

Phosphorus is an essential nutrient for all living organisms owing to its central role in cellular metabolism. Whereas the use of phosphorus in biology is mostly confined to phosphate derivatives, compounds containing reduced forms of phosphorus such as organophosphonates are important in many organisms.1 Prominent examples are phosphonolipids, which are found in a wide variety of species including microorganisms, invertebrates, and vertebrates, and account for as much as 30% of the membrane lipids in Tetrahymena.1–2 The carbon–phosphorus (C–P) linkage is very stable and resistant to chemical hydrolysis, thermal decomposition, and photolysis. The stability of the C–P bond could explain the use of organophosphonates in membrane lipids and exopolysaccharides. A structurally diverse set of organophosphonates and phosphinates are produced as secondary metabolites by bacteria, including the clinically used antibiotic fosfomycin and the herbicides phosphinothricin tripeptide (bialaphos) and phosphonothrixin.3 These represent an underexploited class of bioactive compounds with promise for the treatment of human disease.
Given the chemical stability and biological prevalence of organophosphonates, it is not surprising that nature has evolved specialized enzymatic pathways for degrading these molecules. These pathways are important for the cellular acquisition of phosphorus, which is the growth-limiting nutrient in many ecosystems. Currently four distinct organophosphonate degradation pathways have been identified that can be divided into two mechanistic classes.1b, 4 The first class consists of three hydrolases that are highly specific for phosphonopyruvate, phosphonoacetaldehyde, and phosphonoacetate (Figure 1 A).5 The hydrolysis reactions catalyzed by these enzymes proceed by a similar mechanism, in which the electron-withdrawing β-carbonyl group of the substrates facilitates the heterolytic cleavage of the C–P bond.6 The second pathway utilizes C–P lyase, which is quite promiscuous and cleaves the unactivated C–P bonds of structurally diverse substrates. C–P lyase-catalyzed reactions include the conversion of alkylphosphonates and phenylphosphonate to the corresponding alkanes and benzene (Figure 1 B).1b, 7
Figure 1.
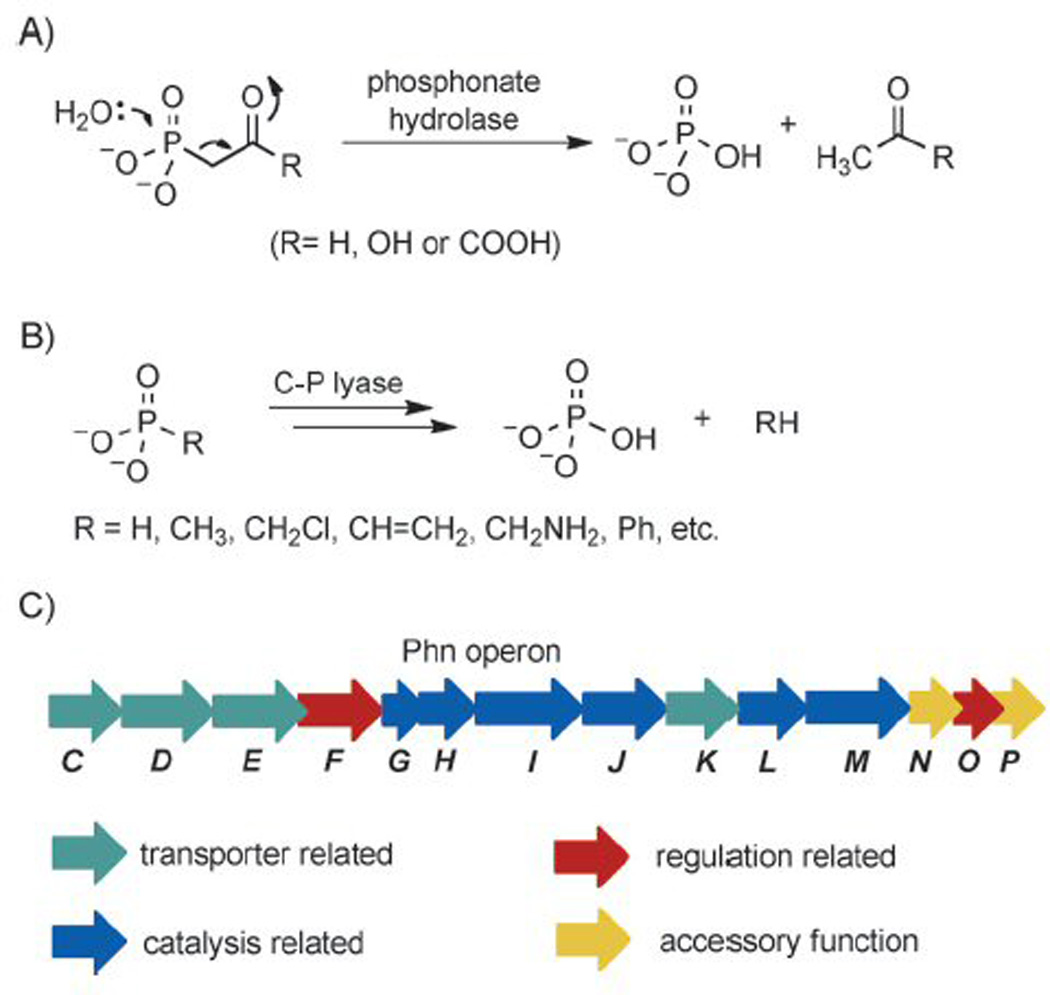
A) Heterolytic cleavage of the C–P bond of substrates with a β-carbonyl group. B) The C–P lyase pathway. C) Organization of the Phn operon of E. coli.
The C–P lyase pathway is widespread among bacteria and is encoded by a 14-cistron phnCDEFGHIJKLMNOP operon (Figure 1 C) that is regulated by the PhoR/PhoB two-component signaling system.4, 8 Sequence analysis and phenotypes of transposon mutants grown on various organophosphonates suggested that PhnC, D and E form a phosphonate transporter, PhnF and PhnO are involved in regulation, and PhnG, H, I, J, K, L and M might form the minimal catalytic cassette for C–P bond cleavage.9 However, despite great efforts since the discovery of the genes of the C–P lyase pathway in the early 1990s,8a–d, 10 the transformations involved remained largely elusive until 2011. This two-decade enigma has now been solved by a series of papers from the Hove-Jensen and Zechel laboratories11 that culminate in the in vitro reconstitution of C–P lyase activity by Raushel and co-workers.12
Attempts in the late 1980s in the Frost laboratory to purify the enzyme(s) responsible for C–P lyase activity were unsuccessful, but did reveal that the pathway possibly involved ribosyl intermediates.7a, 10 Research on the topic remained mostly dormant until 2003, when Hove-Jensen and co-workers identified PhnN as a ribose 1,5-bisphosphokinase involved in an alternative NAD biosynthetic pathway in E. coli.13 When the gene encoding 5-phospho-α-D-ribosyl 1-diphosphate (PRPP) synthase was deleted, PhnN was shown to convert ribose 1,5-bisphosphate to PRPP, thus restoring NAD biosynthesis. A few years later, PhnP was demonstrated to be a metal-dependent hydrolase of the β-lactamase superfamily.14 Screening of a library of putative substrates indicated that PhnP had phosphodiesterase activity with cyclic nucleotides. Although these studies corroborated the early findings of Frost and co-workers regarding the intermediacy of ribose derivatives, how these activities fitted into the C–P lyase pathway was unclear.
The first hint came from the observed accumulation of α-D-ribosyl 1-ethylphosphonate in an E. coli phnJ mutant strain grown on ethylphosphonic acid.11a The authors also showed that 5-phospho-α-D-ribosyl-1,2-cyclic phosphate (PRcP) accumulates in a phnP mutant strain, and suggested that PRcP is formed by C–P bond cleavage of 5-phospho-α-D-ribosyl 1-alkylphosphonates (PRPn) by C–P lyase. They thus determined the back-end of the pathway that allows organisms to funnel the phosphorus atom of organophosphonates into primary metabolism (Scheme 1).
Scheme 1.
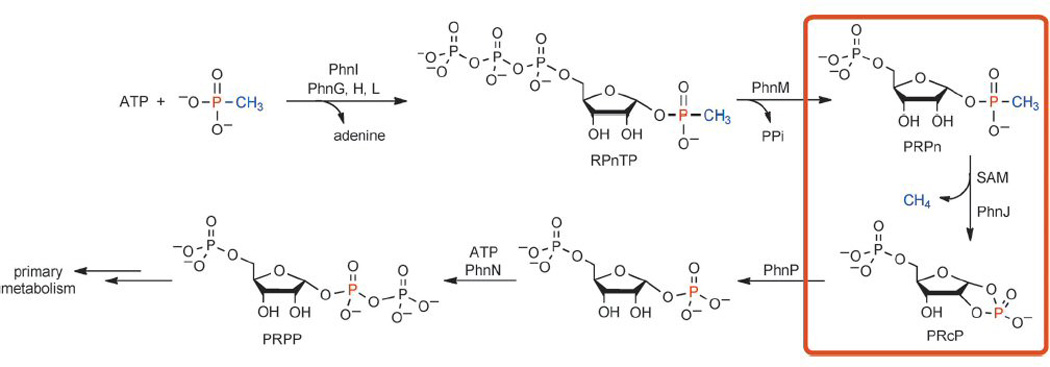
The newly established C–P lyase pathway. The unusual reaction catalyzed by PhnJ is highlighted in the orange box.
Raushel and co-workers came to the same conclusion after reconstituting the front-end of the pathway that makes PRPn.12 Using methyl phosphonate as a model substrate, the authors both reconstituted the C–P lyase activity and revealed key new ribosyl intermediates in the pathway. In a remarkable series of biochemical experiments, PhnI was shown to be a nucleosidase, requiring the presence of PhnG, H, and L to catalyze the conversion of methyl phosphonate and ATP to α-D-ribose-1-methylphosphonate-5-triphosphate (RPnTP) and adenine (Scheme 1). PhnM is a phosphatase, catalyzing the hydrolysis of RPnTP to form PRPn and pyrophosphate (PPi). C–P bond cleavage of PRPn is then achieved by PhnJ and results in methane and PRcP, which is then the substrate of PhnP, as revealed by Zechel, Hove-Jensen and colleagues.11a, b Although PhnJ lacks the CxxxCxxC motif common to most radical S-adenosylmethionine (SAM) enzymes,15 the identification of 5′-deoxyadenosine (dAdoH) and methionine generated in the PhnJ-catalyzed reaction, the presence of four Cys residues in its sequence, and the spectroscopic detection of an [4Fe–4S] cluster strongly suggest that PhnJ belongs to the radical SAM superfamily. Together with the studies on PhnN13 and PhnP,11a, b, 14 these results established the long-awaited C–P lyase pathway (Scheme 1).
In retrospect, it is likely that a combination of factors frustrated previous efforts to study C–P lyase, the most obvious one being the unknown substrate for C–P bond cleavage. Hove-Jensen and co-workers showed that the C–P lyases form a large multienzyme complex that probably consists at least of PhnG4H2I2J2K.11c Indeed, Raushel and colleagues did not observe the desired activity of PhnI unless PhnG, H and L were all present. An additional experimental hurdle could have been the demonstrated instability of many of the enzymes in the pathway. Glutathione S-transferase (GST) fusion enzymes were needed for the soluble expression of many enzymes in the pathway, but the fusion enzymes themselves were inactive. Only by removing the GST fusions in situ were Raushel et al. able to reconstitute activity. Finally, many radical SAM proteins are notoriously sensitive to oxygen, and the impressive strides made in the radical SAM field to improve reconstitution methods since the early studies on C–P lyase undoubtedly contributed to the successful reconstitution of the PhnJ activity. The ability to overcome all these technical obstacles makes the achievement all the more impressive.
Of particular interest in the pathway is the highly unusual transformation catalyzed by PhnJ. Members of the radical SAM enzyme superfamily utilize a [4Fe–4S] cluster to bind SAM and reductively cleave its carbon–sulfur bond to produce a highly reactive 5′-deoxyadenosyl (dAdo) radical, which initiates a wide range of radical reactions relevant to DNA repair, RNA and protein modification, and the biosynthesis of vitamins, coenzymes and antibiotics.15 Although radical SAM enzymes were well known for catalyzing a wide variety of chemical reactions, recent characterization of several complex transformations have indicated that the radical SAM superfamily is even more catalytic diverse than originally appreciated.15–16 The PhnJ-catalyzed reaction (Scheme 1) further expands the catalytic repertoire of radical SAM enzymes.
In light of the general chemistry of radical SAM enzymes, a catalytic mechanism has been proposed for PhnJ,12 a variation of which is shown in Scheme 2. The dAdo radical resulting from SAM cleavage might abstract a hydrogen atom from a protein residue (X1) to yield a protein-based radical.17 This radical could then initiate the homolytic cleavage of the C–P bond to produce a transient methyl radical and a protein–ribosyl adduct. The incipient methyl radical could abstract a hydrogen atom from a hydrogen donor to yield methane. This hydrogen atom donor could be dAdoH or it could be a second protein residue (X2), as illustrated in Scheme 2. Alternatively, given the high reactivity of a methyl radical, C–P bond breakage and methane formation might occur in a concerted fashion, as proposed for methyl coenzyme M reductase (MCR).18 This enzyme utilizes a specialized nickel cofactor, F430, and a thiol cosubstrate to cleave a methyl–sulfur bond and generate methane and a disulfide.19 In PhnJ, the ribosyl moiety would be subsequently released as PRcP, with a hydrogen atom shift from X1 to X2 completing the catalytic cycle. Based on other enzymes that utilize amino acid radicals in catalysis,17 X1 and X2 would likely be cysteine, tyrosine or possibly glycine residues, on which relatively stable radicals can be accommodated. Future spectroscopic and crystallographic studies should provide additional insights into this fascinating enzyme. The new knowledge gained on the C–P lyase pathway might also facilitate future engineering efforts directed at detoxification of man-made organophosphonates, such as detergents and herbicides, that are ubiquitously released to the environment.
Scheme 2.
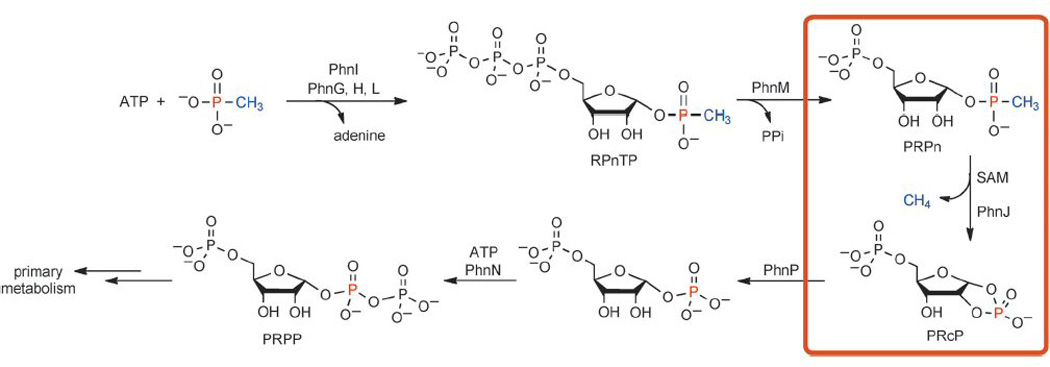
Proposed catalytic mechanism of PhnJ. X1 and X2 are likely to be cysteine, tyrosine or glycine residues.17 The red cube represents a [4Fe–4S] cluster.
Acknowledgements
Our work on phosphonate biosynthesis is supported by the National Institutes of Health (GM077596 to W.A.v.d.D.).
References
- 1.1a Ternan NG, McGrath JW, McMullan G, Quinn JP. World J. Microbiol. Biotechnol. 1998;14:635–647. [Google Scholar]; 1b White AK, Metcalf WW. Annu. Rev. Microbiol. 2007;61:379–400. doi: 10.1146/annurev.micro.61.080706.093357. [DOI] [PubMed] [Google Scholar]
- 2.Kennedy KE, Thompson GA., Jr Science. 1970;168:989–991. doi: 10.1126/science.168.3934.989. [DOI] [PubMed] [Google Scholar]
- 3.Metcalf WW, van der Donk WA. Annu. Rev. Biochem. 2009;78:65–94. doi: 10.1146/annurev.biochem.78.091707.100215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kononova SV, Nesmeyanova MA. Biochemistry–Moscow. 2002;67:184–195. doi: 10.1023/a:1014409929875. [DOI] [PubMed] [Google Scholar]
- 5.Quinn JP, Kulakova AN, Cooley NA, McGrath JW. Environ. Microbiol. 2007;9:2392–2400. doi: 10.1111/j.1462-2920.2007.01397.x. [DOI] [PubMed] [Google Scholar]
- 6.6a Morais MC, Zhang W, Baker AS, Zhang G, Dunaway-Mariano D, Allen KN. Biochemistry. 2000;39:10385–10396. doi: 10.1021/bi001171j. [DOI] [PubMed] [Google Scholar]; 6b Chen CC, Han Y, Niu W, Kulakova AN, Howard A, Quinn JP, Dunaway-Mariano D, Herzberg O. Biochemistry. 2006;45:11491–11504. doi: 10.1021/bi061208l. [DOI] [PubMed] [Google Scholar]; 6c Kulakova AN, Wisdom GB, Kulakov LA, Quinn JP. J. Biol. Chem. 2003;278:23426–23431. doi: 10.1074/jbc.M301871200. [DOI] [PubMed] [Google Scholar]; 6d Kim A, Benning MM, OkLee S, Quinn J, Martin BM, Holden HM, Dunaway-Mariano D. Biochemistry. 2011;50:3481–3494. doi: 10.1021/bi200165h. [DOI] [PMC free article] [PubMed] [Google Scholar]; 6e Agarwal V, Borisova SA, Metcalf WW, van der Donk WA, Nair SK. Chem. Biol. 2011;18:1230–1240. doi: 10.1016/j.chembiol.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.7a Frost JW, Loo S, Cordeiro ML, Li D. J. Am. Chem. Soc. 1987;109:2166–2171. [Google Scholar]; 7b Wackett LP, Shames SL, Venditti CP, Walsh CT. J. Bacteriol. 1987;169:710–717. doi: 10.1128/jb.169.2.710-717.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]; 7c Schowanek D, Verstraete W. Appl. Environ. Microbiol. 1990;56:895–903. doi: 10.1128/aem.56.4.895-903.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]; 7d Cordeiro ML, Pompliano DL, Frost JW. J. Am. Chem. Soc. 1986;108:332–334. [Google Scholar]; 7e Avila LZ, Loo SH, Frost JW. J. Am. Chem. Soc. 1987;109:6758–6764. [Google Scholar]
- 8.8a Chen CM, Ye QZ, Zhu ZM, Wanner BL, Walsh CT. J. Biol. Chem. 1990;265:4461–4471. [PubMed] [Google Scholar]; 8b Metcalf WW, Wanner BL. Gene. 1993;129:27–32. doi: 10.1016/0378-1119(93)90692-v. [DOI] [PubMed] [Google Scholar]; 8c Wanner BL, Boline JA. J. Bacteriol. 1990;172:1186–1196. doi: 10.1128/jb.172.3.1186-1196.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]; 8d Wanner BL, Metcalf WW. FEMS Microbiol. Lett. 1992;100:133–139. doi: 10.1111/j.1574-6968.1992.tb14031.x. [DOI] [PubMed] [Google Scholar]; 8e Yakovleva GM, Kim SK, Wanner BL. Appl. Microbiol. Biotechnol. 1998;49:573–578. doi: 10.1007/s002530051215. [DOI] [PubMed] [Google Scholar]; 8f Huang JL, Su ZC, Xu Y. J. Mol. Evol. 2005;61:682–690. doi: 10.1007/s00239-004-0349-4. [DOI] [PubMed] [Google Scholar]
- 9.9a Metcalf WW, Wolfe RS. J. Bacteriol. 1998;180:5547–5558. doi: 10.1128/jb.180.21.5547-5558.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]; 9b Metcalf WW, Wanner BL. J. Bacteriol. 1993;175:3430–3442. doi: 10.1128/jb.175.11.3430-3442.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Avila LZ, Draths KM, Frost JW. Bioorg. Med. Chem. Lett. 1991;1:51–54. [Google Scholar]
- 11.11a Hove-Jensen B, McSorley FR, Zechel DL. J. Am. Chem. Soc. 2011;133:3617–3624. doi: 10.1021/ja1102713. [DOI] [PubMed] [Google Scholar]; 11b He SM, Wathier M, Podzelinska K, Wong M, McSorley FR, Asfaw A, Hove-Jensen B, Jia ZC, Zechel DL. Biochemistry. 2011;50:8603–8615. doi: 10.1021/bi2005398. [DOI] [PubMed] [Google Scholar]; 11c Jochimsen B, Lolle S, McSorley FR, Nabi M, Stougaard J, Zechel DL, Hove-Jensen B. Proc. Natl. Acad. Sci. USA. 2011;108:11393–11398. doi: 10.1073/pnas.1104922108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kamat SS, Williams HJ, Raushel FM. Nature. 2011;480:570–573. doi: 10.1038/nature10622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hove-Jensen B, Rosenkrantz TJ, Haldimann A, Wanner BL. J. Bacteriol. 2003;185:2793–2801. doi: 10.1128/JB.185.9.2793-2801.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Podzelinska K, He SM, Wathier M, Yakunin A, Proudfoot M, Hove-Jensen B, Zechel DL, Jia Z. J. Biol. Chem. 2009;284:17216–17226. doi: 10.1074/jbc.M808392200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.15a Frey PA, Hegeman AD, Ruzicka FJ. Crit. Rev. Biochem. Mol. Biol. 2008;43:63–88. doi: 10.1080/10409230701829169. [DOI] [PubMed] [Google Scholar]; 15b Booker SJ, Grove TL. F1000 Biol. Rep. 2010;2:52. doi: 10.3410/B2-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.16a Zhang Q, Liu W. J. Biol. Chem. 2011;286:30245–30252. doi: 10.1074/jbc.R111.272690. [DOI] [PMC free article] [PubMed] [Google Scholar]; 16b Zhang Q, van der Donk WA, Liu W. Acc. Chem. Res. 2011 doi: 10.1021/ar200202c. DOI: [DOI] [PMC free article] [PubMed] [Google Scholar]; 16c Challand MR, Driesener RC, Roach PL. Nat. Prod. Rep. 2011;28:1696–1721. doi: 10.1039/c1np00036e. [DOI] [PubMed] [Google Scholar]
- 17.Stubbe J, van der Donk WA. Chem. Rev. 1998;98:705–762. doi: 10.1021/cr9400875. [DOI] [PubMed] [Google Scholar]
- 18.18a Ragsdale SW. Chem. Rev. 2006;106:3317–3337. doi: 10.1021/cr0503153. [DOI] [PubMed] [Google Scholar]; 18b Pelmenschikov V, Blomberg MR, Siegbahn PE, Crabtree RH. J. Am. Chem. Soc. 2002;124:4039–4049. doi: 10.1021/ja011664r. [DOI] [PubMed] [Google Scholar]
- 19.Shima S, Thauer RK. Curr. Opin. Microbiol. 2005;8:643–648. doi: 10.1016/j.mib.2005.10.002. [DOI] [PubMed] [Google Scholar]