

Abstract
The representation of chlorine, bromine, and iodine in aryl halides has been modified in the OPLS-AA and OPLS/CM1A force fields in order to incorporate halogen bonding. The enhanced force fields, OPLS-AAx and OPLS/CM1Ax, have been tested in calculations on gas-phase complexes of halobenzenes with Lewis bases, and for free energies of hydration, densities, and heats of vaporization of halobenzenes. Comparisons with results of MP2/aug-cc-pVDZ(-PP) calculations for the complexes are included. Implementation in the MCPRO software also allowed computation of relative free energies of binding for a series of HIV reverse transcriptase inhibitors via Monte Carlo/free-energy perturbation calculations. The results support the notion that the activity of an unusually potent chloro analog likely benefits from halogen bonding with the carbonyl group of a proline residue.
INTRODUCTION
Halogen bonding is now well recognized to occur commonly between Lewis bases and alkyl or aryl chlorides, bromides, and iodides.1 Depletion of electron density on the backside of the covalent bond to the halogen leads to a “σ-hole” with positive electrostatic potential that can favorably interact with a lone pair of electrons on a heteroatom (Figure 1). The presence of such interactions in protein-ligand complexes has been noted,2 and particularly striking results were recently reported for inhibitors of cathepsin L.3 Naturally, for related computational work including ligand design, it is desirable to properly represent halogen bonding in force fields that are used routinely for molecular modeling and condensed-phase simulations of organic and biomolecular systems. However, the most widely used force fields for biomolecular modeling including AMBER, CHARMM, GROMOS, and OPLS-AA represent electrostatic interactions via a single partial atomic charge on each atom.4 Halogen atoms in alkyl and aryl halides are assigned partial negative charges to be consistent with observed dipole moments, and consequently their electrostatic interactions with heteroatoms bearing lone pairs of electrons as in Figure 1 are all incorrectly repulsive.
Figure 1.

Examples of halogen-bonded complexes; bromobenzene with acetone, water, N-methylacetamide, acetonitrile, trimethylamine, and pyridine. Structures have been optimized with the OPLS-AAx force field.
Limitations of the atomic point-charge model have been recognized since its inception; the obvious way to improve the description of electrostatic potentials is to add additional charged sites.5,6 For example, the addition of charged sites in lone-pair-like positions has proven beneficial in modeling ethyl anion, heterocycles, amines, and water.7 An analogous modification to enable halogen bonding is to add a partial positive charge in the region of the σ-hole along the C-X axis. Initial results using this approach with AMBER force fields have shown that it is possible to improve significantly the geometries and interaction energies for halogen-bonded complexes.8,9 In the present study, implementation and testing of a similar model in the OPLS-AA force field is reported with emphasis on aryl halides. The necessary force field parameters are provided and the testing has included study of gas-phase complexes, free energies of hydration, and pure liquid properties of halobenzenes. Motivation was also enhanced by our recent experimental discovery of catechol diethers as potent anti-HIV agents.10 Compounds 1 – 3 are non-nucleoside inhibitors of HIV reverse transcriptase (NNRTIs) with EC50 values for inhibition of viral replication in human T-cells of 5.0, 3.2 and 0.055 nM, respectively. The possibility that the extreme potency of 3 might arise in part from a halogen bond between the X = Cl substituent and the carbonyl oxygen of Pro95 was raised (Figure 2).10 This issue is also investigated here through free-energy perturbation calculations with and without the extra point charges on the halogens.
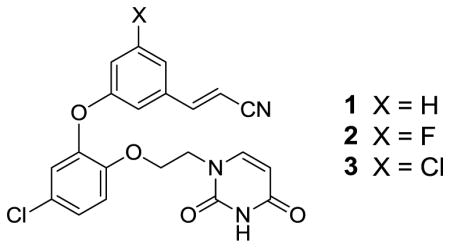
Figure 2.

Computed structure for the complex of 3 with wild-type HIV-1 reverse transcriptase. The possible halogen-bonding interaction with Pro95 is highlighted with the dashed arrow. Carbon atoms of 3 are in yellow. For further details, see ref. 10.
RESULTS AND DISCUSSION
Implementation and Parameterization
The optional addition of the extra sites (X-sites) on chlorine, bromine, and iodine was implemented in BOSS, MCPRO and BOMB, which are used for modeling organic systems, modeling biomolecular systems, and building molecular structures, respectively.11,12 The modeling with the OPLS-AA force field13 includes energy minimizations, and Monte Carlo statistical mechanics simulations for pure liquids and solutions of solutes in any solvent.11,12 The expansion of the force field required definition of new atom types, XC, XB, and XI, for X-sites on chlorine, bromine, and iodine. The force field without the X-sites will continue to be referred to as OPLS-AA and the variant with the X-sites is denoted OPLS-AAx. The X-sites are treated as particles with no mass that are connected to the halogens with stiff bond stretching and angle bending force constants. The addition of the single X-site on a halogen atom is viewed as the simplest modification that can capture the basic features of halogen bonding, while remaining consistent with the form of fixed-charge biomolecular force fields. Consistent with prevailing views,1 the X-sites have not been added to fluorine for fluorobenzenes.
The parameters that were derived and used here are summarized in Tables 1 and 2, which also include the original OPLS-AA parameters for halobenzenes.13 Table 1 contains the partial atomic charges, q, and the Lennard-Jones parameters, σ and ε. Table 2 lists the harmonic bond-stretching and angle-bending parameters. In addition, the torsional energy consists of a two-fold cosine term with V2 = 7.25 kcal/mol for every X-CA-CA-Y quartet, which may be augmented with an improper dihedral angle term with V2 = 5.0 kcal/mol for each CA to reduce out-of-plane bending.13
Table 1.
Non-Bonded Parameters for Halobenzenes
| Atoma | q, e | σ, Å | ε, kcal/mol |
|---|---|---|---|
| OPLS-AA: | |||
| CA | −0.115 | 3.55 | 0.070 |
| HA | 0.115 | 2.42 | 0.030 |
| Fb | −0.220 | 2.85 | 0.061 |
| Cl | −0.180 | 3.40 | 0.300 |
| Br | −0.200 | 3.47 | 0.470 |
| I | −0.100 | 3.75 | 0.600 |
| CA(F) | 0.220 | 3.55 | 0.070 |
| CA(Cl) | 0.180 | 3.55 | 0.070 |
| CA(Br) | 0.200 | 3.55 | 0.070 |
| CA(I) | 0.100 | 3.55 | 0.070 |
| OPLS-AAx: | |||
| Cl | −0.250 | 3.40 | 0.300 |
| XC | 0.075 | 0.0 | 0.0 |
| Br | −0.270 | 3.47 | 0.470 |
| XB | 0.100 | 0.0 | 0.0 |
| I | −0.260 | 3.75 | 0.600 |
| XI | 0.110 | 0.0 | 0.0 |
| CA(Cl) | 0.175 | 3.55 | 0.070 |
| CA(Br) | 0.170 | 3.55 | 0.070 |
| CA(I) | 0.150 | 3.55 | 0.070 |
CA is the OPLS atom type for a carbon atom in a 6-membered aromatic ring and HA is an attached hydrogen.
For difluorobenzenes, qF = −0.20; for penta- and hexa-fluorobenzenes, qF = −0.13.
Table 2.
Bond Stretching and Bond Angle Bending Parameters for Halobenzenes
| Bond | r0 (Å) | kb(kcal/mol- Å2) | Angle | θ0 (deg) | kθ(kcal/mol-rad2) |
|---|---|---|---|---|---|
| CA-HA | 1.080 | 367.0 | CA-CA-CA | 120.0 | 63.0 |
| CA-CA | 1.400 | 469.0 | CA-CA-HA | 120.0 | 35.0 |
| CA-F | 1.354 | 420.0 | CA-CA-F | 120.0 | 80.0 |
| CA-Cl | 1.725 | 300.0 | CA-CA-Cl | 120.0 | 75.0 |
| CA-Br | 1.870 | 300.0 | CA-CA-Br | 120.0 | 75.0 |
| CA-I | 2.080 | 250.0 | CA-CA-I | 120.0 | 75.0 |
| Cl-XC | 1.600 | 600.0 | CA-Cl-XC | 180.0 | 200.0 |
| Br-XB | 1.600 | 600.0 | CA-Br-XB | 180.0 | 200.0 |
| I-XI | 1.800 | 600.0 | CA-I-XI | 180.0 | 200.0 |
As indicated, the charge on the X-sites is 0.075 – 0.110 e, and, as for hydrogens on heteroatoms,13 the Lennard-Jones parameters for the X-sites are zero. The Cl-XC and Br-XB equilibrium distances are 1.60 Å, while the I-XI distance is 1.80 Å. The corresponding distances that have been used previously are in the range 1.4 – 2.4 Å.8,9 A concern with longer distances is that, if the Lennard-Jones parameters for the X-sites are zero, overly close approach to negatively charged sites may occur in a simulation, i.e., the X-site is not sufficiently inside the Lennard-Jones sphere of the halogen. Thus, the present OPLS-AAx model should not be used with TIP5P water7 as the X-sites and water lone-pair sites can fuse; there is no problem with TIP3P and TIP4P as the negative charge is shielded by the Lennard-Jones sphere centered on the oxygen atom.14 If needed, the issue can be avoided by the introduction of small Lennard-Jones parameters for the X-sites. It is noted that the choice8 of r* (= 21/6σ/2) of 1.0 Å and ε = 0.0 has no effect since geometric combining rules are used for ε in the AMBER force fields.15
For more general treatment of organic molecules, we use the OPLS/CM1A force field that features CM1A atomic charges, which are obtained from an AM1 calculation.4c,16 The problem is that OPLS-AA partial charges are not available for all molecules, e.g., those containing the wide range of heterocycles featured in drugs. To implement the X-sites in this case, it was found viable to compute the CM1A charges as usual, then assign a charge of 0.075, 0.10, and 0.11 e to the X sites for chlorine, bromine, and iodine and to add the equivalent negative charge to the halogen. The CM1A charges for neutral molecules are scaled by a factor of 1.14 for the force fields.4c The expected benefit of the OPLS/CM1Ax alternative over OPLS-AAx is better treatment of polarization of the charge on the halogen by substituents in the aryl ring.17 The partial charges on all atoms are influenced by the rest of the molecule with OPLS/CM1Ax; they are not fixed as with OPLS-AAx.
Gas-Phase Complexes
The parameterization initially emphasized reproduction of ab initio quantum mechanical results for gas-phase complexes. A extensive set of results for complexes of acetone with halobenzenes was recently reported by Riley et al.18 They carried out optimizations for planar complexes of chloro-, bromo-, and iodo-benzene and polyfluorinated analogs using MP2/aug-cc-pVDZ calculations with pseudopotentials (-PP) and Boys-Bernardi counterpoise corrections. Fluorination has the expected effect of increasing the σ-hole and increasing the intermolecular attraction.18,19 The MP2 results and those using OPLS-AAx and OPLS/CM1Ax are compared in Table 3. The geometry of a typical complex is illustrated in Figure 1. The interaction energies range from about −1 to −6 kcal/mol, becoming more attractive in progressing from Cl to Br to I and with increasing fluorination. In general the MP2 interaction energies are between the OPLS-AAx and OPLS/CM1Ax results. The mean unsigned error between the MP2 and OPLS-AA ΔE values is 0.68 kcal/mol and it is 0.55 kcal/mol for OPLS/CM1Ax. As the strengths of such interactions are somewhat underestimated with MP2/aug-cc-pVDZ(-PP) calculations,20 the OPLS/CM1Ax results appear to be particularly good. The range of optimal halogen…O distances is remarkably narrow, almost all values are between 3.0 and 3.2 Å. The optimal halogen…O=C angles are also almost all in a small range, 120° – 130°. The exceptions are computed linear values with OPLS/CM1Ax for F5PhBr, m-F2PhI, and F5PhI; however, in all cases the bending potential is very soft.18 For example, the energy difference for the F5PhI complex with the I…O=C angle fixed at 130° and the minimum at 180° is 0.15 kcal/mol with OPLS/CM1Ax.
Table 3.
Computed Interaction Energies (kcal/mol) and Geometries for Aryl Halide - Acetone Complexesa
| Ar-X | OPLS-AAx | OPLS/CM1Ax | MP2b | ||||||
|---|---|---|---|---|---|---|---|---|---|
| −ΔE | r(XO) | θ(XOC) | −ΔE | r(XO) | θ(XOC) | −ΔE | r(XO) | θ(XOC) | |
| PhCl | 1.62 | 3.18 | 119.5 | 2.05 | 3.12 | 122.1 | 1.30 | 3.16 | 118 |
| o-F2PhCl | 1.72 | 3.17 | 118.4 | 2.36 | 3.10 | 122.0 | 2.00 | 3.04 | 119 |
| m-F2PhCl | 1.92 | 3.15 | 121.8 | 2.62 | 3.08 | 127.7 | 1.74 | 3.10 | 120 |
| F5PhCl | 2.04 | 3.14 | 120.8 | 3.13 | 3.05 | 130.2 | 2.63 | 3.00 | 121 |
| PhBr | 2.30 | 3.18 | 122.0 | 2.97 | 3.12 | 125.9 | 2.23 | 3.13 | 122 |
| o-F2PhBr | 2.37 | 3.17 | 120.8 | 3.33 | 3.10 | 126.1 | 3.17 | 2.98 | 123 |
| m-F2PhBr | 2.61 | 3.16 | 124.2 | 3.62 | 3.09 | 137.5 | 2.78 | 3.04 | 123 |
| F5PhBr | 2.69 | 3.15 | 123.1 | 4.25 | 3.06 | 180.0 | 4.09 | 2.91 | 124 |
| PhI | 3.28 | 3.23 | 127.1 | 3.76 | 3.19 | 130.5 | 3.22 | 3.14 | 125 |
| o-F2PhI | 3.29 | 3.23 | 125.5 | 4.20 | 3.17 | 131.9 | 4.71 | 3.00 | 126 |
| m-F2PhI | 3.58 | 3.21 | 130.0 | 4.49 | 3.16 | 180.0 | 4.13 | 3.05 | 128 |
| F5PhI | 3.62 | 3.21 | 128.5 | 5.21 | 3.14 | 180.0 | 5.97 | 2.91 | 128 |
ΔE in kcal/mol; r(XO) in Å; θ(XOC) in deg.; ArX and acetone are constrained to be co-planar; all other geometrical variables were optimized with OPLS.
MP2/aug-cc-pVDZ(-PP) results from ref. 18. A grid search was used such that r(XO) and θ(XOC) were determined only to within 0.1 Å and 2.5°.
The analyses were extended to several additional prototypical complexes, i.e., with water, acetonitrile, trimethylamine, N-methylacetamide (NMA), and pyridine. We performed analogous MP2/aug-cc-pVDZ(-PP) optimizations including the counterpoise corrections in these cases using Gaussian 09.21 A minor difference is that we used full MP2, while the earlier work18,19 used the frozen core approximation. These were complete optimizations for the monomers and complexes with the only constraint being a linear C-halogen…O or N angle, and for NMA the complexes are planar as for acetone. Representative structures are illustrated in Figure 1 and the key results are summarized in Table 4.
Table 4.
Computed Interaction Energies (kcal/mol) and Halogen-O or -N Distances (Å)a
| Complex with | PhCl −ΔE | PhCl r(Cl-O,N) | PhBr −ΔE | PhBr r(Br-O,N) | PhI −ΔE | PhI r(I-O,N) |
|---|---|---|---|---|---|---|
| OPLS-AAx | ||||||
| water (TIP3P) | 1.23 | 3.16 | 2.04 | 3.16 | 3.39 | 3.19 |
| water (TIP4P) | 1.27 | 3.15 | 1.83 | 3.18 | 2.88 | 3.23 |
| NMAb | 0.56 | 3.18 | 1.32 | 3.20 | 2.38 | 3.24 |
| acetonitrile | 0.92 | 3.27 | 1.67 | 3.27 | 2.79 | 3.32 |
| trimethylamine | 2.08 | 3.27 | 3.00 | 3.26 | 4.33 | 3.30 |
| pyridine | 1.93 | 3.22 | 2.72 | 3.23 | 3.87 | 3.29 |
| OPLS/CM1Ax | ||||||
| water (TIP3P) | 1.88 | 3.11 | 2.99 | 3.11 | 4.06 | 3.17 |
| water (TIP4P) | 1.66 | 3.14 | 2.58 | 3.16 | 3.43 | 3.23 |
| NMAb | 1.08 | 3.14 | 2.07 | 3.16 | 2.91 | 3.21 |
| acetonitrile | 1.65 | 3.21 | 2.61 | 3.21 | 3.43 | 3.29 |
| trimethylamine | 2.70 | 3.20 | 3.94 | 3.19 | 5.00 | 3.28 |
| pyridine | 2.50 | 3.18 | 3.53 | 3.19 | 4.43 | 3.27 |
| MP2c | ||||||
| water | 0.47 | 3.23 | 1.18 | 3.18 | 1.87 | 3.24 |
| NMAb | 1.80 | 3.13 | 2.88 | 3.05 | 4.03 | 3.09 |
| acetonitrile | 0.66 | 3.27 | 1.56 | 3.21 | 2.49 | 3.26 |
| trimethylamine | 1.99 | 3.10 | 3.70 | 2.99 | 5.71 | 2.98 |
| pyridine | 1.44 | 3.17 | 2.83 | 3.07 | 4.43 | 3.06 |
As before, the overall accord between the force field and MP2 results appears acceptable. The interactions from the force fields for water may be somewhat too strong, and with NMA too weak. The interactions from the force fields are a little more favorable for TIP3P water than TIP4P water except for PhCl. The order trimethylamine > pyridine > acetonitrile from all methods is consistent with expectations from nitrogen hybridization and basicity. It should be recognized that lower-energy structures without halogen bonds can be found for these complexes. For example, the complex of PhBr with a π-hydrogen-bonded TIP4P water molecule has an interaction energy of −2.90 kcal/mol using OPLS-AAx versus −1.83 kcal/mol for the halogen-bonded alternative, and another energy minimum has an interaction energy of −2.74 kcal/mol with the water molecule bridging between the bromine and an ortho-hydrogen. For PhCl, the π and bridging structures have similar interaction energies, −2.86 and −2.66 kcal/mol. In addition, for NMA with PhBr, five options are shown in Figure 3 with their interaction energies. The coplanar structure from Table 4 with a ΔE of −1.32 kcal/mol is not an energy minimum, while the four additional structures are energy minima. The two π-hydrogen-bonded structures and the last one, which features a hydrogen bond with the bromine, all have interaction energies near −3.9 kcal/mol.
Figure 3.

Five alternative structures for the complex of bromobenzene with N-methylacetamide. The computed interaction energies using OPLS-AAx are shown in kcal/mol.
Liquid State Results
Some liquid state properties were also tested for the OPLS-AAx force field starting with relative free energies of hydration. Free energy calculations were executed for perturbing PhF → PhCl → PhBr → PhI in a periodic cube containing 500 TIP4P water molecules at 25 C and 1 atm. The requisite Metropolis Monte Carlo (MC) simulations were performed using the BOSS 2.9 program with 11 windows of simple overlap sampling (SOS) in the exact manner as previously detailed.22
The results for OPLS-AA22 and OPLS-AAx are summarized in Table 5. For PhF, OPLS-AA and OPLS-AAx are the same since there is no X site. Thus, the difference in the free energies of hydration for PhCl and PhBr with and without the X sites is negligible. The halogen- bonding interactions in these cases are not more favorable than water-PhCl or water-PhBr Lennard-Jones or π-hydrogen-bonding interactions. For example, from the PhBr MC simulations the average total solute-solvent interaction energy (ESX) is −16.1 ± 0.2 kcal/mol for both OPLS-AA and OPLS-AAx. The presence or absence of a halogen-bond has little effect on this total. However, there is some benefit for PhI with OPLS-AAx; the 0.7 kcal/mol lowering of ΔGhyd for PhI improves the accord with experiment. In this case, the potentially stronger halogen bond does seem to help lower the average ESX from −16.2 to −17.4 ± 0.2 kcal/mol in going from OPLS-AA to OPLS-AAx. In viewing configurations from the OPLS-AAx MC simulations, as in Figure 4, there is an increased occurrence of water molecules in the vicinity of the halogens with their oxygens oriented in the manner expected for a halogen bond.
Table 5.
Computed Relative Free Energies of Hydration (kcal/mol)a
| PhX | OPLS-AA | OPLS-AAx | Exptb |
|---|---|---|---|
| PhF | 0.0 | 0.0 | 0.0 |
| PhCl | 0.12 | 0.06 | −0.31 |
| PhBr | −0.45 | −0.39 | −0.64 |
| PhI | −0.21 | −0.91 | −0.94 |
Uncertainties (±σ) in the computed values are ca. 0.05 kcal/mol.
Ref. 23.
Figure 4.
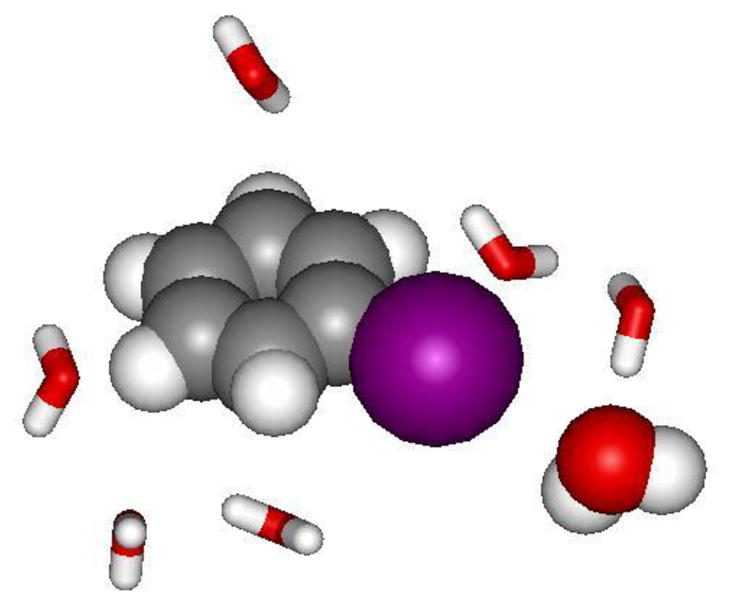
The seven water molecules closest to iodobenzene from the last configuration of a MC simulation that included 500 water molecules in a periodic cube. The iodobenzene and the halogen-bonded water molecule are rendered as space-filling; the I-O distance is 3.79 Å.
MC simulations were also carried out with the BOSS program for the pure liquids of the halobenzenes at 25 C and 1 atm. Standard procedures were followed for N = 267 molecules in a periodic cube with 14 Å spherical cutoffs based on the ring center - ring center separation and with a correction for the Lennard-Jones interactions neglected beyond the cutoff.13a The simulations covered 3 million (M) configurations for equilibration and 3 M configurations for averaging. The principal concern was reproduction of the experimental densities and heats of vaporization as these properties are indicative of proper representation of the size of the molecules and their average intermolecular interactions. Thus, corresponding MC simulations were carried out for the halobenzene monomers in the gas phase in order to compute the heat of vaporization in the standard way, i.e., ΔHvap = E(gas) − E(liq)/N + RT.13 The results are summarized in Table 5. There is little difference between the OPLS-AA and OPLS-AAx results except for a 0.4 kcal/mol improvement for the ΔHvap of bromobenzene with OPLS-AAx. The similarity of the results reflects that the changes introduced by addition of the X-sites are confined to a small region of the total molecular volume. Normal aryl-aryl interactions are expected to dominate (Figure 5). The level of agreement with the experimental data is also typical for OPLS force fields with average errors of ca. 2%.13 Nevertheless, it is important to carry out such checks to make sure that no anomalies arise upon introduction of a new feature such as the X-sites before simulations of more complex systems are undertaken.
Table 5.
Computed Densities and Heats of Vaporization of Pure Liquids at 25 C and 1 atma
| PhX | ρ (g cm−3) | ΔHvap (kcal mol−1) | ||||
|---|---|---|---|---|---|---|
| OPLS-AA | OPLS-AAx | Exptb | OPLS-AA | OPLS-AAx | Exptb | |
| PhF | 1.001 | 1.001 | 1.019 | 7.99 | 7.99 | 8.26 |
| PhCl | 1.103 | 1.101 | 1.101 | 9.85 | 9.71 | 9.79 |
| PhBr | 1.532 | 1.530 | 1.488 | 11.09 | 10.66 | 10.65 |
| PhI | 1.888 | 1.897 | 1.823 | 11.81 | 11.83 | 11.85 |
Uncertainties (±σ) in the computed densities and ΔHvap are ca. 0.002 g cm−3 and 0.04 kcal/mol.
Ref. 24.
Figure 5.
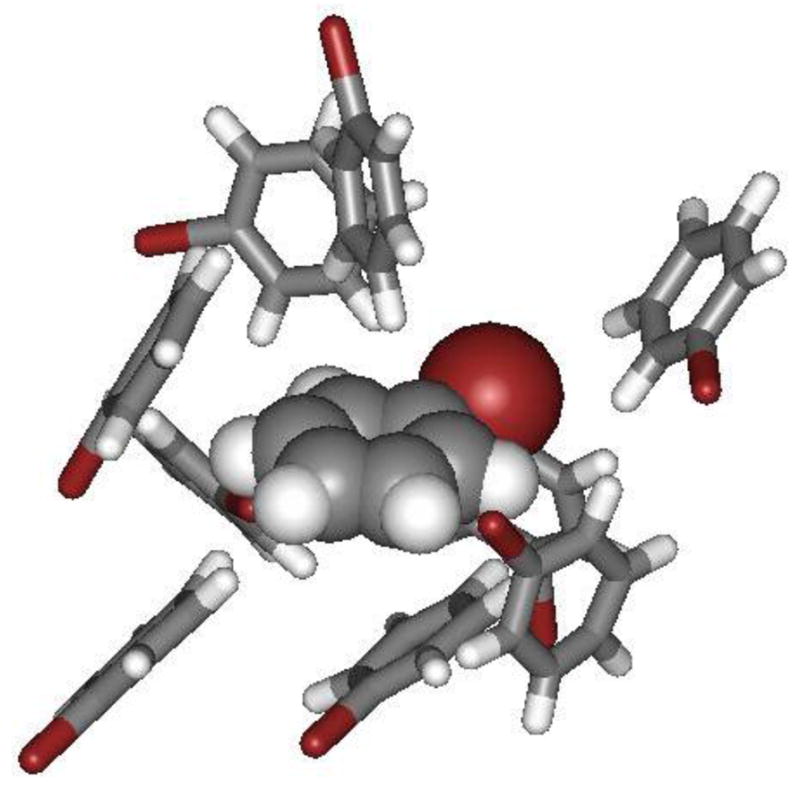
A cluster of the 9 nearest neighbors of the molecule rendered as space-filling from the final configuration of a MC simulation of liquid bromobenzene. Typical aryl-aryl edge-to-face and offset-stacked interactions are present.
Binding of NNRTIs with HIV-RT
The final objective was to investigate the impact of the X-sites on predictions of free energies of binding for 1 – 3 and their bromo (4) and iodo (5) analogs with HIV-1 reverse transcriptase. Extensive FEP calculations were previously performed to help guide the optimization of the initial 5-μM docking hit to yield very potent NNRTIs including 1 – 3.10 The present FEP calculations continue that work and were performed in exactly the same manner.10 Briefly, coordinates of the complexes were constructed starting from the2be2 crystal structure25 using the BOMB program.12 The 175 amino acid residues nearest the ligand were included. The protein is represented with the OPLS/AA force field and the ligands by OPLS/CM1A or OPLS/CM1Ax. The chlorine in the B-ring always had the X-site; the halogen in the A-ring either had it (OPLS/CM1Ax) or did not (OPLS/CM1A). The simulations for the unbound ligands and the complexes used TIP4P water spheres (“caps”) with a 25-Å radius centered on the ligand; ca. 2000 and 1250 water molecules were retained for the unbound and bound MC simulations. 11 windows of simple overlap sampling were used for each FEP calculation with 10-Å residue-based cutoffs.22 Each window consisted of 10–15 M configurations of equilibration and 10–30 M configurations of averaging at 25 C. The ligand and side chains with any atom within ca. 10 Å of the ligand were fully flexible, while, after initial conjugate-gradient relaxation, the protein backbone was kept fixed. The MC/FEP calculations were carried out with MCPRO 2.2.11
Relative free energies of binding were obtained for the complete series, H to I, using both OPLS/CM1A and OPLS/CM1Ax. Two alterative conformers for the bound ligand were considered; the one in Figure 2 with the β-H of the cyanovinyl group proximal to Trp229 and the 180° rotamer with the α-H nearer Trp229, as in Figure 6. The results as summarized for both force fields and both conformers in Table 6. The statistical uncertainties in the results are 0.2 – 0.3 kcal/mol from the fluctuations in the computed averages. This range is reinforced by results from calculations that were repeated from different starting points and from ones that were run in both directions. E.g., for the first conformer, the ΔΔGb result with OPLS/CM1Ax for the 4 → 3 perturbation was −0.18 kcal/mol and for 3 → 4 it was +0.25 kcal/mol.
Figure 6.

Computed alternative structure for the complex of the bromo analog 4 with wild-type HIV-1 reverse transcriptase. In contrast to Figure 2, the α-H of the cyanovinyl group is now closer to Trp229 than the β-H. The Pro95 O – Br distance is 3.52 Å. The structure is from the last configuration of a MC/FEP calculation that included 1246 water molecules; only water molecules within 12 Å of the ligand are shown. Some protein residues have been deleted for clarity.
Table 6.
Computed Relative Free Energies of Binding for NNRTIs 1 – 5 with HIV-RTa
| Cmpd | ΔΔGb (kcal mol−1) | |||
|---|---|---|---|---|
| Fig. 2 Conformer | Fig. 6 Conformer | |||
| OPLS/CM1A | OPLS/CM1Ax | OPLS/CM1A | OPLS/CM1Ax | |
| 1 (X = H) | 0.0 | 0.0 | 0.0 | 0.0 |
| 2 (X = F) | −1.96 | −1.96 | −2.04 | −2.04 |
| 3 (X = Cl) | −4.09 | −5.68 | −4.45 | −6.52 |
| 4 (X = Br) | −4.31 | −5.50 | −4.99 | −6.69 |
| 5 (X = I) | −3.44 | −4.60 | −4.62 | −6.69 |
Uncertainties (±σ) in the computed values are 0.2 – 0.3 kcal/mol.
For the first conformer with both OPLS/CM1A and OPLS/CM1Ax, the binding affinity peaks with the chloro and bromo analogs; however, the binding affinity is 1.2 – 1.6 kcal/mol more attractive for the halogens with the X-sites. In viewing configurations as in Figure 6, it is apparent that the A-ring halogen of 3 – 5 is making closer contacts with the oxygen of Pro95 with the X-sites than without them and that the halogen-bonding is contributing to the more favorable ΔΔGb values. However, a substantial range of halogen – oxygen distance is being sampled, ca. 3.4 – 4.1 Å. The final configuration for 4 from the 5 → 4 FEP calculation in Figure 6 has a Br-O separation of 3.52 Å; in the final configuration for 5, the I-O separation is 3.46 Å. For the second conformer, addition of the X-site enhances the binding affinity by as much as 2.1 kcal/mol for 3. With OPLS/CM1A and OPLS/CM1Ax the binding affinities for 3 – 5 are the same within the statistical uncertainties. At this point, it may be noted that the preferred conformer is unclear, and it may differ for 1 – 5. However, the average protein-ligand interaction energies from the MC simulations are a few kcal/mol lower for the second conformer, which suggests that it is generally favored. The issue can be properly addressed via FEP calculations that interconvert the two conformers.
From a design standpoint, the key messages from these results would be (a) that the chloro analog (3) should be much more active than the fluoro analog (2), and (b) not to expect additional improvement for the bromo and iodo analogs. There is no reason to assume a priori that if a halogen bond is possible with a chloro analog that in a binding or inhibition assay there will be further gains for the bromo and iodo derivatives. Though the optimal halogen bond strength may increase along the halogen series as in Tables 3 and 4, there are complex entropic considerations at play. With increasing halogen size, the conformational freedom of the ligand and the protein may become more restricted, which would then favor the smaller halogen. Hardegger et al. did see improvement in IC50 values from F to I for their series of cathepsin inhibitors, though the gain was a factor of 15 for F to Cl an then only additional factors of 2 for the bromo and iodo analogs.3 It may be noted that these are covalent inhibitors, which provides some restriction to the motion of the covalently linked ligand and protein. In the present case, it is only known currently that the EC50 values from the viral replication assay decline from 5.0 to 3.2 to 0.055 nM in progressing from 1 to 2 to 3.10 The striking increase in potency for the chloro analog coupled with the present FEP results does indicate that there is a contribution from halogen bonding with the oxygen atom of Pro95.
CONCLUSION
Halogen bonding has emerged as an important element of molecular recognition. This is reinforced by the present results for gas-phase complexes. There is no question that halogen bonding needs to be accommodated in force fields that are being used for modeling systems in which such interactions are possible. Otherwise, interactions like those illustrated in Figure 1 are predicted to be repulsive rather than attractive. The need is especially important in drug-design efforts and associated computations of biomolecule-ligand binding since about 50% of compounds that are assayed in drug discovery programs contain a halogen, predominantly chlorine and fluorine.26 In the present study, the implementation of halogen bonding in the OPLS-AA force field has been described for aryl halides. As in other work, the necessary improvement of the electrostatic fields can be achieved by addition of point-charges, in this case, on the backside of the carbon-halogen bond. The modified force field, referred to as OPLS-AAx, and the alternative, which incorporates CM1A atomic charges (OPLS/CM1Ax), were first tested on gas-phase complexes of halobenzenes with Lewis bases. Agreement with results from ab initio MP2/aug-cc-pVDZ(-PP) calculations was found to be reasonable and properly reflective of basic trends of increasing interaction strength with increasing halogen size and electron deficiency of the arylhalide. The new models were further supported through computation of free energies of hydration and pure liquid properties for the halobenzenes. Finally, the results of MC/FEP calculations for relative free energies of binding were obtained for a series of anti-HIV agents that have been suggested to be modulated by halogen bonding. The effect of the addition of halogen bonding was striking, yielding binding enhancements in the 1–2 kcal/mol range.
Acknowledgments
Gratitude is expressed to the National Institutes of Health (GM32136 and AI44616) and the National Foundation for Cancer Research for support of this work. The paper is dedicated to Prof. Wilfred F. van Gunsteren in celebration of his 65th birthday.
Footnotes
The authors declare no competing financial interests.
References
- 1.For a reviews, see: Politzer P, Murray JS, Clark T. Phys Chem Chem Phys. 2010;12:7748–7757. doi: 10.1039/c004189k.Metrangolo P, Murray JS, Pilati T, Politzer P, Resnati G, Terraneo G. Cryst Growth Des. 2011;11:4238–4246.
- 2.Lu Y, Shi T, Wang Y, Yang H, Yan X, Luo X, Jiang H, Zhu W. J Med Chem. 2009;52:2854–2862. doi: 10.1021/jm9000133. [DOI] [PubMed] [Google Scholar]
- 3.Hardegger LA, Kuhn B, Spinnler B, Anselm L, Ecabert R, Stihle M, Gsell B, Thoma R, Diez J, Benz J, Plancher JM, Hartmann G, Banner DW, Haap W, Diederich F. Angew Chem Int Ed. 2011;50:314–318. doi: 10.1002/anie.201006781. [DOI] [PubMed] [Google Scholar]
- 4.For reviews, see: Ponder JW, Case DA. Adv Prot Chem. 2003;66:27–85. doi: 10.1016/s0065-3233(03)66002-x.Oostenbrink C, Villa A, Mark AE, van Gunsteren WF. J Comput Chem. 2004;13:1656–1676. doi: 10.1002/jcc.20090.Jorgensen WL, Tirado-Rives J. Proc Nat Acad Sci USA. 2005;102:6665–6670. doi: 10.1073/pnas.0408037102.
- 5.Auffinger P, Hays FA, Westhof E, Ho PS. Proc Nat Acad Sci USA. 2004;101:16789–16794. doi: 10.1073/pnas.0407607101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Politzer P, Murray JS, Concha MC. J Mol Model. 2008;14:659–665. doi: 10.1007/s00894-008-0280-5. [DOI] [PubMed] [Google Scholar]
- 7.(a) Jorgensen WL, Briggs JM, Gao J. J Am Chem Soc. 1987;109:6857–6858. [Google Scholar]; (b) Jorgensen WL, Briggs JM. J Am Chem Soc. 1989;111:4190–4197. [Google Scholar]; (c) Dixon RW, Kollman PA. J Comput Chem. 1997;18:1632–1646. [Google Scholar]; (d) Mahoney MW, Jorgensen WL. J Chem Phys. 2000;112:8910–8922. [Google Scholar]
- 8.Ibrahim MAA. J Comput Chem. 2011;32:2564–2574. doi: 10.1002/jcc.21836. [DOI] [PubMed] [Google Scholar]
- 9.Rendine S, Pieraccini S, Forni A, Sironi M. Phys Chem Chem Phys. 2011;13:19508–19516. doi: 10.1039/c1cp22436k. [DOI] [PubMed] [Google Scholar]
- 10.Bollini M, Domaoal RA, Thakur VV, Gallardo-Macias R, Spasov KA, Anderson KA, Jorgensen WL. J Med Chem. 2011;54:8582–8591. doi: 10.1021/jm201134m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jorgensen WL, Tirado-Rives J. J Comput Chem. 2005;26:1689–1700. doi: 10.1002/jcc.20297. [DOI] [PubMed] [Google Scholar]
- 12.Jorgensen WL. Acc Chem Res. 2009;42:724–733. doi: 10.1021/ar800236t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Jorgensen WL, Maxwell DS, Tirado-Rives J. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]; (b) Jorgensen WL, Ulmschneider JP, Tirado-Rives J. J Phys Chem B. 2004;108:16264–16270. [Google Scholar]
- 14.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 15.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Jr, Freguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. J Am Chem Soc. 1994;117:5179–5197. [Google Scholar]
- 16.Storer JW, Giesen DJ, Cramer CJ, Truhlar DG. J Comput Aided Mol Design. 1995;9:87–110. doi: 10.1007/BF00117280. [DOI] [PubMed] [Google Scholar]
- 17.Jorgensen WL, Jensen KP, Alexandrova AN. J Chem Theory Comput. 2007;3:1987–1992. doi: 10.1021/ct7001754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Riley KE, Murray JS, Fanfrlik J, Rezac J, Solá RJ, Concha MC, Ramos FM, Politzer P. J Mol Model. 2011;17:3309–3318. doi: 10.1007/s00894-011-1015-6. [DOI] [PubMed] [Google Scholar]
- 19.Riley KE, Murray JS, Politzer P, Concha MC, Hobza P. J Chem Theory Comput. 2009;5:155–163. doi: 10.1021/ct8004134. [DOI] [PubMed] [Google Scholar]
- 20.Riley KE, Hobza P. J Chem Theory Comput. 2008;4:232–242. doi: 10.1021/ct700216w. [DOI] [PubMed] [Google Scholar]
- 21.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision A.02. Gaussian, Inc; Wallingford, CT: 2009. [Google Scholar]
- 22.Jorgensen WL, Thomas LT. J Chem Theory Comput. 2008;4:869–876. doi: 10.1021/ct800011m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abraham MH, Andonian-Haftvan J, Whiting GS, Leo A, Taft RS. J Chem Soc Perkin Trans. 1994;2:1777–1791. [Google Scholar]
- 24.Riddick JA, Bunger WB, Sakano TK. Organic Solvents, Physical Properties and Methods of Purification. 4. II. Wiley; New York: 1986. Techniques of Chemistry. [Google Scholar]
- 25.Himmel DM, Das K, Clark AD, Hughes SH, Benjahad A, Oumouch S, Guillemont J, Coupa S, Poncelet A, Csoka I, Meyer C, Andries K, Nguyen CH, Grierson DS, Arnold E. J Med Chem. 2005;48:7582–7591. doi: 10.1021/jm0500323. [DOI] [PubMed] [Google Scholar]
- 26.(a) Hernandes MZ, Cavalcanti SMT, Moreira DRM, de Azevedo Junior WF, Leite ACL. Curr Drug Targets. 2010;11:303–314. doi: 10.2174/138945010790711996. [DOI] [PubMed] [Google Scholar]; (b) Roughley SD, Jordan AM. J Med Chem. 2011;54:3451–3479. doi: 10.1021/jm200187y. [DOI] [PubMed] [Google Scholar]