

Abstract
Most hereditary nonpolyposis colorectal cancer (HNPCC) patients inherit a defective allele of a mismatch repair (MMR) gene, usually MLH1 or MSH2, resulting in high levels of microsatellite instability (MSIH) in the tumors. Presence of MSI in the normal tissues of mutation carriers has been controversial. Here we directly compare MSI in the peripheral blood leukocyte (PBL) DNA of seven HNPCC patients carrying different types of pathogenic MMR mutations in MLH1 and MSH2 genes with the PBL DNA of normal age-matched controls and of patients with sporadic colorectal cancer (SCRC). Small pool PCR (SP-PCR) was used studying three microsatellite loci for at least 100 alleles each in most samples. The average frequencies of mutant microsatellite fragments in each HNPCC patient (0.04–0.24) were significantly higher (p<0.01) relative to their age-matched normal controls with mutant frequencies (MF) from 0.00 to 0.06, or SCRC patients (MF from 0.01–0.03). The data support the conclusions that higher MF in the PBL DNA of HNPCC patients is real and reproducible, may vary in extent according to the type of germline MMR mutation and the age of the individual, and provide a possible genetic explanation for anticipation in HNPCC families.
Keywords: microsatellite instability, genomic instability, HNPCC, Lynch Syndrome, colorectal cancer, MLH1, MSH2, single molecule PCR, somatic mutation, variants of uncertain significance
Introduction
A high level of microsatellite instability (MSI-H), that is, the frequent production of mutant microsatellite alleles in somatic cells, has been seen in the colorectal tumors of hereditary nonpolyposis colorectal cancer syndrome (HNPCC) patients [Aaltonen et al., 1993; Hampel et al., 2005; Ionov et al., 1993; Lynch and de la Chapelle, 2003; Thibodeau et al., 1993]. In HNPCC, an inherited mutation in an important mismatch repair (MMR) gene, usually either MLH1 (MIM# 120436; NCBI Reference Sequence NM_000249 at chr3:37009983-37067341; GI: 28559089 GenBank build 36.2) or MSH2 (MIM# 609309; NCBI Reference Sequence NM_000251.1; GI:4557760 GenBank build 36.2) [Boland, 2007; Boland and Fishel, 2005; Bronner et al., 1994; de la Chapelle, 2004; Fishel et al., 1993; Kunkel and Erie, 2005; Leach et al., 1993; Li, 2008; Liu et al., 1994; Lynch and de la Chapelle, 2003; Nicolaides et al., 1994; Palombo et al., 1996; Papadopoulos et al. 1994; Parsons et al. 1993], results in loss of MMR function in tissues where the normal allele is lost or silenced [Hemminki et al., 1994; Leach et al., 1993]. MSI-H also occurs in sporadic colorectal cancer (SCRC) where the MLH1 locus has been silenced due to methylation [Kane et al., 1997]. The critical difference between HNPCC and MSI-H SCRC is that only one functional MMR allele is present in the normal somatic tissues of HNPCC patients, whereas SCRC patients have two normal functioning alleles in every normal somatic cell. The hypothesis being tested here is that peripheral blood leukocytes (PBLs) of HNPCC patients, having only one functional copy of the mutated MMR gene in these noncancerous somatic cells, will have a higher frequency of mutant microsatellite fragments than the PBLs of normal controls or SCRC patients having two normal MMR alleles. However, the levels of mutant microsatellite fragments would be too low to detect using conventional methods of MSI testing.
Mixing experiments (mixing different proportions of DNAs from individuals with different microsatellite alleles) have shown that low frequencies (<20–25%) of mutant microsatellite fragments are not detectable in the standard MSI PCR assay because they are overwhelmed by very frequent progenitor alleles in a standard PCR reaction [Coolbaugh-Murphy et al., 2004]. Therefore, the inability to detect subtle MSI in germline DNA of HNPCC patients may be due to insufficient detection efficiency.
To detect lower levels of MSI, Coolbaugh-Murphy et al. [2004] developed a high-throughput modification of a procedure whereby DNA isolated from PBLs was diluted to single genome equivalents (g.e.) and PCR run on multiple pools (over 100) of such DNA. This sensitive and quantitative procedure for detecting mutant microsatellite fragments is called small pool PCR or SPPCR [Monckton and Jeffreys, 1991]. Under these conditions, there is a high probability of trapping, amplifying, detecting, and quantifying mutants produced at levels as low as 1%. In the study of Coolbaugh-Murphy et al. [2004], an HNPCC patient having developed an MSI-H CRC, and carrying a germline MSH2 mutation was shown to have a low (0.063), but significantly higher frequency of mutant fragments in PBL DNA over the six loci in the screen (p<0.01) compared to normal controls. In comparison, a sporadic CRC patient with an MSI-H tumor due to CpG island promoter methylation of the MLH1 gene in the tumor did not display elevated MSI in PBL DNA when compared to the age-matched control [Coolbaugh-Murphy et al., 2004]. The data demonstrate that measurable MSI can be quantified in the PBL DNA of patients carrying a germline MMR gene mutation, and that the MSI is not due to circulating cells from an MSI-H tumor.
Advances in SPPCR and MSI analysis technology [Canzian et al., 1996; Coolbaugh-Murphy et al., 2004] improved its sensitivity to the extent that quantification of increasing MSI with age in the PBL DNA of normal individuals from 1% at 20 years old to 3% at 70 years old [Coolbaugh-Murphy et al., 2005] was attained. Here, we used the method to determine and compare MSI levels in PBL DNA from a number of patients with HNPCC, SCRC, and normal controls. Previous work (presented in depth in the DISCUSSION in relation to our findings) has reported the phenomenon in lymphoblastic cell lines [Parsons et al., 1995] and other systems [Alazzouzi et al., 2005; Chen et al., 2007; Gallinger et al., 2004]. How mutant levels we observed in the PBLs correlate with age and MMR genotype are also presented, and discussed as a possible genetic mechanism to explain anticipation as it has been shown to occur in HNPCC patients [Nilbert et al., 2009].
Materials and Methods
Normal Control DNA
These were selected from 426 anonymized normal control PBL DNAs obtained from the University of Texas M.D. Anderson Blood Bank identified by gender (212 females and 214 males) and age (range: 18–68 year old, with at least eight samples for each year of life). Blood specimens were screened for HIV and hepatitis (by Nucleic Acid Test PCR-NAT analysis) and health questionnaires had been administered to the donors in accordance with American Association of Blood Banks requirements. Specimens chosen did not have a known familial or personal history of cancer. Thirteen normal donor specimens were selected for this study on the basis of matching to HNPCC patients by age and gender. Because of the paucity of older normal controls, one normal was matched for every two older Sporadic CRC patients. These specimens were genotyped at the three loci used for SPPCR analysis, and are listed as normal controls, in Tables 1, 2, and 3, and binned for analysis.
Table 1.
D17S518 Results for Patient H3 to Demonstrate How Data Were Analyzed
| A. D17S518 alleles (mutants in bold italics) present in small pools using normal control N380 PBLs, and PBLs and positive control tumor DNA from MSI-High HNPCC patient H3. | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of pools with the following alleles (bp):a |
|||||||||||||
| Identity | Genotype | <80 | 81 | 83 | 85 | 87 | 89 | 93 | 95 | 97 | 99 | 101 | 103 |
| N380 PBL | 89/93 | 0 | 0 | 0 | 0 | 0 | 116 | 116 | 0 | 0 | 0 | 0 | 0 |
| H3 PBL | 89/95 | 0 | 0 | 0 | 0 | 0 | 35 | 18 | 21 | 0 | 0 | 0 | 0 |
| H3 TU | 89/95 | 2 | 0 | 9 | 11 | 22 | 62 | 4 | 58 | 8 | 12 | 7 | 4 |
| B. Calculation of patient H3 and normal control N380 mutant fragment frequencies (MF) in D17S518 small pools | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Identity | Genotype | Exp.ge1 | Est.ge2 | #pools | Expected alleles3 | Estimated alleles3 | a14 | a24 | Vars5 | MF6 |
| N380 PBL | 89/93 | 0.75 | 2.37 | 128 | 192 | 606 | 116 | 116 | 0 | 0.000 |
| H3 PBL | 89/95 | 0.75 | 0.34 | 128 | 192 | 87 | 35 | 21 | 18 | 0.302 |
| H3 TU | 89/95 | 0.75 | 1.19 | 128 | 192 | 306 | 62 | 58 | 79 | 0.711 |
Chromatograms for an experiment as in Figure 1 were analyzed and compiled into locus specific tables, as in A, showing the observed number of individual variants, bold , and progenitors and their sizes (GeneScore; www.hkasoftware.com/genescore.shtml). The data from all experiments at each locus were compiled into a table, B, which includes the g.e. that was expected and the total number of pools run.). The observed, estimated g.e. for all the data and the estimated total number of alleles seen were calculated. A mutant frequency (MF) and the significance of the MFs between sample and control were calculated from the MF and the bootstrap standard error calculated (SPPCRv.2; www.hkasoftware.com/sppcr.shtml).
No alleles were observed at 91 bp.
Expected genome equivalents (exp. g.e.),
estimated genome equivalents per pool (est. g.e.),
expected and estimated total number of alleles analyzed at that locus (exp. alleles and est. alleles),
numbers of progenitor allele 1 (a1) and allele 2 (a2),
number of variants observed (Vars)
estimated Mutant Frequency (MF). PBL, peripheral blood leukocyte.
Table 2.
Summary of SP-PCR Data for Paired HNPCC Patient and Matched Normal Control PBL DNA at Three Loci
|
D2S123
|
D5S346
|
D17S518
|
Three loci total and avg. |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | PBL Age | n1 | m2 | F3 | p-value4 | n1 | m2 | F3 | p-value4 | n1 | m2 | F3 | p-value4 | n1 | m2 | F3 | p-value4 |
| N40 | 46 | 289 | 5 | 0.017 | >0.05 | 276 | 2 | 0.007 | 257 | 1 | 0.004 | >0.05 | 822 | 8 | 0.010 | <0.01 | |
| H1 | 41 | 277 | 14 | 0.051 | 333 | 17 | 0.051 | <0.01 | 214 | 3 | 0.014 | 824 | 34 | 0.041 | |||
| N296 | 49 | 392 | 5 | 0.013 | <0.05 | 581 | 0 | 0.000 | <0.01 | 409 | 1 | 0.002 | >0.05 | 1382 | 6 | 0.004 | <0.01 |
| H2 | 48 | 205 | 11 | 0.054 | 138 | 25 | 0.181 | 168 | 4 | 0.024 | 511 | 40 | 0.078 | ||||
| N380 | 68 | 270 | 0 | 0.000 | <0.01 | 384 | 0 | 0.000 | <0.01 | 582 | 0 | 0.000 | <0.01 | 1236 | 0 | 0.000 | <0.01 |
| H3 | 71 | 53 | 24 | 0.453 | 79 | 11 | 0.139 | 87 | 18 | 0.207 | 219 | 53 | 0.242 | ||||
| N378 | 36 | 172 | 3 | 0.017 | <0.05 | 141 | 10 | 0.071 | <0.01 | 178 | 5 | 0.028 | <0.01 | 491 | 18 | 0.037 | <0.01 |
| H4 | 36 | 121 | 12 | 0.099 | 51 | 24 | 0.471 | 130 | 23 | 0.177 | 302 | 59 | 0.195 | ||||
| N170 | 46 | 142 | 3 | 0.021 | <0.05 | 65 | 1 | 0.015 | <0.01 | 173 | 2 | 0.012 | >0.05 | 380 | 6 | 0.016 | <0.05 |
| H5 | 46 | 151 | 10 | 0.066 | 99 | 8 | 0.081 | 191 | 3 | 0.016 | 441 | 21 | 0.048 | ||||
| N374 | 59 | 83 | 3 | 0.036 | >0.05 | 90 | 2 | 0.022 | <0.01 | 128 | 8 | 0.063 | <0.01 | 301 | 13 | 0.043 | <0.01 |
| H6 | 59 | 155 | 1 | 0.006 | 94 | 17 | 0.181 | 126 | 37 | 0.294 | 375 | 55 | 0.147 | ||||
| N20 | 46 | 267 | 1 | 0.004 | <0.05 | 381 | 5 | 0.013 | <0.01 | 219 | 9 | 0.041 | <0.05 | 867 | 15 | 0.017 | <0.01 |
| H75 | 42 | 241 | 18 | 0.075 | 142 | 8 | 0.056 | 164 | 17 | 0.104 | 547 | 43 | 0.079 | ||||
Estimated number of alleles (n),
number of observed mutants (m),
mutant frequency (F) = m/n,
p-value = z-score significance,
patient H7 was previously published as “Patient B” [Coolbaugh-Murphy et al., 2004].
PBL, peripheral blood leukocyte;
HNPCC, hereditary nonpolyposis colorectal cancer.
Table 3.
Summary of SP-PCR Data for Sporadic CRC Patient, Matched Normal Control, and HNPCC Positive Control PBL DNA at Three Loci
|
D2S123
|
D5S346
|
D17S518
|
3 loci total and avg. |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | PBL Age | n1 | m2 | F3 | p-value4 | n1 | m2 | F3 | p-value4 | n1 | m2 | F3 | p-value4 | n1 | m2 | F3 | p-value4 |
| N264 | 66 | 88 | 5 | 0.057 | 79 | 1 | 0.013 | 95 | 0 | 0.000 | 262 | 6 | 0.023 | ||||
| S1 | 65 | 478 | 5 | 0.010 | >0.05 | 259 | 1 | 0.004 | >0.05 | 332 | 5 | 0.015 | <0.05 | 1069 | 11 | 0.010 | >0.05 |
| S2 | 70 | 190 | 4 | 0.021 | >0.05 | 123 | 4 | 0.033 | >0.05 | 252 | 0 | 0.000 | >0.05 | 565 | 8 | 0.014 | >0.05 |
| H2 or H35 | 48, 71 | 31 | 16 | 0.516 | <0.01 | 38 | 6 | 0.158 | <0.05 | 176 | 32 | 0.182 | <0.01 | 245 | 54 | 0.220 | <0.01 |
| N380 | 68 | 132 | 5 | 0.038 | 259 | 6 | 0.023 | 177 | 2 | 0.011 | 568 | 13 | 0.023 | ||||
| S3 | 75 | 109 | 2 | 0.018 | >0.05 | 183 | 8 | 0.044 | >0.05 | 149 | 3 | 0.020 | >0.05 | 441 | 13 | 0.029 | >0.05 |
| S4 | 80 | 198 | 8 | 0.016 | >0.05 | 411 | 3 | 0.007 | >0.05 | 248 | 5 | 0.020 | >0.05 | 1157 | 16 | 0.014 | >0.05 |
| H2 or H35 | 48, 71 | 62 | 8 | 0.129 | <0.05 | 53 | 10 | 0.189 | <0.01 | 104 | 9 | 0.087 | <0.05 | 219 | 27 | 0.123 | <0.01 |
| N343 | 67 | 102 | 0.020 | 89 | 0 | 0.000 | 83 | 0 | 0.000 | 274 | 0.007 | ||||||
| S5 | 72 | 517 | 4 | 0.008 | >0.05 | 253 | 6 | 0.024 | <0.01 | 358 | 2 | 0.006 | >0.05 | 1128 | 12 | 0.011 | >0.05 |
| S6 | 68 | 59 | 5 | 0.085 | >0.05 | 300 | 5 | 0.017 | <0.05 | 75 | 3 | 0.040 | <0.01 | 434 | 13 | 0.030 | <0.05 |
| H2 or H35 | 48, 71 | 65 | 14 | 0.215 | <0.05 | 35 | 6 | 0.171 | <0.01 | 96 | 14 | 0.146 | <0.01 | 196 | 34 | 0.173 | <0.01 |
| N343 | 67 | 22 | 1 | 0.045 | 48 | 3 | 0.063 | 28 | 0 | 0.000 | 98 | 4 | 0.041 | ||||
| S7 | 76 | 281 | 0 | 0.000 | >0.05 | 66 | 2 | 0.030 | >0.05 | 246 | 3 | 0.012 | <0.05 | 593 | 5 | 0.008 | >0.05 |
| H2 or H35 | 48, 71 | 21 | 4 | 0.190 | <0.05 | 35 | 10 | 0.286 | <0.05 | 46 | 7 | 0.152 | <0.05 | 102 | 21 | 0.206 | <0.01 |
| N406 | 67 | 128 | 7 | 0.055 | 179 | 7 | 0.039 | 150 | 6 | 0.040 | 457 | 20 | 0.044 | ||||
| S86 | 73 | 130 | 5 | 0.038 | >0.05 | 123 | 0 | 0.000 | >0.05 | 144 | 6 | 0.042 | >0.05 | 397 | 11 | 0.028 | >0.05 |
Estimated number of alleles (n),
number of observed mutants (m),
mutant frequency (F) = m/n,
p-value = z-score significance,
for positive controls: H3 for D2S123 and D17S518; H2 for D5S346,
patient S8 was previously published as “Patient C” [Coolbaugh-Murphy et al., 2004]. CRC, colorectal cancer.
HNPCC Patient DNA
The subjects included seven HNPCC patients from The University of Texas M.D. Anderson Cancer Center's Hereditary Nonpolyposis Colorectal Cancer Registry. They were chosen because they met Amsterdam or Bethesda Criteria, and all were classified either MSI-L or -H based on traditional PCR analysis of their tumor DNA. Details on identification, classification, and recruitment of these individuals are described elsewhere [Chen et al., 2007; Frazier et al., 2001]. MMR gene mutations in the germline of these patients were determined at a laboratory certified by the Clinical Laboratory Improvement Act. Informed consent was obtained from all participants and was approved by an institutional review board. These patients are listed in Table 4 as H1 through H7 along with data on gender, ages, and pathological MMR mutations. The mutations were annotated with the aid of the Mutalyzer program (http://eu.liacs.nl/mutalyzer/1.0.4/; as described by Wildeman et al., 2008). Three each were MLH1 and MSH2 germline mutation carriers and harbored pathologic missense, deletion, and splice variants. Patient H1 alone was originally classified as MSI-L, and subsequent MAPP-MMR analysis of his missense mutation (MLH1 D591G) indicated a borderline deleterious score of 3.5 by MAPP-MMR [Chao et al., 2008; Stone and Sidow, 2005] (MAPP-MMR is a software program for classification of MLH1/MSH2 missense variants with multivariate analysis of protein polymorphisms in mismatch repair genes; http://mappmmr.blueankh.com/Impact.php). In contrast to this score, the other missense variants seen in patients H2 (MLH1 C39Y) and H6 (MSH2 G683R) produced deleterious MAPP-MMR scores of 14.69 and 44.00, respectively.
Table 4.
HNPCC Patients in This Study
| Pt. ID | Age at PBL sample | Gender | Gene | Germline MMR mutation | Mutation classification |
|---|---|---|---|---|---|
| H1 | 41 | m | MLH1 | ex 16 NM_000249.2:c.l772A>G NP_000240.1:p.Asp591Gly | Missense |
| H2 | 48 | f | MLH1 | ex 1 NM_000249.2:c.116G>A NP_000240.1:p.Cys39Tyr | Missense/splice |
| H3 | 71 | m | MLH1 | ex 8 NM_000249.2:c.676C>T NP_000240.1:p.Arg226* | Nonsense/stop |
| H4 | 36 | m | MSH2 | Ex5-int 4 NM_000251.1:c.861-1G>A | Splice |
| H5 | 46 | f | MSH2 | ex 13 NM_000251.1:c.2113delG NP_000242.1:p.Val705Trpfs*5 | Deletion/frameshift |
| H6 | 59 | m | MSH2 | ex 12 NM_000251.1:c.2047G>A NP_000242.1:p.Gly683Arg | Missense |
| H7a | 42 | m | MSH2 | NM_000251.1 :unknown | IHC (−) MSH2 |
Demographic data for the 7 HNPCC patients is shown. Patients are identified as H1 through H7, along with age at time of PBL sampling, gender, mismatch repair gene, and germline mutation associated with disease, and the mutation's classification. Nucleotide numbering reflects cDNA numbering with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence, according to journal guidelines (www.hgvs.org/mutnomen). The initiation codon is codon 1.
Patient H7 was previously identified as Patient B in Coolbaugh et al. [2004]. Mutation nomenclature verified by Mutalyzer at http://eu.liacs.n1/mutalyzer/l.0.4/.
The HNPCC patient H7 in Table 4 was previously described as Patient B [Coolbaugh-Murphy et al., 2004], a patient who developed CRC at the age of 42. He is part of a kindred meeting the most stringent Amsterdam criteria for HNPCC, and his tumor displayed negative immunostaining for MSH2, as well as classical MSI by traditional PCR. Although a specific MSH2 mutation has not yet been found in this kindred, the syndrome as described identifies the patient as having Muir-Torre Syndrome (a subtype of HNPCC) and a carrier of a germline mutation for CRC predisposition.
Sporadic Colorectal Cancer (SCRC) Patient DNA
Seven SCRC patients were selected from a consecutive series of CRC patients that were collected through an institutional resource at the UT MDACC (TexGen). TexGen patients provide blood samples for DNA extraction and are also administered a questionnaire. CRC patients were selected for this study based on their lack of familial cancer history, and older age of onset of CRC. As shown in Table 5, PBL DNA samples from six males and two females (S1–S8) ranging in age from 65 to 80 years were analyzed. The DNAs were subjected to end-to-end sequencing of each of the exons of the MLH1 and MSH2 genes, including the intron–exon boundaries. In addition, DNA samples isolated from tumors of each of these patients were tested for promoter methylation of the MLH1 gene and MSI. Seven out of eight of the tumors were MSS by traditional PCR and did not demonstrate methylation. Sporadic patient C is the eighth SCRC (S8) patient listed in Table 5. He was previously described [Coolbaugh-Murphy et al., 2004], and was a 74-year-old patient with no family history of CRC. His tumor DNA displayed CpG island promoter methylation of the MLH1 gene and was MSI-H [Frazier et al., 2003].
Table 5.
Sporadic Colorectal Cancer Patients Used in This Study
| Patient ID# | Age at PBL sample | Gender |
|---|---|---|
| S1 | 65 | m |
| S2 | 70 | m |
| S3 | 75 | f |
| S4 | 80 | f |
| S5 | 72 | m |
| S6 | 68 | m |
| S7 | 76 | m |
| S8a | 73 | m |
Demographic data for the eight sporadic CRC patients is shown. Patients are identified as S1 through S8, along with age at PBL sampling and gender.
Patient S8 was previously identified as Patient C in Coolbaugh et al. [2004]. PBL, peripheral blood leukocyte.
SPPCR Methodology
We have previously published the details of this method, which we have developed [Coolbaugh-Murphy et al., 2004]. Essential aspects are summarized here. DNA was extracted from 10-ml blood samples drawn from the study subjects and controls in Vacutainer tubes containing EDTA (Becton Dickinson Vacutainer System, San Jose, CA, USA) by using the AUTOPURE LS Automated DNA Purification Instrument (Gentra Systems, Inc., Minnerapolis, MN, USA) according to the manufacturer's instructions. Upon receipt of the DNA samples, genotyping was carried out by standard PCR for the three dinucleotide microsatellite loci in the study, D2S123, D5S346, and D17S518. The multiplexed PCR products were genotyped on the ABI 3100 (ABI, Foster City, CA, USA). The DNAs were precisely quantitated by determining the O.D. 260 readings using a Pharmacia Spectrophotometer UltraSpec III (Piscataway, NJ, USA). They were then diluted to approximately single diploid genome levels in 0.1 × TE. The DNAs were initially quantified by SPPCR at the D2S123 locus at 1, 10, and 100 genome equivalents to determine the amount of amplifiable DNA present in the samples. For the analysis of the HNPCC samples, multiple heminested SPPCRs were conducted on sets of two DNAs consisting of one unrelated, age- and gender-matched normal control PBL DNA and the patient's PBL DNA. For the analysis of the sporadic CRC samples, because of the dearth of older normal control samples, one age-matched normal control PBL DNA was examined alongside two sporadic and one HNPCC PBL DNA (as a positive control). Approximately 100 alleles distributed over 96–112 PCR replicates per sample were amplified at each locus, with the three-to-four DNAs on one 384-well tray per locus. The MWG Biotech RoboSeq 4204 S robot with an onboard Primus-HT 384 thermocycler (MWG Biotech, High Point, NC, USA) was used for the setup and amplification of the initial “outer” PCR, after which they were diluted using the Qiagen BioRapidPlate and Twister I robots (Valencia, CA, USA). The secondary “inner” PCR was also distributed using the Qiagen robot. The secondary PCR was also amplified using another MWG Biotech Dualblock Primus-HT 384 thermocycler (High Point, NC,USA). The RapidPlate was again used to multiplex all the loci's 384-well trays for each sample set, resulting in 384 wells, each containing the SP-PCR products of multiple loci. The trays of multiplexed SP-PCR products were submitted to the UT-MDACC DNA Analysis Core Facility where they were analyzed on an ABI 3100 capillary system with GeneScan software. The dyes used on the loci for this project were 6-FAM and NED on the primers, and ROX for the internal size standard on the ABI. The primers and dyes used for each locus were previously described [Coolbaugh-Murphy et al., 2004, 2005]. All PCRs were set up, amplified, diluted, and rearrayed in dedicated clean-rooms, with robots contained in Air Clean 600 PCR Workstations (ISC BioExpress, Kaysville, UT, USA), using dedicated pipetmen, tips, tubes, trays, reagents, etc., to minimize contamination potential.
Data Analysis
After ABI GeneScan analysis, the raw data were printed out as chromatograms to be scored for allele counts and variants. The data element consisted of whether or not each fragment was seen in every small pool. A model in which the number of alleles in replicate pools had a Poisson distribution, and in which particular allele frequencies constituted a fixed proportion of the total has been described [Coolbaugh-Murphy et al., 2004]. Maximum likelihood estimates of the mean number of alleles in each pool and the frequencies of each allele along with the standard error of each frequency were calculated by computer program. The in-house programs of GeneScore (www.hkasoftware.com/genescore.shtml) and SPPCRv.2 (www.hkasoftware.com/sppcr.shtml) were used for these functions and to calculate p-values and 95% confidence intervals). A logit transformation was necessary for a linear regression analysis to plot age against mutant frequencies, as described in Coolbaugh-Murphy et al. [2005].
Figure 1 and Table 1 demonstrate how we proceed from raw chromatograms through counting alleles, estimating number of analyzed alleles, and then estimating the mutant frequency and significance. For this demonstration, we show data from HNPCC patient H3, who not only has a nonsense/stop mutation in MLH1, but also showed MSI in PBL DNA by SPPCR.
Figure 1.
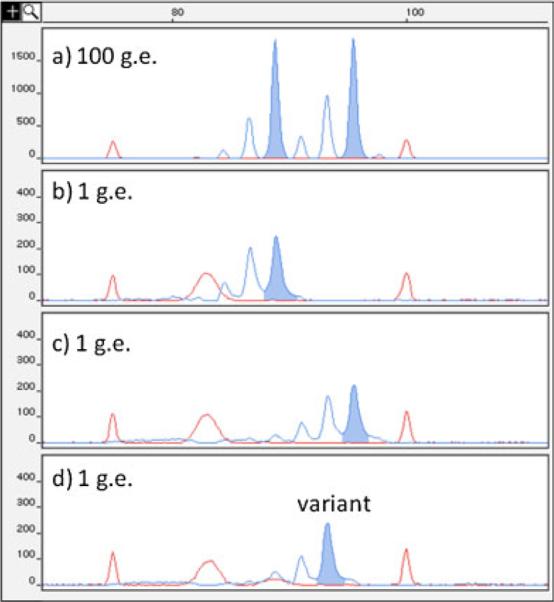
Representative small pool PCR chromatograms of patient H3 PBL at the D17S518 locus. Patient H3 at D17S518 is an (89, 95) basepair (bp) heterozygote, demonstrated by the presence of both alleles in the 100 genome equivalent genotype in a. Red peaks are internal size standards at 75 and 100 bp. Also seen is a nonspecific electrophoretic band in the size standard mix near 85 bp. After single-molecule PCR, the two progenitors at this locus are seen separated into individual wells in b (89 bp) and c (95 bp). A variant is observed alone in d (93 bp), separated from either progenitor allele.
Figure 1 shows representative chromatograms from SPPCR at the D17S518 locus on patient H-3 PBL DNA. Red peaks are internal size standards at 75 and 100 basepairs. Panel a shows 100 genome equivalent (g.e.) genotyping reactions with 89/95 bp heterozygous alleles at D17S518. Panels b and c show the segregation of individual D17S518 89 or 95 bp alleles into those wells after single molecule PCR, whereas panel d shows just a single D17S518 93 bp variant allele. Panel d demonstrates the isolation of a mutant alone in the well under small pool conditions.
Table 1A and B shows representative data from the analysis of tumor and normal peripheral blood lymphocyte DNA from Patient H3 and its normal control (N380) at the D17S518 dinucleotide repeat. SPPCR data are shown in Table 1A as the number of variant (bolded italics) and progenitor alleles seen for each PCR fragment size in the total pools analyzed in patient H3 PBL and the simultaneously examined normal control PBL DNA and positive control patient tumor DNA (TU). Then, shown in Table 1B, are the tabulated number of pools, expected genome equivalents (exp. g.e.), estimated genome equivalents (est. g.e.) per pool, expected and estimated total number of alleles (exp. alleles and est. alleles) analyzed at that locus, as well as the numbers of progenitor allele 1 (a1) and allele 2 (a2). Also shown are the number of variants observed (Vars), and the estimated Mutant Frequency (MF) for those tissues at that locus.
The statistical significance of differences in frequencies were assessed by using the asymptotic normality of the estimates. The estimated frequency divided by its standard deviation is approximately Gaussian. The approximation approaches perfection as the number of small pools increases—this is the meaning of asymptotic. Thus, for example, to determine the statistical significance of the difference in frequency between control and patient, the following calculation was performed. Z_c was the estimated mutant frequency in the control divided by its standard; Z_p was the same for the patient. If the difference in frequencies were due entirely to chance (i.e., the means do not differ), then Z_p-Z_c was distributed as a Gaussian distribution with mean zero and standard deviation the square root of 2. The p-value was then obtained from tables of the Gaussian distribution. In particular, if Z_p–Z_c>2.326 then the difference was significant at the 0.05 level; if Z_p–Z_c>3.289 then the difference was significant at the 0.01 level. The computer program did not combine estimates over alleles. The frequency over the combined alleles, f, was estimated as the binomial mean, the total number of mutants seen, m (totaled over the alleles considered) divided by the total number of alleles examined, n. This was justified because f was small so the probability of two or more mutants in one small pool is tiny. The standard deviation of f is the binomial standard deviation, that is, the square root of f×(1−f).
Results
Quantification of MSI in the PBL DNA of HNPCC Patients Containing Pathogenic Mutations in MSH2 or MLH1
By use of the SPPCR technology, one can count the number of progenitor or “normal” microsatellite locus alleles present in the small pools tested from a sample from an individual and also count the number of mutant alleles, enabling the determination of the frequency of mutant alleles in each sample at each locus tested. These data for the three microsatellite loci in the screen are presented in Table 2 for the PBL DNA from seven HNPCC patients (patients H1–H7) as well as from PBL DNA of their six age-matched controls run at the same time on the same plates. For each patient/control set at each locus, the total number of alleles tested (n), number of mutant alleles observed (m), the frequency of mutant alleles (f), and the Z-score significance of the difference between the mutant frequencies in the control versus the HNPCC patient were recorded. There were 21 direct such comparisons (seven patients × three loci) and in 20 of them there was a higher frequency of mutant alleles in the HNPCC PBL DNA. The probability of such a result in the absence of a contributing effect in the HNPCC patients (when a higher mutant frequency in the normals should occur in 1/2 of the 21 comparisons) is p = 10.5 × 10−6. however, in only 16 comparisons (at a level of p<0.05) of the mutant frequencies the Z-score indicated a significant difference (at a level of p<0.05), with 11 of these 16 comparisons at a level of p<0.01. We attribute this to an insufficient number of alleles tested at any one locus, resulting in a sampling error for a low-frequency event. Therefore, the mean mutant allele frequencies over all three loci for each patient set (control and patient PBL DNA) were calculated and are also in Table 2. For all seven patients there was a significantly higher mean frequency of mutants across all three loci. These data suggest that patients carrying a deleterious germline mutation in a MMR gene have a significant level of MSI in their nontumor (constitutive) tissue.
MSI in the PBLs of SCRC Patients
It could be argued that the above result was not related to the patients' carrying germline MMR mutations but due to a general genome instability in the constitutive tissues of CRC patients. Therefore, the same SPPCR analysis was conducted on the PBL DNA of eight sporadic CRC (SCRC) patients and the PBL DNA of their age-matched controls. These data are present in Table 3. For each assay set (normal control and two patients) there were also included samples from one of the PBL DNAs of HNPCC patients H2 or H3. These latter samples had high mutant frequencies for certain loci (H3 for D2S123 and D17S518, H2 for D5S346) and served as positive controls for MSI as well as verifying that the high levels of MSI seen in those patients for those loci (Table 2) were reproducible. As in Table 2 for the HNPCC patients, for each patient/control set at each locus, we record the total number of alleles tested (n), number of mutant alleles observed (m), the frequency of mutant alleles (f), and the Z-score significance of the difference between the mutant frequencies in the control versus the SCRC patient.
There were 24 direct comparisons of the frequencies of mutant alleles in normal control versus SCRC PBL DNA. None of the overall weighted average mutant frequencies were significantly higher in the SCRC PBLs compared to the normal control PBLs at the p<0.01 level. Consequently, there appears to be an absence of a contributing effect to MSI in the PBL DNA of SCRC patients because higher mutant frequencies in the normal individuals occurred in approximately half of the 24 comparisons. However, there were occasional low, but significant, mutant frequencies in SCRC PBL DNA than controls (patient S6 at D5S346 and D17S518 and patient S7 at D17S518). The difference in S6 is also reflected in the three locus-weighted average frequency. However, these differences are attributable to the low numbers of control amplifications we had in those samples because the mutant frequency range in those SCRC DNAs at those loci was in the normal range of 3% for individuals at that age (68 years old). We therefore conclude that unlike the PBL DNA from HNPCC patients carrying pathogenic germline mutations in one of their major MMR genes, the MSI level in SCRC patient PBL DNA is no higher than in age-matched normal controls.
The repeatability of the significantly higher mutant frequencies in PBL DNA from HNPCC patients H2 and H3, compared to the levels in controls, was confirmed in this same set of experiments. Patient H2, used as the positive control at the D5S346 locus, had a mutant frequency of 0.181 at that locus initially (Table 2). Sample from H2 was used as positive control four times for D5S346 in the SCRC cases, and the frequencies were 0.158, 0.189, 0.171, and 0.286 (Table 3) for a mean of 0.199. Similar significant results were obtained when Patient H3 PBL DNA was used as the positive control for loci D2S123 and D17S518. Mutant frequencies at D2S123 were 0.516, 0.129, 0.215, and 0.190 for a significant weighted mean of 0.235 compared to an initial mutant frequency of 0.453; whereas at D17S518 frequencies were 0.182, 0.087, 0.146, and 0.152 (Table 3) for a mean of 0.147, compared to an initial frequency of 0.207.
In addition, six out of seven normal controls demonstrated a total of 19 one repeat deletions and 12 one repeat insertions. The majority of variants seen in seven normal PBL sets are deletions (45/65 variants), with more than 1/3 being one repeat deletions (19/45).
We conclude that the phenomenon of higher mutant frequency in the PBL DNA of patients carrying MMR gene mutations considered to be the genetic basis of HNPCC is real and repeatable.
Heterogeneity of MSI in PBL DNA of HNPCC Patients
Although the mean frequencies of microsatellite mutant fragments in PBL DNA over all three loci in the screen in each HNPCC patient were higher than in their age-matched controls, there was a wide variation in those frequencies amongst the patients, that is, from 0.04 to 0.20. Because MSI has been shown to increase with age in normal individuals [Coolbaugh-Murphy et al., 2005; Slebos et al., 2008], the contribution of age to that heterogeneity was evaluated by plotting MSI against age. Added to the normal controls in this analysis were the data from the normal individuals studied previously [Coolbaugh-Murphy et al., 2005] bringing the total studied to 23 (Fig. 2). Mutant frequencies from the PBL DNA of the sporadic CRC patients were not included because they are not in the same age range but are only in the oldest age category. The resultant positive slope of mutant frequency with age in the PBL DNA of normal individuals is no different than it was in the original study and further supports that earlier observation that MSI increases with age in normal individuals. A much more dramatically increased MSI with age in the PBLs of the HNPCC patients than in controls can partially account for the heterogeneity seen in the HNPCC PBLs. However, the effect of aging cannot explain the extraordinary, and repeatedly high, mutant frequency in the PBL DNA of 36 year old patient H4 who had an MSH2 splice mutation as the basis of his MSI.
Figure 2.

Comparison of PBL mutant frequencies from normal controls and MSI-H HNPCC patients (by age in years). The squares represent the normal controls used in this study, and those from our previously published aging study (MAD 126:1051, 2005). The circles represent the MSI-H HNPCC patient PBL DNAs used in this study and the patient previously published, (Genomics 84:419, 2004.) The lower line on the graph is the linear regression line for the normal controls. Its equation is the bottom one in the upper left corner. The top equation is for the upper linear regression line for the MSI-H patients, circles. The numbers associated with the circles are the patient IDs.
Discussion
Advantages and Limitation of SPPCR Quantification of MSI
The SPPCR method makes it possible to quantify frequencies of mutant fragments at individual microsatellite loci, and therefore it provides a sensitive measure of MSI. Its sensitivity derives from the ability of the method to detect mutant microsatellite fragments that would otherwise be obscured by progenitor alleles when using traditional PCR. This is most clearly demonstrated in Figure 1, where the progenitor alleles are 89 and 95 bp for D17S518. In panel d a mutant allele of 93 bp is detected alone in the well. Being infrequent, these mutant alleles would never be resolved unless trapped with few alleles in a small pool. As indicated in Table 1, many mutants are in this category (one repeat unit smaller than a progenitor) and would all be missed if not for SPPCR. At the same time, the mutant frequencies observed are underestimates. This is particularly true for a locus that is heterozygous with progenitor fragments being one repeat unit apart. Then, for instance, a one-epeat unit deletion mutant of the larger progenitor would be indistinguishable from the smaller progenitor and not be scored as a mutant. Finally, although the system is quantitative because individual mutants can be counted, mutation frequencies cannot be precisely determined because one cannot determine whether the same size mutants are the result of different mutational events, or the same event in which the mutant has been replicated. Also, because the mutants are not measured according to any time event (numbers of replications, cell divisions, or days in culture), mutation rates cannot be derived from such data. However, increased mutant frequencies observed over controls are most logically considered a function of increased mutation frequency and rate.
This approach has enabled us to identify mutant fragments in noncancerous tissue (e.g., PBL) in individuals carrying a germline MMR mutation. Due to the stochastic nature of the mutation process, it is possible that even though there is increased probability for mutation, any one locus may not have had the mutational event occur at a sufficiently early stage of tissue development for mutant fragments to be present in enough descendant cells to be detected above background, even by SPPCR. Using an average mutant frequency identified by SPPCR over three loci, we quantified the low, but increasing with age, levels of MSI in PBL DNA in normal individuals [Coolbaugh-Murphy et al., 2005]. That same approach was effective here.
MSI in Constitutive (Nontumor) Tissue
Using standard PCR, MSI had been detected in normal tissues of children with neurofibromatosis Type 1 symptoms. However, those subjects were homozygous for mutations in the MMR gene MLH1 [Gallinger et al. 2004; Vilkki et al.2001; Wang et al.1999]. Our interest here is the ability to detect MSI in normal tissues heterozygous for germline MMR mutations. Gallinger et al. [Chen et al. 2007; Gallinger et al. 2004] employed SPPCR and found 2.5–2.7% MSI at the mononucleotide repeat BAT26 in the PBLs of the mother of one of the MLH1 homozygous children. The lower level of MSI than seen in the studies reported here may very well have been due the underestimation of mutants of mononucleotide repeats discussed earlier [Coolbaugh-Murphyetal.,2005]. MSI in the PBL of MMR mutation carriers was observed using a PCR cloning approach [Alazzouzi et al., 2005]. In that study, 5.6% of BAT26 clones had nonprogenitor fragments, whereas no variant fragments were detected in clones from family members who did not carry a MMR mutation. The cloning method introduced isolated single fragments into clones that could be quantified; however, the single locus assay, and the fact that the procedure introduces selection steps, make estimations of the in vivo mutant frequencies problematical. The inspiration for our study was the use of a form of SPPCR in which MSI was detected in DNA from Epstein-Barr virus transformed lymphoblastoid cells from some HNPCC patients [Parsons et al.,1995]. In analyzing PBLs directly we mitigated any selection events associated with cloning or cell transformation or any impact viral infection may have had on MSI because MSI in human cells had been seen to be promoted by the hepatitis C core protein [Naganuma et al., 2004].
Bases for the Heterogeneity of Elevated MSI in Constitutive Tissues of MMR Mutation Carriers
Parsons et al. [1995] reported that only some (three of six) HNPCC patients had MSI variation in lymphoblastoid cell line DNA. Similarly, we saw much higher MSI (reproducibly) in some HNPCC patients rather than others. Although this was partially explainable by the age effect in MSI (higher in older people) [Coolbaugh-Murphy et al., 2005], it is likely that the nature of the MMR germline mutation in the family is a factor. Recent developments in the in silico evaluation of missense MMR mutations and the function of the protein complex are useful to consider relative to our observations. The use of MAPP-MMR [Chao et al., 2008] demonstrates that highly pathogenic missense variants produce a high score, as seen in the score of 44 for Patient H6's MSH2 G683R variant. When compared to the score of 3.5 for patient H1's MLH1 D591G variant, it would not be too surprising to see that different variants have varying severity of effect. This idea is supported functionally when looking at the microsatellite mutant frequencies in these same patients' PBL DNAs. The patient with the more severe MMR gene mutation had higher levels of MSI in PBL DNA (H6's MF of 14% as contrasted with H1's 4%). This is consistent with the observation of Parsons et al. [1995] that two of their three patients with MSI in their lymphoblastoid cells were sibs who shared the same PMS2 mutation. Although it was suggested [Nicolaides et al., 1998] that the mutation lead to a dominant negative effect resulting in MSI in constitutive tissue, any decrease in repair efficiency even in only one of two alleles may impact MSI and resultant carcinogenesis.
Significance of Elevated MSI in Constitutive Tissues of MMR Mutation Carriers
Although it would be useful for this approach to be applied at the clinical level, due to the heterogeneity discussed above, it is not possible to unambiguously score HNPCC from the PBL DNA. Where PBL MSI in on the high end it would be possible because the levels in controls are so low. However, false negatives at the lower levels of HNPCC patients and false positives at the higher levels in older normal individuals preclude such a clinical application. Considering the labor intensiveness of the small pool procedures, there was never any intention or claim to make this a clinical test. The purpose here was to show that the elevated MSI in nontumor tissue was a real phenomenon, and elucidation of the mechanism (haploinsuffciency, dominant/negative effect, modifier genes and mutations, etc.) and targets of such non-Mendellian mutations may allow genetic testing to identify who is at risk for early onset HNPCC or even which noncarriers still have increased cancer risk in HNPCC families.
Importantly, our finding that mutations in MMR genes can result in increased MSI in constitutive cells is of particular significance as a result of a recent report of anticipation in HNPCC [Nilbert et al., 2009]. There, patients with pathogenic MMR mutations developed cancer at earlier ages of onset and with greater severity in successive generations. Our finding suggests that instability can occur in other tissues of HNPCC patients including germ cells. Therefore, individuals carrying germline mutations in MMR genes might well be expected to experience low levels of MSI in germ cells and pass on mutant alleles to offspring at a higher frequency than individuals not having such germline mutations. These mutations passed on to HNPCC offspring would be new non-Mendellian variants that could be detected in the germline DNA of the offspring. Some of the mutations could convey an increased risk for early onset cancer, thereby explaining the mechanism of genetic anticipation in HNPCC.
When anticipation is seen in neuromuscular disease its molecular basis presents as expansions at trinucleotide repeats of a single locus [Caskey et al., 1992]. In comparison, in HNPCC, the mutations are not likely to occur at any single locus; rather, the mutations would occur at numerous locations across the genome in which MMR had not been carried out and which now contain mutant sequences. An increased number of such germline mutations throughout the genome with each successive generation is seen as a reasonable mechanism to influence disease penetrance, age of onset, disease progression, response to treatment or other aspects of carcinogenesis [Nilbert et al., 2009]. This becomes a hypothesis that can be tested now that we have data verifying that MMR is not carried out efficiently in the constitutive cells of HNPCC patients.
Acknowledgments
The authors thank TexGen Research and the Center for Clinical and Translational Sciences (CCTS) of the University of Texas Health Science Center at Houston for their kind support and permission to use its data and samples. This work was supported by the National Cancer Institute [grants CA112508 to M.J.S., CA070759 to M.L.F., CA57730 -R25T cancer prevention postdoctoral fellowship to M.C.M., CA-16672NIH Cancer Center Support Grant (DNA Analysis Facility and Biospecimen Extraction Resource) to J. Mendelsohn, TexGen]; and the Rosalie B. Hite Cancer Research Fellowship (to M.C.M.).
References
- Aaltonen LA, Peltomaki P, Leach F, Sistonen P, Pylkkanen SM. Clues to the pathogenesis of familial colorectal cancer. Science. 1993;260:812. doi: 10.1126/science.8484121. [DOI] [PubMed] [Google Scholar]
- Alazzouzi H, Domingo E, Gonzalez S, Blanco I, Armengol M, Espin E, Plaja A, Schwartz S, Capella G, Schwartz S., Jr Low levels of microsatellite instability characterize MLH1 and MSH2 HNPCC carriers before tumor diagnosis. Hum Mol Genet. 2005;14:235–239. doi: 10.1093/hmg/ddi021. [DOI] [PubMed] [Google Scholar]
- Boland CR. Clinical uses of microsatellite instability testing in colorectal cancer: an ongoing challenge. J Clin Oncol. 2007;25:754–756. doi: 10.1200/JCO.2006.09.4607. [DOI] [PubMed] [Google Scholar]
- Boland CR, Fishel R. Lynch syndrome: form, function, proteins, and basketball. Gastroenterology. 2005;129:751–755. doi: 10.1016/j.gastro.2005.05.067. [DOI] [PubMed] [Google Scholar]
- Bronner CE, Baker SM, Morrison PT, Warren G, Smith LG, Lescoe MK, Kane M, Earabino C, Lipford L, Lindblom A. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature. 1994;368:258–261. doi: 10.1038/368258a0. [DOI] [PubMed] [Google Scholar]
- Canzian F, Salovaara R, Hemminki A, Kristo P, Chadwick RB, Aaltonen LA, de la Chapelle A. Semiautomated assessment of loss of heterozygosity and replication error in tumors. Cancer Res. 1996;56:3331–3337. [PubMed] [Google Scholar]
- Caskey CT, Pizzuti A, Fu Y-H, Fenwick RG, Nelson DL. Triplet repeat mutations in human disease. Science. 1992;256:784. doi: 10.1126/science.1589758. [DOI] [PubMed] [Google Scholar]
- Chao EC, Velasquez JL, Witherspoon MS, Rozek LS, Peel D, Ng P, Gruber SB, Watson P, Rennert G, Anton-Culver H, Lynch H, Lipkin SM. Accurate classification of MLH1/MSH2 missense variants with multivariate analysis of protein polymorphisms-mismatch repair (MAPP-MMR) Hum Mutat. 2008;29:852–860. doi: 10.1002/humu.20735. [DOI] [PubMed] [Google Scholar]
- Chen J, Sen S, Amos CI, Wei C, Jones JS, Lynch P, Frazier ML. Association between Aurora-A kinase polymorphisms and age of onset of hereditary nonpolyposis colorectal cancer in a Caucasian population. Mol Carcinog. 2007;46:249–256. doi: 10.1002/mc.20283. [DOI] [PubMed] [Google Scholar]
- Coolbaugh-Murphy M, Maleki A, Ramagli L, Frazier M, Lichtiger B, Monckton DG, Siciliano MJ, Brown BW. Estimating mutant microsatellite allele frequencies in somatic cells by small-pool PCR. Genomics. 2004;84:419–430. doi: 10.1016/j.ygeno.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Coolbaugh-Murphy MI, Xu J, Ramagli LS, Brown BW, Siciliano MJ. Microsatellite instability (MSI) increases with age in normal somatic cells. Mech Age Dev. 2005;126:1051–1059. doi: 10.1016/j.mad.2005.06.005. [DOI] [PubMed] [Google Scholar]
- de la Chapelle A. Genetic predisposition to colorectal cancer. Nat Rev Cancer. 2004;4:769–780. doi: 10.1038/nrc1453. [DOI] [PubMed] [Google Scholar]
- Fishel R, Lescoe MK, Rao MR, Copeland NG, Jenkins NA, Garber J, Kane M, Kolodner R. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer [Erratum appears in Cell 1994. 77:167] Cell. 1993;75:1027–1038. doi: 10.1016/0092-8674(93)90546-3. [DOI] [PubMed] [Google Scholar]
- Frazier ML, O'Donnell FT, Kong S, Gu X, Campos I, Luthra R, Lynch PM, Amos CI. Age-associated risk of cancer among individuals with N-acetyltransferase 2 (NAT2) mutations and mutations in DNA mismatch repair genes. Cancer Res. 2001;61:1269–1271. [PubMed] [Google Scholar]
- Frazier ML, Xi L, Zong J, Viscofsky N, Rashid A, Wu EF, Lynch PM, Amos CI, Issa JP. Association of the CpG island methylator phenotype with family history of cancer in patients with colorectal cancer. Cancer Res. 2003;63:4805–4808. [PubMed] [Google Scholar]
- Gallinger S, Aronson M, Shayan K, Ratcliffe EM, Gerstle JT, Parkin PC, Rothenmund H, Croitoru M, Baumann E, Durie RP, Weksberg R, Pollett A, Riddell RH, Ngan BY, Cutz E, Lagarde AE, Chan HS. Gastrointestinal cancers and neurofibromatosis type 1 features in children with a germline homozygous MLH1 mutation. Gastroenterology. 2004;126:576–585. doi: 10.1053/j.gastro.2003.11.008. [DOI] [PubMed] [Google Scholar]
- Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, Nakagawa H, Sotamaa K, Prior TW, Westman J, Panescu J, Fix D, Lockman J, Comeras I, de la Chapelle A. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer) N Engl J Med. 2005;352:1851–1860. doi: 10.1056/NEJMoa043146. [DOI] [PubMed] [Google Scholar]
- Hemminki A, Peltomäki P, Mecklin J-P, Jaärvinen H, Salovaara R, Nyström-Lahti M, de la Chapelle A, Aaltonen LA. Loss of the wild type MLH1 gene is a feature of hereditary nonpolyposis colorectal cancer. Nat Genet. 1994;8:405–410. doi: 10.1038/ng1294-405. [DOI] [PubMed] [Google Scholar]
- Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993;363:558–561. doi: 10.1038/363558a0. [DOI] [PubMed] [Google Scholar]
- Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup M, Kolodner R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57:808–811. [PubMed] [Google Scholar]
- Kunkel TA, Erie DA. DNA Mismatch Repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- Leach FS, Nicolaides NC, Papadopoulos N, Liu B, Jen J, Parsons R, Peltomaki P, Sistonen P, Aaltonen LA, Nystrom-Lahti M. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell. 1993;75:1215–1225. doi: 10.1016/0092-8674(93)90330-s. [DOI] [PubMed] [Google Scholar]
- Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18:85–98. doi: 10.1038/cr.2007.115. [DOI] [PubMed] [Google Scholar]
- Liu B, Parsons RE, Hamilton SR, Petersen GM, Lynch HT, Watson P, Markowitz S, Willson JK, Green J, de la Chapelle A, Kinzler KW, Vogelstein B. hMSH2 mutations in hereditary nonpolyposis colorectal cancer kindreds. Cancer Res. 1994;54:4590–4594. [PubMed] [Google Scholar]
- Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348:919. doi: 10.1056/NEJMra012242. [DOI] [PubMed] [Google Scholar]
- Monckton DG, Jeffreys AJ. Minisatellite “isoallele” discrimination in pseudohomozygotes by single molecule PCR and variant repeat mapping. Genomics. 1991;11:465–467. doi: 10.1016/0888-7543(91)90158-b. [DOI] [PubMed] [Google Scholar]
- Naganuma A, Dansako H, Nakamura T, Nozaki A, Kato N. Promotion of microsatellite instability by hepatitis C virus core protein in human nonneoplastic hepatocyte cells. Cancer Res. 2004;64:1307–1314. doi: 10.1158/0008-5472.can-03-2992. [DOI] [PubMed] [Google Scholar]
- Nicolaides NC, Littman SJ, Modrich P, Kinzler KW, Vogelstein B. A naturally occurring hPMS2 mutation can confer a dominant negative mutator phenotype. Mol Cell Biol. 1998;18:1635–1641. doi: 10.1128/mcb.18.3.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaides NC, Papadopoulos N, Liu B, Wei YF, Carter KC, Ruben SM, Rosen CA, Haseltine WA, Fleischmann RD, Fraser CM. Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature. 1994;371:75–80. doi: 10.1038/371075a0. [DOI] [PubMed] [Google Scholar]
- Nilbert M, Timshel S, Bernstein I, Larsen K. Role for genetic anticipation in Lynch syndrome. J Clin Oncol. 2009;27:360–364. doi: 10.1200/JCO.2008.16.1281. [DOI] [PubMed] [Google Scholar]
- Palombo F, Iaccarino I, Nakajima E, Ikejima M, Shimada T, Jiricny J. hMutSbeta, a heterodimer of hMSH2 and hMSH3, binds to insertion/deletion loops in DNA. Curr Biol. 1996;6:1181–1184. doi: 10.1016/s0960-9822(02)70685-4. [DOI] [PubMed] [Google Scholar]
- Papadopoulos N, Nicolaides NC, Wei YF, Ruben SM, Carter KC. Mutation of a mutL homolog in hereditary colon cancer. Science. 1994;263:1625. doi: 10.1126/science.8128251. [DOI] [PubMed] [Google Scholar]
- Parsons R, Li GM, Longley M, Modrich P, Liu B, Berk T, Hamilton SR, Kinzler KW, Vogelstein B. Mismatch repair deficiency in phenotypically normal human cells. Science. 1995;268:738–740. doi: 10.1126/science.7632227. [DOI] [PubMed] [Google Scholar]
- Parsons R, Li GM, Longley MJ, Fang WH, Papadopoulos N, Jen J, de la Chapelle A, Kinzler KW, Vogelstein B, Modrich P. Hypermutability and mismatch repair deficiency in RER1 tumor cells. Cell. 1993;75:1227–1236. doi: 10.1016/0092-8674(93)90331-j. [DOI] [PubMed] [Google Scholar]
- Slebos RJ, Li M, Vadivelu S, Burkey BB, Netterville JL, Sinard R, Gilbert J, Murphy B, Chung CH, Shyr Y, Yarbrough WG. Microsatellite mutations in buccal cells are associated with aging and head and neck carcinoma. Br J Cancer. 2008;98:619–626. doi: 10.1038/sj.bjc.6604198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone EA, Sidow A. Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome Res. 2005;15:978–986. doi: 10.1101/gr.3804205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibodeau SN, Bren G, Schaid D. Microsatellite instability in cancer of the proximal colon. Science. 1993;260:816–819. doi: 10.1126/science.8484122. [DOI] [PubMed] [Google Scholar]
- Vilkki S, Tsao JL, Loukola A, Poyhonen M, Vierimaa O, Herva R, Aaltonen LA, Shibata D. Extensive somatic microsatellite mutations in normal human tissue. Cancer Res. 2001;61:4541–4544. [PubMed] [Google Scholar]
- Wang Q, Lasset C, Desseigne F, Frappaz D, Bergeron D. Neurofibromatosis and early onset of cancers in hMLH1-deficient children. Cancer Res. 1999;59:294. [PubMed] [Google Scholar]
- Wildeman M, van Ophuizen E, den Dunnen JT, Taschner PE. Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. Hum Mutat. 2008;29:6–13. doi: 10.1002/humu.20654. [DOI] [PubMed] [Google Scholar]