. doi: 10.1016/j.biopsych.2010.09.038. [DOI] [PubMed] [Google Scholar]
- Erickson SL, Lewis DA. Postnatal development of parvalbumin- and GABA transporter-immunoreactive axon terminals in monkey prefrontal cortex. J Comp Neurol. 2002;448:186–202. doi: 10.1002/cne.10249. [DOI] [PubMed] [Google Scholar]
- Ernst M, Romeo RD, Andersen SL. Neurobiology of the development of motivated behaviors in adolescence: a window into a neural systems model. Pharmacol Biochem Behav. 2009;93:199–211. doi: 10.1016/j.pbb.2008.12.013. [DOI] [PubMed] [Google Scholar]
- Floresco SB, Seamans JK, Phillips AG. Selective roles for hippocampal, prefrontal cortical, and ventral striatal circuits in radial-arm maze tasks with or without a delay. J Neurosci. 1997;17:1880–1890. doi: 10.1523/JNEUROSCI.17-05-01880.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogtay N, Vyas NS, Testa R, Wood SJ, Pantelis C. Age of onset of schizophrenia: perspectives from structural neuroimaging studies. Schizophr Bull. 2011;37:504–513. doi: 10.1093/schbul/sbr030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman-Rakic PS. Cellular basis of working memory. Neuron. 1995;14:477–485. doi: 10.1016/0896-6273(95)90304-6. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS. The physiological approach: functional architecture of working memory and disordered cognition in schizophrenia. Biol Psychiatry. 1999;46:650–661. doi: 10.1016/s0006-3223(99)00130-4. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Burgos G, Fish KN, Lewis DA. GABA neuron alterations, cortical circuit dysfunction and cognitive deficits in schizophrenia. Neural Plast. 2011;2011:723184. doi: 10.1155/2011/723184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunze HC, Rainnie DG, Hasselmo ME, Barkai E, Hearn EF, McCarley RW, Greene RW. NMDA-dependent modulation of CA1 local circuit inhibition. J Neurosci. 1996;16:2034–2043. doi: 10.1523/JNEUROSCI.16-06-02034.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai K, Yoshioka H, Kihara M, Hasegawa K, Sakamoto T, Sawada T, Fushiki S. Inhibiting neuronal migration by blocking NMDA receptors in the embryonic rat cerebral cortex: a tissue culture study. Brain Res Dev Brain Res. 1999;114:63–67. doi: 10.1016/s0165-3806(99)00019-x. [DOI] [PubMed] [Google Scholar]
- Holcomb HH, Lahti AC, Medoff DR, Weiler M, Tamminga CA. Sequential regional cerebral blood flow brain scans using PET with H2(15)O demonstrate ketamine actions in CNS dynamically. Neuropsychopharmacology. 2001;25:165–172. doi: 10.1016/S0893-133X(01)00229-9. [DOI] [PubMed] [Google Scholar]
- Holcomb HH, Lahti AC, Medoff DR, Cullen T, Tamminga CA. Effects of noncompetitive NMDA receptor blockade on anterior cingulate cerebral blood flow in volunteers with schizophrenia. Neuropsychopharmacology. 2005;30:2275–2282. doi: 10.1038/sj.npp.1300824. [DOI] [PubMed] [Google Scholar]
- Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27:11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover WB, Vertes RP. Anatomical analysis of afferent projections to the medial prefrontal cortex in the rat. Brain Struct Funct. 2007;212:149–179. doi: 10.1007/s00429-007-0150-4. [DOI] [PubMed] [Google Scholar]
- Ishikawa A, Nakamura S. Convergence and interaction of hippocampal and amygdalar projections within the prefrontal cortex in the rat. J Neurosci. 2003;23:9987–9995. doi: 10.1523/JNEUROSCI.23-31-09987.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson ME, Homayoun H, Moghaddam B. NMDA receptor hypofunction produces concomitant firing rate potentiation and burst activity reduction in the prefrontal cortex. Proc Natl Acad Sci U S A. 2004;101:8467–8472. doi: 10.1073/pnas.0308455101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–1308. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- Jay TM, Witter MP. Distribution of hippocampal CA1 and subicular efferents in the prefrontal cortex of the rat studied by means of anterograde transport of Phaseolus vulgaris-leucoagglutinin. J Comp Neurol. 1991;313:574–586. doi: 10.1002/cne.903130404. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Roth RH. The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology. 1999;20:201–225. doi: 10.1016/S0893-133X(98)00060-8. [DOI] [PubMed] [Google Scholar]
- Kantrowitz JT, Javitt DC. N-methyl-d-aspartate (NMDA) receptor dysfunction or dysregulation: the final common pathway on the road to schizophrenia? Brain Res Bull. 2010;83:108–121. doi: 10.1016/j.brainresbull.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, Amminger GP, Aguilar-Gaxiola S, Alonso J, Lee S, Ustün TB. Age of onset of mental disorders: a review of recent literature. Curr Opin Psychiatry. 2007;20:359–364. doi: 10.1097/YCO.0b013e32816ebc8c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney JW, Davis CN, Tabarean I, Conti B, Bartfai T, Behrens MM. A specific role for NR2A-containing NMDA receptors in the maintenance of parvalbumin and GAD67 immunoreactivity in cultured interneurons. J Neurosci. 2006;26:1604–1615. doi: 10.1523/JNEUROSCI.4722-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss T, Hoffmann WE, Scott L, Kawabe TT, Milici AJ, Nilsen EA, Hajós M. Role of thalamic projection in NMDA receptor-induced disruption of cortical slow oscillation and short-term plasticity. Front Psychiatry. 2011;2:14. doi: 10.3389/fpsyt.2011.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koss WA, Franklin AD, Juraska JM. Delayed alternation in adolescent and adult male and female rats. Dev Psychobiol. 2011;53:724–731. doi: 10.1002/dev.20543. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Jr, Charney DS. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Anand A, Moghaddam B. Effects of NMDA receptor antagonists: implications for the pathophysiology of schizophrenia. Arch Gen Psychiatry. 2002;59:663–664. doi: 10.1001/archpsyc.59.7.663. [DOI] [PubMed] [Google Scholar]
- Labonte B, Bambico FR, Gobbi G. Potentiation of excitatory serotonergic responses by MK-801 in the medial prefrontal cortex. Naunyn Schmiedebergs Arch Pharmacol. 2009;380:383–397. doi: 10.1007/s00210-009-0446-4. [DOI] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci. 2004;5:793–807. doi: 10.1038/nrn1519. [DOI] [PubMed] [Google Scholar]
- Mcdonald AJ. Glutamate and aspartate immunoreactive neurons of the rat basolateral amygdala: colocalization of excitatory amino acids and projections to the limbic circuit. J Comp Neurol. 1996;365:367–379. doi: 10.1002/(SICI)1096-9861(19960212)365:3<367::AID-CNE3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Moghaddam B, Adams BW. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science. 1998;281:1349–1352. doi: 10.1126/science.281.5381.1349. [DOI] [PubMed] [Google Scholar]
- Nakazawa K, Zsiros V, Jiang Z, Nakao K, Kolata S, Zhang S, Belforte JE. GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology. 2012;62:1574–1583. doi: 10.1016/j.neuropharm.2011.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Institute of Mental Health–National Advisory Mental Health Council. Transformative neurodevelopmental research in mental illness. Report of the National Advisory Mental Health Council's Workshop. 2008. http://www.nimh.nih.gov/about/advisory-boards-and-groups/namhc/neurodevelopment_workgroup_report.pdf.
- O'Donnell P. Adolescent onset of cortical disinhibition in schizophrenia: insights from animal models. Schizophr Bull. 2011;37:484–492. doi: 10.1093/schbul/sbr028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paus T, Keshavan M, Giedd JN. Why do many psychiatric disorders emerge during adolescence? Nat Rev Neurosci. 2008;9:947–957. doi: 10.1038/nrn2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. New York: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Phillips ML, Drevets WC, Rauch SL, Lane R. Neurobiology of emotion perception. I. The neural basis of normal emotion perception. Biol Psychiatry. 2003;54:504–514. doi: 10.1016/s0006-3223(03)00168-9. [DOI] [PubMed] [Google Scholar]
- Powell SB, Sejnowski TJ, Behrens MM. Behavioral and neurochemical consequences of cortical oxidative stress on parvalbumin-interneuron maturation in rodent models of schizophrenia. Neuropharmacology. 2012;62:1322–1331. doi: 10.1016/j.neuropharm.2011.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SG, Williams GV, Goldman-Rakic PS. Destruction and creation of spatial tuning by disinhibition: GABAA blockade of prefrontal cortical neurons engaged by working memory. J Neurosci. 2000;20:485–494. doi: 10.1523/JNEUROSCI.20-01-00485.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rujescu D, Bender A, Keck M, Hartmann AM, Ohl F, Raeder H, Giegling I, Genius J, McCarley RW, Möller HJ, Grunze H. A pharmacological model for psychosis based on N-methyl-d-aspartate receptor hypofunction: molecular, cellular, functional and behavioral abnormalities. Biol Psychiatry. 2006;59:721–729. doi: 10.1016/j.biopsych.2005.08.029. [DOI] [PubMed] [Google Scholar]
- Sawaguchi T, Matsumura M, Kubota K. Delayed response deficit in monkeys by locally disturbed prefrontal neuronal activity by bicuculline. Behav Brain Res. 1988;31:193–198. doi: 10.1016/0166-4328(88)90023-x. [DOI] [PubMed] [Google Scholar]
- Sawaguchi T, Matsumura M, Kubota K. Delayed response deficits produced by local injection of bicuculline into the dorsolateral prefrontal cortex in Japanese macaque monkeys. Exp Brain Res. 1989;75:457–469. doi: 10.1007/BF00249897. [DOI] [PubMed] [Google Scholar]
- Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459:698–702. doi: 10.1038/nature07991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani MR, Moghaddam B. Transient N-methyl-d-aspartate receptor blockade in early development causes lasting cognitive deficits relevant to schizophrenia. Biol Psychiatry. 2005;57:433–436. doi: 10.1016/j.biopsych.2004.11.031. [DOI] [PubMed] [Google Scholar]
- Swanson LW. A direct projection from Ammon's horn to prefrontal cortex in the rat. Brain Res. 1981;217:150–154. doi: 10.1016/0006-8993(81)90192-x. [DOI] [PubMed] [Google Scholar]
- Szabadics J, Lorincz A, Tamás G. Beta and gamma frequency synchronization by dendritic GABAergic synapses and gap junctions in a network of cortical interneurons. J Neurosci. 2001;21:5824–5831. doi: 10.1523/JNEUROSCI.21-15-05824.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, O'Donnell P. Dopamine modulation of prefrontal cortical interneurons changes during adolescence. Cereb Cortex. 2007a;17:1235–1240. doi: 10.1093/cercor/bhl034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, O'Donnell P. D2 dopamine receptors recruit a GABA component for their attenuation of excitatory synaptic transmission in the adult rat prefrontal cortex. Synapse. 2007b;61:843–850. doi: 10.1002/syn.20432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, Kasanetz F, Kargieman L, Riquelme LA, Murer MG. Cortical slow oscillatory activity is reflected in the membrane potential and spike trains of striatal neurons in rats with chronic nigrostriatal lesions. J Neurosci. 2001;21:6430–6439. doi: 10.1523/JNEUROSCI.21-16-06430.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, Chambers RA, Lipska BK. The neonatal ventral hippocampal lesion as a heuristic neurodevelopmental model of schizophrenia. Behav Brain Res. 2009;204:295–305. doi: 10.1016/j.bbr.2008.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, Caballero A, Dec A, Cass DK, Simak N, Sunu E, Park MJ, Blume SR, Sammut S, Park DJ, West AR. Inhibition of striatal soluble guanylyl cyclase-cGMP signaling reverses basal ganglia dysfunction and akinesia in experimental parkinsonism. PLoS One. 2011;6:e27187. doi: 10.1371/journal.pone.0027187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner CP, DeBenedetto D, Ware E, Stowe R, Lee A, Swanson J, Walburg C, Lambert A, Lyle M, Desai P, Liu C. Postnatal exposure to MK801 induces selective changes in GAD67 or parvalbumin. Exp Brain Res. 2010;201:479–488. doi: 10.1007/s00221-009-2059-z. [DOI] [PubMed] [Google Scholar]
- Uhlhaas PJ, Singer W. Neural synchrony in brain disorders: relevance for cognitive dysfunctions and pathophysiology. Neuron. 2006;52:155–168. doi: 10.1016/j.neuron.2006.09.020. [DOI] [PubMed] [Google Scholar]
- Uhlhaas PJ, Singer W. The development of neural synchrony and large-scale cortical networks during adolescence: relevance for the pathophysiology of schizophrenia and neurodevelopmental hypothesis. Schizophr Bull. 2011;37:514–523. doi: 10.1093/schbul/sbr034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlhaas PJ, Linden DE, Singer W, Haenschel C, Lindner M, Maurer K, Rodriguez E. Dysfunctional long-range coordination of neural activity during Gestalt perception in schizophrenia. J Neurosci. 2006;26:8168–8175. doi: 10.1523/JNEUROSCI.2002-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volman V, Behrens MM, Sejnowski TJ. Downregulation of parvalbumin at cortical GABA synapses reduces network gamma oscillatory activity. J Neurosci. 2011;31:18137–18148. doi: 10.1523/JNEUROSCI.3041-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CZ, Yang SF, Xia Y, Johnson KM. Postnatal phencyclidine administration selectively reduces adult cortical parvalbumin-containing interneurons. Neuropsychopharmacology. 2008;33:2442–2455. doi: 10.1038/sj.npp.1301647. [DOI] [PubMed] [Google Scholar]
- Wang HX, Gao WJ. Prolonged exposure to NMDAR antagonist induces cell-type specific changes of glutamatergic receptors in rat prefrontal cortex. Neuropharmacology. 2012;62:1808–1822. doi: 10.1016/j.neuropharm.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XJ, Buzsáki G. Gamma oscillation by synaptic inhibition in a hippocampal interneuronal network model. J Neurosci. 1996;16:6402–6413. doi: 10.1523/JNEUROSCI.16-20-06402.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong EH, Kemp JA, Priestley T, Knight AR, Woodruff GN, Iversen LL. The anticonvulsant MK-801 is a potent N-methyl-d-aspartate antagonist. Proc Natl Acad Sci U S A. 1986;83:7104–7108. doi: 10.1073/pnas.83.18.7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood J, Kim Y, Moghaddam B. Disruption of prefrontal cortex large scale neuronal activity by different classes of psychotomimetic drugs. J Neurosci. 2012;32:3022–3031. doi: 10.1523/JNEUROSCI.6377-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi D, Zhang W, Wang HX, Stradtman GG, Gao WJ. Dizocilpine (MK-801) induces distinct changes of N-methyl-d-aspartic acid receptor subunits in parvalbumin-containing interneurons in young adult rat prefrontal cortex. Int J Neuropsychopharmacol. 2009;12:1395–1408. doi: 10.1017/S146114570900042X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Behrens MM, Lisman JE. Prolonged exposure to NMDAR antagonist suppresses inhibitory synaptic transmission in prefrontal cortex. J Neurophysiol. 2008;100:959–965. doi: 10.1152/jn.00079.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]