

Abstract
Brain-derived neurotrophic factor (BDNF) plays critical roles in many aspects of brain functions, including cell survival, differentiation, development, learning and memory. Aberrant BDNF expression has also been implicated in numerous neurological disorders. Thus, significant effort has been made to understand how BDNF transcription as well as translation is regulated. Interestingly, the BDNF gene structure suggests that multiple promoters control its transcription, leading to the existence of distinct mRNA species. Further, the long- and short-tail of the 3’un-translated region may dictate different sub-cellular BDNF mRNA targeting and translational responses following neuronal stimulation. This review aims to summarize the main findings that demonstrate how neuronal activities specifically up-regulate the transcription and translation of unique BDNF transcripts. We also discuss some of the recent reports that emphasize the epigenetic regulation of BDNF transcription.
Keywords: Brain-derived neurotrophic factor, calcium-responsive element, cAMP-responsive element, intracellular signaling, neuroplasticity, transcription and translation
Introduction
Like other neurotrophins, brain-derived neurotrophic factor (BDNF) was initially identified for its role in neuron proliferation, neurogenesis, differentiation and degeneration [1,2]. Its function in regulating activity-dependent neuronal modification has also been demonstrated. For example, BDNF regulates both memory formation and long-term potentiation (LTP), an activity-dependent strengthening of synaptic efficacy [3]. Two independent lines of BDNF mutant mice show severe impairments in LTP at the CA1 synapses in hippocampus [4,5]. Importantly, BDNF heterozygous mutants show similar defective LTP to that of homozygous mice, indicating that full level of BDNF is required. Furthermore, these synaptic defects in the mutant mice are rescued by either acute application of exogenous recombinant BDNF or by virus-based over-expression of BDNF [5,6]. Because the homozygous null mutants display shorter life span, memory formation has been mainly investigated with heterozygous mutants [7,8]. Consistently, BDNF heterozygous mutants show impairments in the hippocampus-dependent paradigms, including Morris water maze [7] and contextual fear conditioning [8]. Another independent group used forebrain-specific BDNF homozygous mutants, and found severe spatial memory defects in the Morris water maze test [9]. Moreover, infusion of BDNF antisense oligonucleotides or anti-BDNF antibodies also impairs spatial memory [10,11].
How does BDNF support activity-dependent modification of synapses and brain function? It has been suggested that the induction of LTP requires BDNF release from the presynaptic vesicles [12,13]. Further, BDNF may promote vesicle docking at the presynaptic active zone [14]. However, the postsynaptic function is also suggested by that postsynaptic BDNF secretion triggered by a spike-timing protocol at the single-cell level is required for the long-term structural change of spines [15]. These data implicate that presynaptic and postsynaptic BDNF release may differentially facilitate the induction and the maintenance phase of LTP, respectively. Mechanistic studies have indicated that several signaling pathways may be activated upon BDNF binding to its receptor TrkB, a receptor tyrosine kinase [16]. For example, BDNF-dependent TrkB activation may potentiate the glutamate function by promoting glutamate release [17,18] or increasing the open probability of NMDA receptor (NMDAR) [19,20]. Additionally, BDNF elevates the expression and regulates the trafficking of both NMDA and AMPA receptors, which are essential steps for the induction and maintenance of LTP [21-23].
Interestingly, BDNF is not only required for many aspects of activity-dependent plasticity and brain function, its expression is also triggered by neuronal activity both in vitro and in vivo. This is consistent with the notion that gene transcription and new protein synthesis are required for both LTP and memory formation [3,24]. Through the investigation on how activity-dependent intracellular signaling and transcription factors regulate BDNF expression, we have also achieved better understanding on gene-environment interaction. This review will mainly focus on transcriptional and translational regulation of BDNF.
Structure of BDNF gene
BDNF transcription is significantly induced by Ca2+ and neural activity. In cultured neurons, calcium influx through L type voltage gated calcium channel (L-VGCC) [25] or NMDAR [26,27] robustly increases BDNF mRNA that may last for at least 6 hrs. Electrical activity, such as high frequency stimulation that induces LTP, also triggers BDNF transcription in brain slices [28]. In live animals, induction of BDNF transcription has been observed after training of contextual learning [29], physical exercise [30], exposure to novel environment [31], chronic exposure to drugs of abuse [32], as well as kainic acid (KA)-induced seizure [33].
Intriguingly, the activity-dependent BDNF transcription only produces certain BDNF mRNA isoforms [33,34]. BDNF gene consists of at least eight 5’ non-coding exons (i.e. from exon I to VIII) and one 3’ coding exon (i.e. exon IX). By using 5’ rapid amplification of cDNA ends (RACE), bioinformatics, RT-PCR and sequencing analysis, Aid and colleagues identified nine distinct transcriptional initiation sites: eight at the beginning of the individual 5’ non-coding exon and one in the intron proceeding exon IX [35]. This indicates that nine different promoters control BDNF transcription. Because of the existence of a splicing donor site 3’ to each of the exons, a single non-coding exon will join exon IX following RNA splicing. The discovery of exon IXA, alternative splice donor sites within exon II, and two poly-adenylation sites in the 3’ un-translated region of exon IX predicts that there are at least 22 isoforms of BDNF mRNA [33,35] (Figure 1). Because only exon IX contains the coding region, all these transcripts will be translated to a single species of BDNF polypeptide. Such sophisticated gene structure may fine-tune its dynamic transcriptional regulation in different cell types and by different neuronal activity. For example, fear conditioning increases BDNF exon I and IV in hippocampus (but only exon IV in the CA1 region [34,36]) and amygdale [37,38], whereas fear memory extinction elevates BDNF exon I and IV in prefrontal cortex [39]. Administration of cocaine enhances the activity of exon II promoter [40]. The existence of different non-coding exons and different versions of the 3’ UTR (dictated by the different poly-adenylation sites) may cause different sub-cellular localization [41-43].
Figure 1.
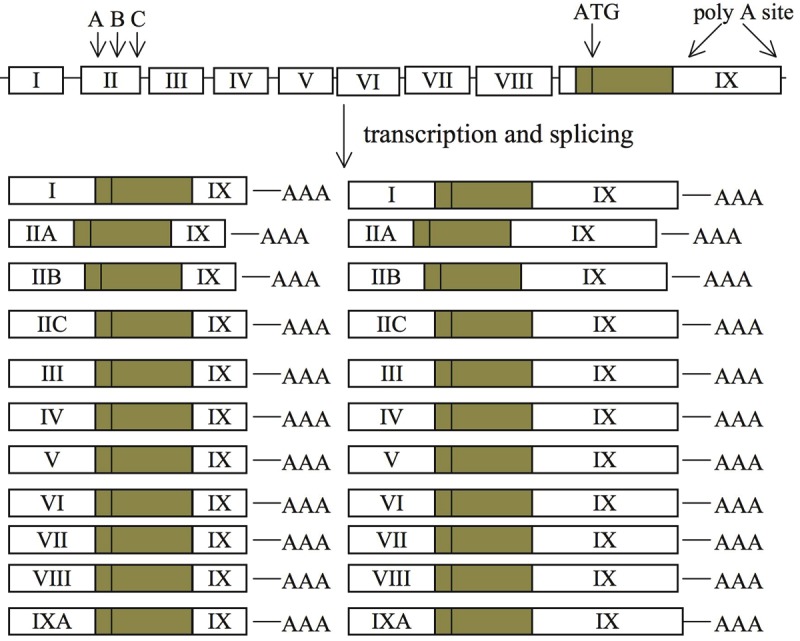
The structure of mouse and rat BDNF gene. BDNF gene consists of eight 5’ exons (I-VIII) and one 3’ exon (IX). Introns in the gene are presented as straight horizontal lines. The protein-coding region is shown as a dark box, and the non-coding exon regions are shown as open boxes. Three alternative splicing sites (A, B and C) in exon II and the two poly-adenylation sites (polyA site) are indicated with arrows. Exon IXA is the 5’ extended coding exon. Adapted from Aid et al., 2007 [35].
There are multiple Ca2+-responsive elements in the BDNF promoter IV
Among all promoters that control the transcription of distinct exons, the regulatory feature of promoter IV is most thoroughly investigated. In cortical neurons, exon IV-containing BDNF mRNA (this is according to the new designation; it was originally referred to as exon III.) is the major form induced by neuronal activity [25,27,44]. Interestingly, BDNF exon IV transcription is also up-regulated by BDNF itself both in vitro and in vivo [45,46]. Investigations on this specific transcript suggest that the 170-base pair (bp) 5’ flanking sequence of exon IV contributes as the regulatory region for calcium-mediated BDNF IV transcription. Within this regulatory region, three calcium responsive elements (i.e. CaRE1, 2 and 3) have been identified (Figure 2).
Figure 2.
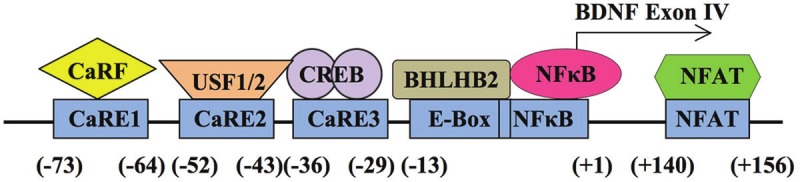
The arrangement of functional cis-elements and the corresponding transcription factors in BDNF promoter IV. The relative location of the sequences to the transcription initiation site (+1) is labeled. The E-Box and the NFκB sequence have one base pair overlap.
CaRE1 is located at -73 to -64 relative to the exon IV transcriptional initiation site (Figure 2), which is conserved in rat, mouse and human BDNF gene. By using the yeast one hybrid screening, Tao et al. [47] identified a novel transcription activator CaRF (calcium responsive factor) that can bind the CaRE1 sequence. Recently, McDowell and colleagues generated mice lacking CaRF DNA binding domain, and found that BDNF IV mRNA level is reduced in the mutant cortex [48]. The mutant mice also show disrupted remote memory and impaired extinction. Interestingly, activity profiles of CaRE1 and CaRF appear to be different. For example, the Ca2+-dependent CaRE1-mediated transcription depends on both L-VGCC and NMDAR. The CaRF-mediated transcription only depends on L-VGCC but not NMDAR [27]. Further, the L-VGCC-dependent activation of CaRE1-mediated transcription requires PKA (protein kinase A), CaMKI (calmodulin-dependent protein kinase I), and CaMKIV (calmodulin-dependent protein kinase IV) activity. In contrast, CaRF activation requires MEK, PI3K and CaMKII activity [27]. It is suggested that, in addition to CaRF, other transcriptional regulators may also exist in the CaRE1/CaRF complex. Another possibility is that the CaRF activation and its binding to CaRE1 may be differentially regulated by different intracellular signaling molecules.
CaRE2 locates at 52 to 43 bp upstream of exon IV transcriptional initiation site (Figure 2). It has been identified as an E-box element, which may be recognized and bound by members of the basic helix-loop-helix (bHLH) family. Chen et al. has demonstrated that homo- or heterodimer of USF1 (upstream stimulatory factor 1, a bHLH protein) and USF2 regulates BDNF transcription in a L-VGCC dependent manner in cultured DIV 5 (days in vitro 5) neurons [49]. Surprisingly, the activation of L-VGCC or NMDAR fails to stimulate CaRE2-mediated transcription in DIV 11 neurons [27], suggesting possible developmental influences.
CaRE3 locates between nucleotide -36 and -29 relative to transcriptional initiation site of exon IV. The CaRE3 sequence resembles the consensus sequence of cAMP response element (CRE) (Figure 2). Electrophoretic mobility shift assays (EMSA) demonstrate that CaRE3 binds CRE binding protein (CREB) in vitro [25]. Several lines of evidence also suggest regulatory effects of CREB on CaRE3-mediated transcription in live neurons. First, the activity of CaRE3 and CRE is similarly regulated by the same Ca2+-stimulated protein kinases [27]. Secondly, constitutive active CREB (i.e. CREB-VP16) upregulates both CaRE3- and CRE-mediated transcription in cultured neurons [27]. Furthermore, disruption of CREB binding by mutating CaRE3 sequence abolishes activity-induced BDNF IV transcription [25,50]. Consistent with that BDNF transcription is responsive to neuronal activity, CREB activity can be up-regulated by calcium influx through either L-VGCC and NMDAR, by high frequency stimulation [51], and by learning-related trainings (such as contextual fear conditioning and passive avoidance) [52]. Similar to the effects caused by the reduction of BDNF, lack of CREB results in defective LTP and memory formation [53,54].
In addition to the CaREs, accumulating evidence has also implicated the function of other cis-elements in regulating BDNF IV transcription (Figure 2). An NFκB (nuclear factor κB) site and a class B E-box [55,56] exist in a 22 bp segment spanning from -21 to +1 of the initiation site. Although the function of NFkB binding is unclear, the NMDA-stimulated BDNF IV transcription does require the intact NFkB site. The class B E-box is bound by BHLH2, a suppressor in the bHLH family. The reduced BHLH2 binding after NMDAR activation is needed for the upregulation of BDNF IV. Furthermore, both the basal and KA-induced level of BDNF IV are increased in BHLH2 knockout (KO) mice. Moreover, the existence of a composite NFAT/MEF2 (nuclear factor of activated T cell/ myocyte enhancer factor 2) consensus sequence is identified at the +140-156 region. The NMDAstimulated BDNF IV mRNA up-regulation is decreased by silencing NFATc4, and increased by overexpressing NFATc4. This indicates that NFATc4 positively regulates BDNF IV transcription [57]. Through interacting with NFATc4, a transcription factor MEF2 [58], which recruits histone deacetylases (HDAC) also regulates the activity of promoter IV [59]. Additionally, the persistent up-regulation of BDNF IV following neuronal stimulation may require Npas4 [67].
Regulation of BDNF transcription by Ca2+-stimulated intracellular signaling
Because BDNF is an immediate early gene, its transcriptional up-regulation depends on the pre-existing molecules, and does not require any de novo protein synthesis. The causal function of calcium influx following the activation of L-VGCC and NMDAR suggests that Ca2+-stimulated protein kinase activity may regulate the activation of the relevant transcription factors. It is well known that elevation of intracellular calcium leads to the activation of Ras/ERK1/2 [60], cAMP/PKA [61] and CaM kinase pathway [62]. These Ca2+-stimulated kinases may, in turn, stimulate both CREB and CaRF. Although PKA was identified as the main kinase for CREB initially, ERK1/2 may play a major role for the persistent phosphorylation of CREB in neurons [63,64]. CaMKII may have dual effects on CREB, because it may phosphorylate both Ser 133 and 142 of CREB. While the phosphorylation at Ser 133 activates CREB-mediated transcription, the phosphorylation at Ser 142 inhibits the interaction between CREB and its co-activator CBP (CREB binding protein) [65]. Furthermore, the CaMKIV-dependent phosphorylation of CBP at Ser-301 [66] may trigger its histone acetyltransferase (HAT) activity, which modifies histone and chromatin structure, recruits RNA polymerase II complex, and acts as an adaptor for other transcriptional factors (e.g. calcium-responsive trans-activator or CREST) [67-70]. Further, calcineurin-dependent CREST-CBP interaction facilitates CRE-mediate transcription [71]. Sequence analysis predicts that CaRF may be phosphorylated by ERK1/2, CaMKII, and PKC (protein kinase C) [47]. Experimentally, CaRF-mediated transcription in cortical neuron requires the activity of MEK, PI3K, and CaMKII [27].
To thoroughly examine the function of the major Ca2+-stimulated protein kinases, we studied the regulation of BDNF IV mRNA and promoter IV activity in cultured cortical neurons [27]. We found that the previously defined promoter IV activity and BDNF IV mRNA level share similar regulatory properties. They are both up-regulated by the activation of L-VGCC or NMDAR. Inhibition of MEK or CaMK activity decreased the basal level as well as Ca2+-stimulated increase of both promoter IV activity and BDNF IV mRNA. PKA or PI3K activity is only required for Ca2+-stimulated up-regulation of both promoter IV and BDNF IV mRNA. Further investigation with dominant negative CaMK constructs show that CaMKI is required for the basal and Ca2+-stimulated activity of promoter IV. CaMKII and CaMKIV are required for the Ca2+-stimulated but not basal activity of promoter IV. Interestingly, the L-VGCC- and NMDAR-dependent activation of the CaREs may involves different Ca2+-stimulated protein kinases. Specifically, the L-VGCC- but not NMDAR-mediated CaRE1 activation requires PKA, CaMKI, and CaMKIV activity. The NMDAR- but not L-VGCC-mediated CaRE3 activation requires CaMKI activity.
In addition to the major Ca2+-stimulated protein kinases, other Ca2+-stimulated intracellular molecules may also regulate activity-dependent BDNF transcription through ERK1/2 and CREB. The activity of type 1 and 8 adenylyl cyclases (AC1 and AC8) can be stimulated by calcium, leading to Ca2+-stimulated production of cAMP and activation of PKA [72,73]. Mice lacking both AC1 and AC8 are defective in LTP and hippocampus-dependent memory formation [72,73]. The impairments may be due to the lack of learning-induced ERK1/2 and CREB activation in the mutant hippocampus [74]. This is consistent with that CREB activation requires PKA as well as ERK1/2, whose activity may be stimulated by PKA and cAMP through Rap1 [75-77]. The function of Ca2+-stimulated ACs (i.e. AC1 and 8) in regulating CREB activity suggests their involvement in BDNF transcription. We trained wild type (WT) and AC1/AC8 double knockout (DKO) mice with contextual fear conditioning. Quantitative RT-PCR revealed that, comparing to the shock only group, pairing shock with the context caused significant increase of BDNF exon I in WT hippocampus (Figure 3A). The level of exon IV remained unchanged after training. There was no training-induced BDNF mRNA up-regulation in AC1/AC8 DKO mice (Figure 3B). This is consistent with that exon I may be more sensitive to activity. Six-hour physical exercise elevates exon I transcription but not other exons in the hippocampus; longer-time exercise up-regulates the transcription of multiple exons [78]. We are aware that previous studies demonstrated that BDNF exon IV is up-regulated in the hippocampus following contextual fear conditioning [34,36]. The contrary may result from different animal models (e.g. rat vs. mouse) and different experimental conditions. Mechanistically, due to the existence of CRE in promoter I [79] (Figure 3C), AC1 and AC8 may regulate activity-dependent exon I transcription through CREB [74]. It is interesting to note that the training-induced upregulation of another CREB target gene c-fos was also ablated in AC1/AC8 DKO mice (Figure 3A and 3B).
Figure 3.
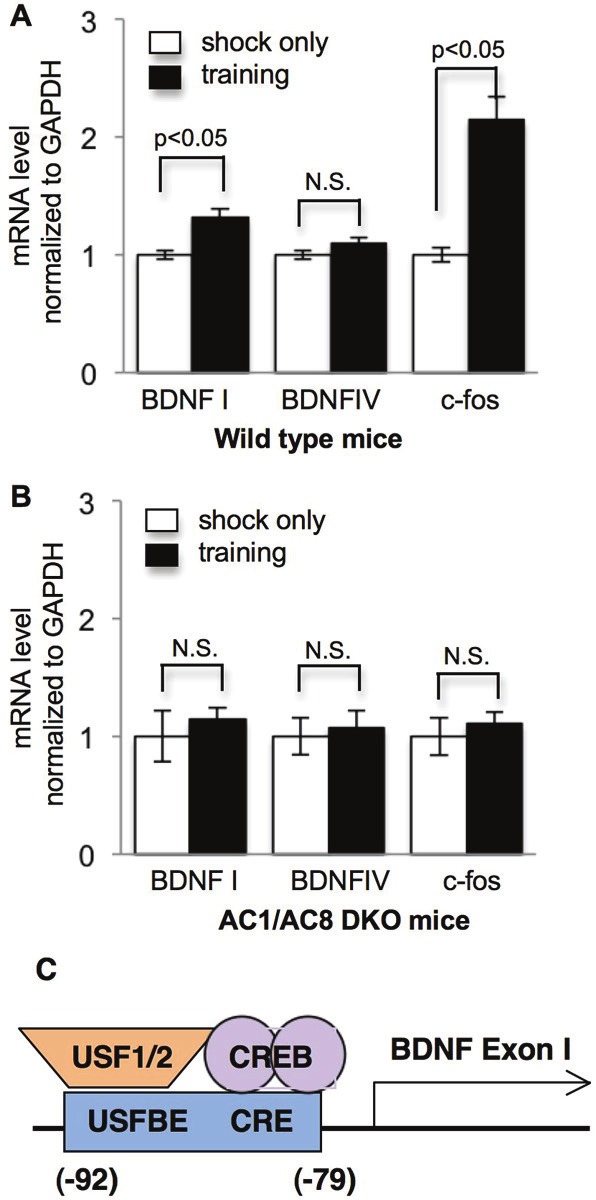
Ca2+-stimulated adenylyl cyclase activity is required for learning-induced BDNF exon I transcription. Two- to three-month old wild type (n = 21) and AC1/AC8 DKO (n = 12) mice were trained by contextual fear conditioning. Animals were introduced to the contextual chamber, and allowed to explore for 2 min, after which 3 mild electric foot shocks (0.7 mA, 2 sec duration) were delivered with 20 sec intervals. Animals remained in the chamber for 1 min after the last shock, and were then returned to their home cages. The control animals only received 3 shocks without exposure to the contextual chamber. Hippocampi were collected 2 hours after training. The procedures have been approved by the Institutional Animal Care and Use Committee at Michigan State University. The mRNA levels of BDNF exon I, BDNF exon IV, and c-fos were determined by quantitative RT-PCR, and normalized to GADPH mRNA level. The primers used for BDNF exon I were ACTCAAAGGGAAACGTGTCTC (forward) and GCCTTCATGCAACCGAAGTA (reverse); the primers used for exon IV were CTCCGCCATGCAATTTCCAC (forward) and GCCTTCATGCAACCGAAGTA (reverse); the primers used for c-fos were AGCCTTTCCTACTACCATCC (forward) and ATTCCGGCACTTGGCTGCAG (reverse); the primers used for GADPH were TCCATGACAACTTTGGCATTGTGG (forward) and GTTGCTGTTGAAGTCGCAGGAGAC (reverse). A. BDNF exon I and c-fos but not BDNF exon IV are significantly increased after contextual training. B. The up-regulation of BDNF exon I and c-fos is abolished in AC1/AC8 DKO mice. N.S.: not significant. C. the arrangement of cis-elements and the corresponding transcription factors in BDNF promoter I. The relative location of the cis-elements to the transcription initiation site (+1) is labeled. The sequences of USFBE and CRE overlap.
Epigenetic regulation of BDNF transcription
Beside the regulation by transcriptional factors, accumulating evidence indicates that epigenetic mechanisms including both chromatin remodeling and DNA methylation also regulate BDNF transcription. Changes in chromatin structure results from the post-translational modification on histone proteins by acetylation, methylation, and phosphorylation [80]. A recent study first demonstrates the role of HDAC2 (histone deacetylase 2) in synaptic plasticity and memory formation. HDAC2 knockout mice show enhanced memory formation, whereas HDAC2 over-expression animals show learning deficits [81]. Additionally, a decrease in lysine 9 dimethylation of histone 3 (H3) is essential for NMDA-induced BDNF exon I transcription in hippocampal neurons [82]. It is interesting and important to note that the activity-dependent modification of chromatin structure is not universal, but rather occurs in histones associated with specific genes. For example, memory extinction protocol increases the acetylation of H4 in chromatins at BDNF promoter IV but not other exons [39]. Acetylation of H3 is uniquely up-regulated at promoter IV following contextual fear conditioning [34]. Environmental enrichment specifically up-regulates the methylation of H3 lysine 4 at promoter III and IV, but down-regulates lysine 9 methylation of H3 at promoter IV [83]. As described earlier, CBP is a CREB co-activator and confers acetyltransferase (HAT) activity. Mice with CBP mutation show reduced histone acetylation along with impaired LTP and memory retention [84,85]. Notably, the inheritable disorder Rubinstein-Taybi syndrome is caused by CBP mutation, and HDAC inhibitor rescued memory and synaptic deficits in an animal model [86]. Although deficiency of CBP leads to histone hypoacetylation, the expression level of BDNF is surprisingly intact in the CBP mutant mice [86]. However, overexpression of CBP increases BDNF expression and rescues memory deficits in an animal model of Alzheimer’s disease [87].
DNA methylation is another important epigenetic mechanism for BDNF transcription and synaptic plasticity. For example, membrane depolarization decreases expression of Dnmt1 (DNA methyltransferase I) and Dnmt3a in cultured cortical neurons and consequently unmethylates the CpG islands (which contain a high frequency of a cytosine residue followed by a guanine residue; the methylation occurs on the cytosine residue) in promoter I and IV, which at least partially contribute to activity-dependent BDNF mRNA up-regulation [88,89]. In vivo, contextual learning also decreases methylation at BDNF promoters [34]. It appears that the global level of methylation reduction and BDNF up-regulation are only transient in the hippocampus after contextual learning. In contrast, DNA hypermethylation in dorsomedial prefrontal cortex consisting of anterior cingulate and prelimbic cortices, persists for as long as one month, and may be crucial for the storage of remote memory [90]. Notably, although inhibition of Dnmt results higher BDNF expression, applying Dnmt inhibitors or genetically deleting Dnmt1 and 3a in forebrain impairs synaptic plasticity and results in learning deficits [34,88,91]. There are two possible interpretations: 1) other Dnmt targets are involved and function as negative regulators of memory formation (e.g. protein phosphatase 1 [92]), and 2) the maintenance of basal DNA methylation is more essential for the de novo signals to modify brain structure.
In general, DNA hypermethylation leads to the reduction of transcription, possibly due to that the addition of methyl group on cytosine reduces the binding and assembly of transcription machinery. Another consequence of DNA methylation is to facilitate the recruitment of MBD (methyl-binding domain)-containing proteins. For BDNF IV promoter, the occupancy of the methyl-CpG binding protein 2 (MeCP2) undergoes dynamic changes following neuronal stimulation. Two independent research groups have revealed novel insight of how neuronal activity changes the interplay between MeCP2 and BDNF promoter IV in cultured cortical neurons [93,94]. By using chromatin immunoprecipitation assay, both groups found that the occupancy of MeCP2 was more predominant at promoter IV than at other promoters of BDNF. Further, they both showed that membrane depolarization reduced DNA methylation in promoter IV along with up-regulation of exon IV transcription. Consequently, MeCP2 dissociated from promoter IV following membrane depolarization. Martinowich et al. [94] further demonstrated that engineered methylation of specific cytosine sites (at -109, -66, -35, and -24) was sufficient to decrease promoter IV activity. Consistently, overall hypomethylation in Dnmt1 mutant neurons showed higher expression level of BDNF mRNA. Chen et al. [93] identified another mechanism that caused MeCP2 dissociation from promoter IV. Membrane depolarization triggered MeCP2 phosphorylation at Ser 421, which lowers the binding affinity even when the target sequence in promoter IV is highly methylated.
Intriguingly, these two teams reported that different methylated CpG sites changed in promoter IV. While Martinowich et al. found decrease in cytosine methylation at -111, -109, and -24 sites after membrane depolarization [94], Chen et al. observed the major change at -148 site [93]. It is also apparent that such epigenetic regulation on BDNF transcription may vary depending on the type of neuron and stimulation. For example, Lubin et al. demonstrated that contextual fear conditioning induced significant decrease of DNA methylation in promoter I and IV along with tremendous increase of methylation in promoter VI in the hippocampus [34]. Within promoter IV, bisulfite-sequencing analysis identified at least 12 CpG sites, whose methylation was down regulated following contextual training. To add another layer of complication, examination of gene expression patterns in hypothalamus lacking or overexpressing MeCP2 revealed that the majority of genes, including BDNF, are activated rather than suppressed by MeCP2 [95], whereas BDNF exon IV mRNA is expressed higher in cultured Mecp2 null neurons [93]. Moreover, Zhou et al. suppressed endogenous wild type MeCP2 and expressed MeCP2 S421A (the mutation abolishes the activity-dependent MeCP2 phosphorylation) in cultured neurons, and observed lower BDNF mRNA induction [96], supporting the findings of Chen et al.. However, this mechanism is challenged by in vivo studies. Li and colleagues generated MeCP2 S421A/S424A mice and found higher expression of BDNF exon IV as well as enhanced plasticity and learning [97]. One possibility is that MeCP2 may associate with transcription activators such as CREB1 and up-regulate gene expression [95]. The controversial conclusions from in vitro cultures and live animals also suggest that MeCP2 regulates gene expression in a more systematic pattern involving both excitatory and inhibitory neurons [98].
Translational regulation of BDNF through the 3’ un-translated region
Due to two poly-adenylation sites (Figure 1), either a long 2.85 kb or a short 0.35 kb 3’ UTR is present in BDNF transcripts [33]. A recent study has found that the expression ratio of long to short version of BDNF mRNA shows variation in different brain regions. For example, the relative abundance of the long transcript is higher in the cortex and lower in the brain stem [41]. Further, mRNA with short 3’ UTR is localized within soma; long mRNA is detected in both soma and dendrite. In mouse mutants that only express mRNA with short 3’ UTR, BDNF transcript is absent in dendrites. Strikingly, the mutants only show impaired LTP at the dendritic synapses but not at the somatic synapses. This study, consistent with previous findings [99], suggests that local protein synthesis of dendrite-targeted mRNAs is required for certain aspects of neuroplasticity. It would be more revealing if learning and memory are examined with the 3’ UTR mutant mice.
The study by Lau et al. suggests another aspect of the BDNF 3’ UTR function [100]. Robust expression of luciferase in hippocampal neurons is observed when the short but not long 3’ UTR is present in the reporter construct. Consistently, BDNF transcript with long 3’ UTR is mainly associated with the translational inactive ribonucleoprotein particles. Thus, it is suggested that the long 3’ UTR suppresses translation, and the basal BDNF level is maintained mostly by the translation of short 3’ UTR transcript. Because the long 3’ UTR transcript is targeted to dendrite, this specific mRNA may be involved in activity-dependent translational upregulation. Two lines of evidence have demonstrated this possibility. First, stimulation with tetraethylammonium, which is sufficient to induce LTP, increases the expression of a reporter gene attached to long but not short 3’ UTR. Second, the presence of long BDNF transcript is remarkably increased in polyribosomeassociated complexes, and decreased in translational inactive complexes after pilocarpine-induced seizure. Identification of the key cis-elements and trans-factors that are responsible for dendritic targeting and activity-dependent translation shall further advance the field.
In summary, the dynamic and multi-level regulation of BDNF expression has demonstrated how cellular signaling and genomic programs enable excitable cells (i.e. neurons) to cope with the environmental changes. Importantly, altered expression of BDNF and mutations of many of its transcription regulators are implicated in a variety of neurological disorders including Rubinstein-Taybi syndrome [101], Rett syndrome [102], depression [103], neurodegeneration [104,105], addiction [106], schizophrenia [107], and bipolar disease [108]. It is hoped that better understanding on BDNF function and regulation may benefit therapeutic development.
Acknowledgements
This study was supported by NIH grants (MH076906 and MH093445 to HW). Xianju Zhou was supported by a postdoctoral fellowship from American Heart Association (10POST4450000 to XZ). Due to the space limit, we apologize that this review may have missed many significant literatures.
References
- 1.Barde YA. Neurotrophins: a family of proteins supporting the survival of neurons. Prog Clin Biol Res. 1994;390:45–56. [PubMed] [Google Scholar]
- 2.Connor B, Dragunow M. The role of neuronal growth factors in neurodegenerative disorders of the human brain. Brain Res Brain Res Rev. 1998;27:1–39. doi: 10.1016/s0165-0173(98)00004-6. [DOI] [PubMed] [Google Scholar]
- 3.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 4.Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci U S A. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- 6.Korte M, Griesbeck O, Gravel C, Carroll P, Staiger V, Thoenen H, Bonhoeffer T. Virus-mediated gene transfer into hippocampal CA1 region restores long-term potentiation in brain-derived neurotrophic factor mutant mice. Proc Natl Acad Sci U S A. 1996;93:12547–12552. doi: 10.1073/pnas.93.22.12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Linnarsson S, Bjorklund A, Ernfors P. Learning deficit in BDNF mutant mice. Eur J Neurosci. 1997;9:2581–2587. doi: 10.1111/j.1460-9568.1997.tb01687.x. [DOI] [PubMed] [Google Scholar]
- 8.Liu IY, Lyons WE, Mamounas LA, Thompson RF. Brain-derived neurotrophic factor plays a critical role in contextual fear conditioning. J Neurosci. 2004;24:7958–7963. doi: 10.1523/JNEUROSCI.1948-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gorski JA, Balogh SA, Wehner JM, Jones KR. Learning deficits in forebrain-restricted brain-derived neurotrophic factor mutant mice. Neuroscience. 2003;121:341–354. doi: 10.1016/s0306-4522(03)00426-3. [DOI] [PubMed] [Google Scholar]
- 10.Ma YL, Wang HL, Wu HC, Wei CL, Lee EH. Brain-derived neurotrophic factor antisense oligonucleotide impairs memory retention and inhibits long-term potentiation in rats. Neuroscience. 1998;82:957–967. doi: 10.1016/s0306-4522(97)00325-4. [DOI] [PubMed] [Google Scholar]
- 11.Mu JS, Li WP, Yao ZB, Zhou XF. Deprivation of endogenous brain-derived neurotrophic factor results in impairment of spatial learning and memory in adult rats. Brain Res. 1999;835:259–265. doi: 10.1016/s0006-8993(99)01592-9. [DOI] [PubMed] [Google Scholar]
- 12.Jia Y, Gall CM, Lynch G. Presynaptic BDNF promotes postsynaptic long-term potentiation in the dorsal striatum. J Neurosci. 2010;30:14440–14445. doi: 10.1523/JNEUROSCI.3310-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zakharenko SS, Patterson SL, Dragatsis I, Zeitlin SO, Siegelbaum SA, Kandel ER, Morozov A. Presynaptic BDNF required for a presynaptic but not postsynaptic component of LTP at hippocampal CA1-CA3 synapses. Neuron. 2003;39:975–990. doi: 10.1016/s0896-6273(03)00543-9. [DOI] [PubMed] [Google Scholar]
- 14.Pozzo-Miller LD, Gottschalk W, Zhang L, Mc-Dermott K, Du J, Gopalakrishnan R, Oho C, Sheng ZH, Lu B. Impairments in high-frequency transmission, synaptic vesicle docking, and synaptic protein distribution in the hippocampus of BDNF knockout mice. J Neurosci. 1999;19:4972–4983. doi: 10.1523/JNEUROSCI.19-12-04972.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tanaka J, Horiike Y, Matsuzaki M, Miyazaki T, Ellis-Davies GC, Kasai H. Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines. Science. 2008;319:1683–1687. doi: 10.1126/science.1152864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamada K, Nabeshima T. Brain-derived neurotrophic factor/TrkB signaling in memory processes. J Pharmacol Sci. 2003;91:267–270. doi: 10.1254/jphs.91.267. [DOI] [PubMed] [Google Scholar]
- 17.Takei N, Numakawa T, Kozaki S, Sakai N, Endo Y, Takahashi M, Hatanaka H. Brain-derived neurotrophic factor induces rapid and transient release of glutamate through the non-exocytotic pathway from cortical neurons. J Biol Chem. 1998;273:27620–27624. doi: 10.1074/jbc.273.42.27620. [DOI] [PubMed] [Google Scholar]
- 18.Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- 19.Levine ES, Crozier RA, Black IB, Plummer MR. Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing N-methyl-D-aspartic acid receptor activity. Proc Natl Acad Sci U S A. 1998;95:10235–10239. doi: 10.1073/pnas.95.17.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin SY, Wu K, Levine ES, Mount HT, Suen PC, Black IB. BDNF acutely increases tyrosine phosphorylation of the NMDA receptor subunit 2B in cortical and hippocampal postsynaptic densities. Brain Res Mol Brain Res. 1998;55:20–27. doi: 10.1016/s0169-328x(97)00349-5. [DOI] [PubMed] [Google Scholar]
- 21.Caldeira MV, Melo CV, Pereira DB, Carvalho R, Correia SS, Backos DS, Carvalho AL, Esteban JA, Duarte CB. Brain-derived neurotrophic factor regulates the expression and synaptic delivery of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor subunits in hippocampal neurons. J Biol Chem. 2007;282:12619–12628. doi: 10.1074/jbc.M700607200. [DOI] [PubMed] [Google Scholar]
- 22.Caldeira MV, Melo CV, Pereira DB, Carvalho RF, Carvalho AL, Duarte CB. BDNF regulates the expression and traffic of NMDA receptors in cultured hippocampal neurons. Mol Cell Neurosci. 2007;35:208–219. doi: 10.1016/j.mcn.2007.02.019. [DOI] [PubMed] [Google Scholar]
- 23.Nakata H, Nakamura S. Brain-derived neurotrophic factor regulates AMPA receptor trafficking to post-synaptic densities via IP3R and TRPC calcium signaling. FEBS Lett. 2007;581:2047–2054. doi: 10.1016/j.febslet.2007.04.041. [DOI] [PubMed] [Google Scholar]
- 24.Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294:1030–1038. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- 25.Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 26.Tabuchi A, Nakaoka R, Amano K, Yukimine M, Andoh T, Kuraishi Y, Tsuda M. Differential activation of brain-derived neurotrophic factor gene promoters I and III by Ca2+ signals evoked via L-type voltage-dependent and N-methyl-D-aspartate receptor Ca2+ channels. J Biol Chem. 2000;275:17269–17275. doi: 10.1074/jbc.M909538199. [DOI] [PubMed] [Google Scholar]
- 27.Zheng F, Zhou X, Luo Y, Xiao H, Wayman G, Wang H. Regulation of brain-derived neurotrophic factor exon IV transcription through calcium responsive elements in cortical neurons. PloS one. 2011;6:e28441. doi: 10.1371/journal.pone.0028441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patterson SL, Grover LM, Schwartzkroin PA, Bothwell M. Neurotrophin expression in rat hippocampal slices: a stimulus paradigm inducing LTP in CA1 evokes increases in BDNF and NT-3 mRNAs. Neuron. 1992;9:1081–1088. doi: 10.1016/0896-6273(92)90067-n. [DOI] [PubMed] [Google Scholar]
- 29.Hall J, Thomas KL, Everitt BJ. Rapid and selective induction of BDNF expression in the hippocampus during contextual learning. Nat Neurosci. 2000;3:533–535. doi: 10.1038/75698. [DOI] [PubMed] [Google Scholar]
- 30.Neeper SA, Gomez-Pinilla F, Choi J, Cotman C. Exercise and brain neurotrophins. Nature. 1995;373:109. doi: 10.1038/373109a0. [DOI] [PubMed] [Google Scholar]
- 31.Young D, Lawlor PA, Leone P, Dragunow M, During MJ. Environmental enrichment inhibits spontaneous apoptosis, prevents seizures and is neuroprotective. Nat Med. 1999;5:448–453. doi: 10.1038/7449. [DOI] [PubMed] [Google Scholar]
- 32.Wang L, Lv Z, Hu Z, Sheng J, Hui B, Sun J, Ma L. Chronic cocaine-induced H3 acetylation and transcriptional activation of CaMKIIalpha in the nucleus accumbens is critical for motivation for drug reinforcement. Neuropsychopharmacology. 2010;35:913–928. doi: 10.1038/npp.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Timmusk T, Palm K, Metsis M, Reintam T, Paalme V, Saarma M, Persson H. Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron. 1993;10:475–489. doi: 10.1016/0896-6273(93)90335-o. [DOI] [PubMed] [Google Scholar]
- 34.Lubin FD, Roth TL, Sweatt JD. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J Neurosci. 2008;28:10576–10586. doi: 10.1523/JNEUROSCI.1786-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aid T, Kazantseva A, Piirsoo M, Palm K, Timmusk T. Mouse and rat BDNF gene structure and expression revisited. J Neurosci Res. 2007;85:525–535. doi: 10.1002/jnr.21139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fuchikami M, Yamamoto S, Morinobu S, Takei S, Yamawaki S. Epigenetic regulation of BDNF gene in response to stress. Psychiatry Investig. 2010;7:251–256. doi: 10.4306/pi.2010.7.4.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rattiner LM, Davis M, Ressler KJ. Differential regulation of brain-derived neurotrophic factor transcripts during the consolidation of fear learning. Learn Mem. 2004;11:727–731. doi: 10.1101/lm.83304. [DOI] [PubMed] [Google Scholar]
- 38.Ou LC, Gean PW. Transcriptional regulation of brain-derived neurotrophic factor in the amygdala during consolidation of fear memory. Mol Pharmacol. 2007;72:350–358. doi: 10.1124/mol.107.034934. [DOI] [PubMed] [Google Scholar]
- 39.Bredy TW, Wu H, Crego C, Zellhoefer J, Sun YE, Barad M. Histone modifications around individual BDNF gene promoters in prefrontal cortex are associated with extinction of conditioned fear. Learn Mem. 2007;14:268–276. doi: 10.1101/lm.500907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumar A, Choi KH, Renthal W, Tsankova NM, Theobald DE, Truong HT, Russo SJ, Laplant Q, Sasaki TS, Whistler KN, Neve RL, Self DW, Nestler EJ. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron. 2005;48:303–314. doi: 10.1016/j.neuron.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 41.An JJ, Gharami K, Liao GY, Woo NH, Lau AG, Vanevski F, Torre ER, Jones KR, Feng Y, Lu B, Xu B. Distinct role of long 3’ UTR BDNF mRNA in spine morphology and synaptic plasticity in hippocampal neurons. Cell. 2008;134:175–187. doi: 10.1016/j.cell.2008.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chiaruttini C, Sonego M, Baj G, Simonato M, Tongiorgi E. BDNF mRNA splice variants display activity-dependent targeting to distinct hippocampal laminae. Mol Cell Neurosci. 2008;37:11–19. doi: 10.1016/j.mcn.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 43.Pattabiraman PP, Tropea D, Chiaruttini C, Tongiorgi E, Cattaneo A, Domenici L. Neuronal activity regulates the developmental expression and subcellular localization of cortical BDNF mRNA isoforms in vivo. Mol Cell Neurosci. 2005;28:556–570. doi: 10.1016/j.mcn.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 44.Zheng F, Wang H. NMDA-mediated and self-induced bdnf exon IV transcriptions are differentially regulated in cultured cortical neurons. Neurochem Int. 2009;54:385–392. doi: 10.1016/j.neuint.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wibrand K, Messaoudi E, Havik B, Steenslid V, Lovlie R, Steen VM, Bramham CR. Identification of genes co-upregulated with Arc during BDNF-induced long-term potentiation in adult rat dentate gyrus in vivo. Eur J Neurosci. 2006;23:1501–1511. doi: 10.1111/j.1460-9568.2006.04687.x. [DOI] [PubMed] [Google Scholar]
- 46.Yasuda M, Fukuchi M, Tabuchi A, Kawahara M, Tsuneki H, Azuma Y, Chiba Y, Tsuda M. Robust stimulation of TrkB induces delayed increases in BDNF and Arc mRNA expressions in cultured rat cortical neurons via distinct mechanisms. J Neurochem. 2007;103:626–636. doi: 10.1111/j.1471-4159.2007.04851.x. [DOI] [PubMed] [Google Scholar]
- 47.Tao X, West AE, Chen WG, Corfas G, Greenberg ME. A calcium-responsive transcription factor, CaRF, that regulates neuronal activity-dependent expression of BDNF. Neuron. 2002;33:383–395. doi: 10.1016/s0896-6273(01)00561-x. [DOI] [PubMed] [Google Scholar]
- 48.McDowell KA, Hutchinson AN, Wong-Goodrich SJ, Presby MM, Su D, Rodriguiz RM, Law KC, Williams CL, Wetsel WC, West AE. Reduced cortical BDNF expression and aberrant memory in Carf knock-out mice. J Neurosci. 2010;30:7453–7465. doi: 10.1523/JNEUROSCI.3997-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen WG, West AE, Tao X, Corfas G, Szentirmay MN, Sawadogo M, Vinson C, Greenberg ME. Upstream stimulatory factors are mediators of Ca2+-responsive transcription in neurons. J Neurosci. 2003;23:2572–2581. doi: 10.1523/JNEUROSCI.23-07-02572.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hong EJ, McCord AE, Greenberg ME. A biological function for the neuronal activity-dependent component of Bdnf transcription in the development of cortical inhibition. Neuron. 2008;60:610–624. doi: 10.1016/j.neuron.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Impey S, Mark M, Villacres EC, Poser S, Chavkin C, Storm DR. Induction of CRE-mediated gene expression by stimuli that generate longlasting LTP in area CA1 of the hippocampus. Neuron. 1996;16:973–982. doi: 10.1016/s0896-6273(00)80120-8. [DOI] [PubMed] [Google Scholar]
- 52.Impey S, Smith DM, Obrietan K, Donahue R, Wade C, Storm DR. Stimulation of cAMP response element (CRE)-mediated transcription during contextual learning. Nat Neurosci. 1998;1:595–601. doi: 10.1038/2830. [DOI] [PubMed] [Google Scholar]
- 53.Yin JC, Wallach JS, Del Vecchio M, Wilder EL, Zhou H, Quinn WG, Tully T. Induction of a dominant negative CREB transgene specifically blocks long-term memory in Drosophila. Cell. 1994;79:49–58. doi: 10.1016/0092-8674(94)90399-9. [DOI] [PubMed] [Google Scholar]
- 54.Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- 55.Jiang X, Tian F, Du Y, Copeland NG, Jenkins NA, Tessarollo L, Wu X, Pan H, Hu XZ, Xu K, Kenney H, Egan SE, Turley H, Harris AL, Marini AM, Lipsky RH. BHLHB2 controls Bdnf promoter 4 activity and neuronal excitability. J Neurosci. 2008;28:1118–1130. doi: 10.1523/JNEUROSCI.2262-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lipsky RH, Xu K, Zhu D, Kelly C, Terhakopian A, Novelli A, Marini AM. Nuclear factor kappaB is a critical determinant in N-methyl-D-aspartate receptor-mediated neuroprotection. J Neurochem. 2001;78:254–264. doi: 10.1046/j.1471-4159.2001.00386.x. [DOI] [PubMed] [Google Scholar]
- 57.Vashishta A, Habas A, Pruunsild P, Zheng JJ, Timmusk T, Hetman M. Nuclear factor of activated T-cells isoform c4 (NFATc4/NFAT3) as a mediator of antiapoptotic transcription in NMDA receptor-stimulated cortical neurons. J Neurosci. 2009;29:15331–15340. doi: 10.1523/JNEUROSCI.4873-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109(Suppl):S67–79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- 59.Flavell SW, Cowan CW, Kim TK, Greer PL, Lin Y, Paradis S, Griffith EC, Hu LS, Chen C, Greenberg ME. Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science. 2006;311:1008–1012. doi: 10.1126/science.1122511. [DOI] [PubMed] [Google Scholar]
- 60.Sweatt JD. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol. 2004;14:311–317. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 61.Wang H, Storm DR. Calmodulin-regulated adenylyl cyclases: cross-talk and plasticity in the central nervous system. Mol Pharmacol. 2003;63:463–468. doi: 10.1124/mol.63.3.463. [DOI] [PubMed] [Google Scholar]
- 62.Wayman GA, Lee YS, Tokumitsu H, Silva AJ, Soderling TR. Calmodulin-kinases: modulators of neuronal development and plasticity. Neuron. 2008;59:914–931. doi: 10.1016/j.neuron.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu GY, Deisseroth K, Tsien RW. Activity-dependent CREB phosphorylation: convergence of a fast, sensitive calmodulin kinase pathway and a slow, less sensitive mitogenactivated protein kinase pathway. Proc Natl Acad Sci U S A. 2001;98:2808–2813. doi: 10.1073/pnas.051634198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron. 1998;21:869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 65.Parker D, Jhala US, Radhakrishnan I, Yaffe MB, Reyes C, Shulman AI, Cantley LC, Wright PE, Montminy M. Analysis of an activator:coactivator complex reveals an essential role for secondary structure in transcriptional activation. Mol cell. 1998;2:353–359. doi: 10.1016/s1097-2765(00)80279-8. [DOI] [PubMed] [Google Scholar]
- 66.Impey S, Fong AL, Wang Y, Cardinaux JR, Fass DM, Obrietan K, Wayman GA, Storm DR, Soderling TR, Goodman RH. Phosphorylation of CBP mediates transcriptional activation by neural activity and CaM kinase IV. Neuron. 2002;34:235–244. doi: 10.1016/s0896-6273(02)00654-2. [DOI] [PubMed] [Google Scholar]
- 67.Greer PL, Greenberg ME. From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron. 2008;59:846–860. doi: 10.1016/j.neuron.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 68.Impey S, Goodman RH. CREB signalingtiming is everything. Sci STKE. 2001;2001:pe1. doi: 10.1126/stke.2001.82.pe1. [DOI] [PubMed] [Google Scholar]
- 69.Bedford DC, Kasper LH, Fukuyama T, Brindle PK. Target gene context influences the transcriptional requirement for the KAT3 family of CBP and p300 histone acetyltransferases. Epigenetics. 2010;5:9–15. doi: 10.4161/epi.5.1.10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sakamoto K, Karelina K, Obrietan K. CREB: a multifaceted regulator of neuronal plasticity and protection. J Neurochem. 2011;116:1–9. doi: 10.1111/j.1471-4159.2010.07080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Qiu Z, Ghosh A. A calcium-dependent switch in a CREST-BRG1 complex regulates activity-dependent gene expression. Neuron. 2008;60:775–787. doi: 10.1016/j.neuron.2008.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wong ST, Athos J, Figueroa XA, Pineda VV, Schaefer ML, Chavkin CC, Muglia LJ, Storm DR. Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent longterm memory and late phase LTP. Neuron. 1999;23:787–798. doi: 10.1016/s0896-6273(01)80036-2. [DOI] [PubMed] [Google Scholar]
- 73.Zhang M, Storm DR, Wang H. Bidirectional synaptic plasticity and spatial memory flexibility require Ca2+-stimulated adenylyl cyclases. J Neurosci. 2011;31:10174–10183. doi: 10.1523/JNEUROSCI.0009-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sindreu CB, Scheiner ZS, Storm DR. Ca2+-stimulated adenylyl cyclases regulate ERKdependent activation of MSK1 during fear conditioning. Neuron. 2007;53:79–89. doi: 10.1016/j.neuron.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stork PJ, Schmitt JM. Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol. 2002;12:258–266. doi: 10.1016/s0962-8924(02)02294-8. [DOI] [PubMed] [Google Scholar]
- 76.Grewal SS, Fass DM, Yao H, Ellig CL, Goodman RH, Stork PJ. Calcium and cAMP signals differentially regulate cAMP-responsive element-binding protein function via a Rap1-extracellular signal-regulated kinase pathway. J Biol Chem. 2000;275:34433–34441. doi: 10.1074/jbc.M004728200. [DOI] [PubMed] [Google Scholar]
- 77.Altschuler DL, Peterson SN, Ostrowski MC, Lapetina EG. Cyclic AMP-dependent activation of Rap1b. The Journal of biological chemistry. 1995;270:10373–10376. doi: 10.1074/jbc.270.18.10373. [DOI] [PubMed] [Google Scholar]
- 78.Oliff HS, Berchtold NC, Isackson P, Cotman CW. Exercise-induced regulation of brain-derived neurotrophic factor (BDNF) transcripts in the rat hippocampus. Brain Res Mol Brain Res. 1998;61:147–153. doi: 10.1016/s0169-328x(98)00222-8. [DOI] [PubMed] [Google Scholar]
- 79.Tabuchi A, Sakaya H, Kisukeda T, Fushiki H, Tsuda M. Involvement of an upstream stimulatory factor as well as cAMP-responsive element-binding protein in the activation of brain-derived neurotrophic factor gene promoter I. J Biol Chem. 2002;277:35920–35931. doi: 10.1074/jbc.M204784200. [DOI] [PubMed] [Google Scholar]
- 80.Bird A. Perceptions of epigenetics. Nature. 2007;447:396–398. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 81.Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tian F, Hu XZ, Wu X, Jiang H, Pan H, Marini AM, Lipsky RH. Dynamic chromatin remodeling events in hippocampal neurons are associated with NMDA receptor-mediated activation of Bdnf gene promoter 1. J Neurochem. 2009;109:1375–1388. doi: 10.1111/j.1471-4159.2009.06058.x. [DOI] [PubMed] [Google Scholar]
- 83.Kuzumaki N, Ikegami D, Tamura R, Hareyama N, Imai S, Narita M, Torigoe K, Niikura K, Takeshima H, Ando T, Igarashi K, Kanno J, Ushijima T, Suzuki T. Hippocampal epigenetic modification at the brain-derived neurotrophic factor gene induced by an enriched environment. Hippocampus. 2011;21:127–132. doi: 10.1002/hipo.20775. [DOI] [PubMed] [Google Scholar]
- 84.Barrett RM, Malvaez M, Kramar E, Matheos DP, Arrizon A, Cabrera SM, Lynch G, Greene RW, Wood MA. Hippocampal focal knockout of CBP affects specific histone modifications, long-term potentiation, and long-term memory. Neuropsychopharmacology. 2011;36:1545–1556. doi: 10.1038/npp.2011.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Korzus E, Rosenfeld MG, Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42:961–972. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Alarcon JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A. Chromatin acetylation, memory, and LTP are impaired in CBP+/- mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42:947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 87.Caccamo A, Maldonado MA, Bokov AF, Majumder S, Oddo S. CBP gene transfer increases BDNF levels and ameliorates learning and memory deficits in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2010;107:22687–22692. doi: 10.1073/pnas.1012851108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Levenson JM, Roth TL, Lubin FD, Miller CA, Huang IC, Desai P, Malone LM, Sweatt JD. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J Biol Chem. 2006;281:15763–15773. doi: 10.1074/jbc.M511767200. [DOI] [PubMed] [Google Scholar]
- 89.Sharma RP, Tun N, Grayson DR. Depolarization induces downregulation of DNMT1 and DNMT3a inprimary cortical cultures. Epigenetics. 2008;3:74–80. doi: 10.4161/epi.3.2.6103. [DOI] [PubMed] [Google Scholar]
- 90.Miller CA, Gavin CF, White JA, Parrish RR, Honasoge A, Yancey CR, Rivera IM, Rubio MD, Rumbaugh G, Sweatt JD. Cortical DNA methylation maintains remote memory. Nat Neurosci. 2010;13:664–666. doi: 10.1038/nn.2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Feng J, Zhou Y, Campbell SL, Le T, Li E, Sweatt JD, Silva AJ, Fan G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci. 2010;13:423–430. doi: 10.1038/nn.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53:857–869. doi: 10.1016/j.neuron.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 93.Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- 94.Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- 95.Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, Hu L, Steen JA, Weitz CJ, Greenberg ME. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52:255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Li H, Zhong X, Chau KF, Williams EC, Chang Q. Loss of activity-induced phosphorylation of MeCP2 enhances synaptogenesis, LTP and spatial memory. Nature neuroscience. 2011;14:1001–1008. doi: 10.1038/nn.2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Guy J, Cheval H, Selfridge J, Bird A. The role of MeCP2 in the brain. Annu Rev Cell Dev Biol. 2011;27:631–652. doi: 10.1146/annurev-cellbio-092910-154121. [DOI] [PubMed] [Google Scholar]
- 99.Bramham CR, Wells DG. Dendritic mRNA: transport, translation and function. Nat Rev Neurosci. 2007;8:776–789. doi: 10.1038/nrn2150. [DOI] [PubMed] [Google Scholar]
- 100.Lau AG, Irier HA, Gu J, Tian D, Ku L, Liu G, Xia M, Fritsch B, Zheng JQ, Dingledine R, Xu B, Lu B, Feng Y. Distinct 3’UTRs differentially regulate activity-dependent translation of brain-derived neurotrophic factor (BDNF) Proc Natl Acad Sci U S A. 2010;107:15945–15950. doi: 10.1073/pnas.1002929107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RC, Masuno M, Tommerup N, van Ommen GJ, Goodman RH, Peters DJ, et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348–351. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- 102.Autry AE, Monteggia LM. Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol Rev. 2012;64:238–258. doi: 10.1124/pr.111.005108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Duman RS, Aghajanian GK. Synaptic dysfunction in depression: potential therapeutic targets. Science. 2012;338:68–72. doi: 10.1126/science.1222939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Milnerwood AJ, Raymond LA. Early synaptic pathophysiology in neurodegeneration: insights from Huntington’s disease. Trends neurosci. 2010;33:513–523. doi: 10.1016/j.tins.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 105.Allen SJ, Watson JJ, Dawbarn D. The neurotrophins and their role in Alzheimer’s disease. Curr Neuropharmacol. 2011;9:559–573. doi: 10.2174/157015911798376190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Russo SJ, Mazei-Robison MS, Ables JL, Nestler EJ. Neurotrophic factors and structural plasticity in addiction. Neuropharmacology. 2009;56(Suppl 1):73–82. doi: 10.1016/j.neuropharm.2008.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lu B, Martinowich K. Cell biology of BDNF and its relevance to schizophrenia. Novartis Found Symp. 2008;289:119–129. doi: 10.1002/9780470751251.ch10. discussion 129-135, 193-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Barnett JH, Smoller JW. The genetics of bipolar disorder. Neuroscience. 2009;164:331–343. doi: 10.1016/j.neuroscience.2009.03.080. [DOI] [PMC free article] [PubMed] [Google Scholar]