

Abstract
We developed a method to translate DNA sequences into densely functionalized nucleic acids by using T4 DNA ligase to mediate the DNA-templated polymerization of 5′-phosphorylated trinucleotides containing a wide variety of appended functional groups. This polymerization proceeds sequence specifically along a DNA template and can generate polymers of at least 50 building blocks (150 nucleotides) in length with remarkable efficiency. The resulting single-stranded highly modified nucleic acid is a suitable template for primer extension using deep vent (exo-) DNA polymerase, thereby enabling the regeneration of template DNA. We integrated these capabilities to perform iterated cycles of in vitro translation, selection, and template regeneration on libraries of modified nucleic acid polymers.
The in vitro selection of nucleic acids has given rise to DNA and RNA receptors and catalysts for a broad range of applications including therapeutics and diagnostics.1 Despite their ability to fold into well-defined globular structures with binding or catalytic activities, researchers have speculated that the limited chemical functionality present within natural nucleic acids explains the dominance of proteins over nucleic acids among biological receptors and catalysts.2 The sequence-specific incorporation of a much wider range of chemical functionality into a nucleic acid polymer therefore could enable the discovery of functional nucleic acids with chemical properties, selectivities, or activity levels exceeding those of known DNAs and RNAs.
All previously reported strategies for the sequence-specific incorporation of non-natural chemical functionality throughout a nucleic acid polymer rely on DNA and RNA polymerases to incorporate modified nucleoside triphosphates.3 While effective at generating modified nucleic acids, this approach generally limits the number of different sequence-defined functional groups to four in ssDNA3,4 and eight in dsDNA,3j while limiting the size and nature of the functional groups to those compatible with the polymerase active site. Generating sequence-defined nucleic acid polymers having more than four different non-natural functional groups requires a strategy beyond traditional polymerase-based methods.
We hypothesized that a DNA ligase-catalyzed, DNA-templated polymerization of short 5′-phosphorylated oligonucleotides containing various modifications might provide access to highly functionalized sequence-defined polymers. We chose T4 DNA ligase on the basis of its high stability, sequence specificity, and known compatibility with short oligonucleotide substrates.5,6 While the tolerance of T4 DNA ligase for accepting modified DNA has not been extensively characterized, modifications at the ligation site that do not block ligase activity have been reported for both the ligated strands and the template strand.7
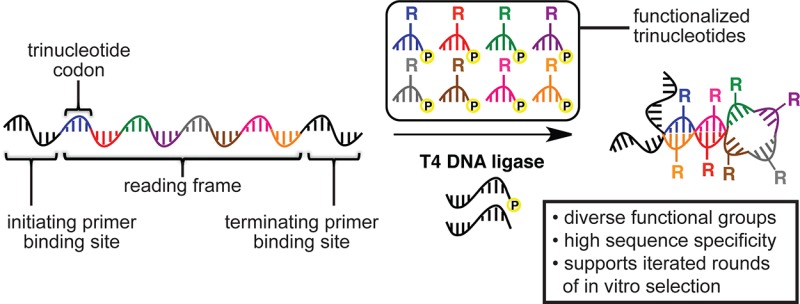
Inspired by the trinucleotide coding system used during ribosomal translation of mRNA into proteins, we chose 5′-phosphorylated trinucleotides as the functionalized monomers to be polymerized by T4 DNA ligase. Such a system would enable up to 64 different modifications to be incorporated sequence specifically throughout a nucleic acid polymer. Although the shortest T4 DNA ligase substrate previously reported was a pentamer,6a we hypothesized that optimization of ligation conditions might enable the polymerization of modified trinucleotides. Because T4 DNA ligase is known to be inefficient at ligating substrates separated by a gap when hybridized to a template,8 we envisaged the translation process occurring within a reading frame defined by a 5′-phosphorylated initiation primer and a nonphosphorylated termination primer (Figure 1a).
Figure 1.
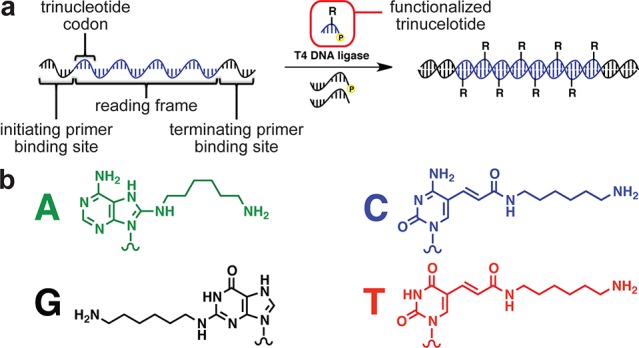
(a) General strategy for the translation of DNA into functionalized DNA using T4 DNA ligase-mediated sequence-defined polymerization of functionalized trinucleotides. (b) Amine-linked bases that were elaborated into functionalized trinucleotides.
The role of the initiation primer is to specify the beginning of the reading frame, while the role of the termination primer is to specify the end of the reading frame and preclude the formation of blunt- or cohesive-end ligation byproducts. Between the two primer-binding sites on the template are the set of codons that specify the sequence of the functionalized nucleic acid polymer.
First we characterized and optimized the ability of T4 DNA ligase to polymerize unfunctionalized trinucleotide substrates on a template encoding eight consecutive trinucleotides. Although standard DNA ligase reaction conditions did not generate full-length product efficiently, we found that the inclusion of the molecular crowding reagent PEG 60009 could significantly enhance polymerization, resulting in >95% yield of full-length product. Further, we observed that the addition of 0.1 mg/mL of BSA greatly improved the activity of T4 DNA ligase for reactions beyond 2 h, presumably due to the superior stability of the enzyme over extended periods of time in the presence of BSA.5f Despite the low melting temperatures of the trinucleotides used during these experiments (all substrate Tms were <10 °C), the polymerization of the trinucleotides was found to be high yielding up to 30 °C, highlighting the efficiency of T4 DNA ligase at ligating the trinucleotides under the optimized conditions (Figure S7). Polymerizations proceeded most efficiently at 25 °C over 12 h using 20 U/μL T4 DNA ligase, 0.1 mg/mL BSA, 1 mM ATP, 1 μM DNA template, and 4 μM/codon trinucleotide.
Next we synthesized functionalized 5′-phosphorylated trinucleotides using standard automated oligonucleotide synthesis with commercially available amine-modified nucleoside phosphoramidites and 5′-phosphorylation reagents. The amine groups served as a chemical handle to install various functional groups using well-established bioconjugation chemistries, such as amine addition to cyclic anhydrides, isothiocyanate, and activated esters (Figure 1b).10 Functionalized 5′-phosphorylated trinucleotides were readily generated in high yield following purification by reverse-phase HPLC.
To characterize the ability of T4 DNA ligase to tolerate functionalization at different positions on the substrates, we prepared a series of trinucleotides containing an amine-modified nucleobase at either the first (5′-end), middle, or third nucleotide position (3′-end). T4 DNA ligase exhibited a high tolerance for substitutions at the first nucleobase and incomplete polymerization resulting when substitutions were at the third nucleobase. Interestingly, no observable polymerization occurred when modifications were at the second position (Figure S8). In addition, when functional groups were linked to the Watson–Crick face at any of the three nucleotide positions, T4 DNA ligase also failed to effect polymerization. The crude strand-separated product from the polymerization of a trinucleotide substrate containing an amine group at the 5′ position on a 24-mer template encoding eight consecutive substrates was characterized by ESI-LC-MS and found to be consistent with the expected mass of the desired full-length product (calculated mass: 15771.1 Da, observed mass: 15770.6 Da).
We then challenged the optimized polymerization system with a wide variety of trinucleotide substrates bearing functional groups at the 5′ position (Figure 2a). The functional groups included hydrogen-bond donors and acceptors, Brønsted acids and bases, several alkyl and aryl hydrophobic groups, and a metal chelator. In total, eight different groups were each linked to a different trinucleotide sequence. Each substrate was polymerized using the T4 DNA ligase-mediated polymerization along a homo-octameric template (24-mer coding region) and then analyzed by nondenaturing PAGE (Figure 2b). All modified trinucleotides were polymerized very efficiently, yielding full-length products of the expected molecular weights in >95% yield.
Figure 2.
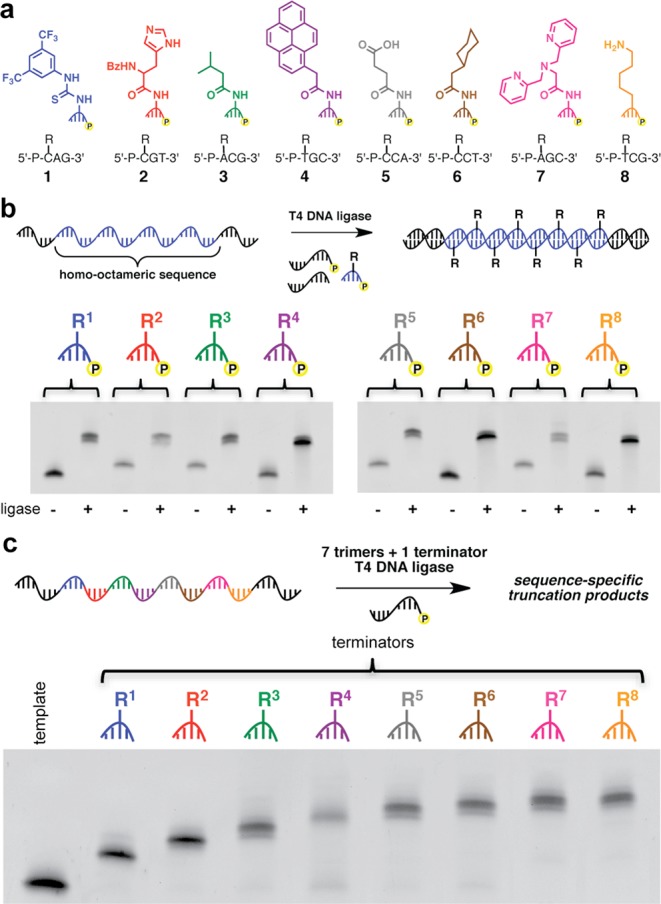
(a) Structure and sequence of functionalized trinucleotides. (b) Nondenaturing PAGE analysis of products from polymerization of the functionalized trinucleotides in (a) along their corresponding homo-octameric codon templates. (c) Nondenaturing PAGE analysis of sequence specificity using a chain-termination method.
To evaluate the sequence specificity of polymerization, we used a chain-termination strategy.11 Since T4 DNA ligase requires 5′-phosphates to continue the templated polymerization of trinucleotides, incorporating trinucleotides that lack a 5′-phosphate into the reaction should terminate the polymerization, resulting in truncation products. If T4 DNA ligase incorporates these terminator substrates in a sequence-specific manner, then chain-termination should occur only at the codon specifying the nonphosphorylated trinucleotide. If sequence specificity is poor, then polymerization should generate nucleic acid polymers of undesired length either by misincorporation of a nonterminator substrate opposite the terminator codon or by nonspecific incorporation of the terminator substrate at nonterminator codons. We used a hetero-octameric template containing all eight codons flanked by 12-nt initiation and termination primer-binding sites. Seven 5′-phosphorylated trinucleotides linked to diverse functional groups and one functionalized trinucleotide lacking a 5′-phosphate were polymerized along the template, and the reaction products were analyzed by nondenaturing PAGE (Figure 2c). One predominant product at the anticipated molecular weight was observed for each chain-termination reaction, indicating that incorporation of all eight functionalized substrates along a mixed-codon template proceeds with a high degree of sequence specificity.
We next sought to assess the efficiency of polymerization along templates of increasing sequence length (Figure 3). Templates containing reading frames ranging from 10 to 50 codons (30–150 nucleotides) encoding the isopropyl-modified trinucleotide building block flanked by 12 nt primer sets were synthesized. The polymerizations were performed at 16 °C for 12 h using the conditions described above. Full-length products were generated in high yields (>80%) for all reactions tested, highlighting the highly efficient nature of this polymerization system.
Figure 3.
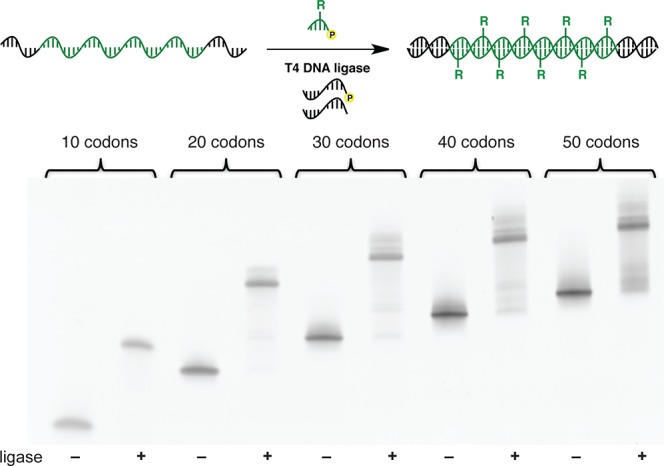
Nondenaturing PAGE analysis of the polymerization of isopropyl-modified trinucleotide 3 along homomeric templates of increasing length from 10 to 50 codons (30–150 coding nucleotides).
In order for this modified nucleic acid translation system to enable the in vitro selection of functionalized polymers, the polymer must be either linked to its encoding template in a manner that does not impede polymer folding,11b or must be amenable to primer extension to regenerate the template as canonical DNA (conceptually analogous to reverse translation). Recent efforts have demonstrated that family B DNA polymerases are particularly suited for primer extension along modified nucleic acid templates.3i Encouraged by these findings, we examined a panel of polymerases for their ability to use our highly functionalized modified DNA product as a template to generate an unmodified DNA strand. We generated a hetero-octameric polymer of modified trinucleotides and separated the functionalized polymer strand from the biotinylated template. The polymer contained each functional group once and was flanked by 18-nt primers.
Consistent with previous findings,3 we observed that deep vent (exo-) polymerase performed primer extension to generate a canonical DNA strand on this highly functionalized polymer template in excellent yield after 30 min at 70 °C (Figure S9). To characterize the sequence specificity of the primer extension, we sequenced the PAGE-purified product of the deep vent (exo-) primer extension reaction. The DNA sequence that was generated from the primer extension process using deep vent (exo-) was identical by Sanger sequencing to that of the initial template, demonstrating that both the translation and primer extension processes occurred with high sequence specificity (Figure S10).
To test a full cycle of translation, selection, template regeneration, and amplification in a library format, we sought to select a modified DNA containing a pharmacophore that binds carbonic anhydrase II from a translated library of highly functionalized DNA (Figure 4a). We generated a library of DNA templates using split-and-pool synthesis, which encoded seven trinucleotide substrates (1–7, Figure 2a) across eight coding positions. The theoretical complexity of this library is 5.8 × 106. Next, we performed a translation and mock selection using a solution containing this template library and 1/5.8 × 106th of one equivalent of a positive control template that uniquely encodes the carbonic anhydrase II inhibitor Gly-Leu-4-carboxybenzene-sulfonamide12 attached to substrate 8 and that contains an MluI restriction site to monitor the enrichment of the sequence by digestion and PAGE analysis. Following four iterated rounds of translation, strand separation, selection, primer extension, and amplification, the positive control library member was enriched >2.5 × 107-fold (Figure 4b), demonstrating the capability of this system to support highly effective iterative cycles of in vitro selection on modified nucleic acid polymers.
Figure 4.

(a) Complete cycle of ligase-mediated translation, strand separation, selection, primer extension, and template amplification for functionalized nucleic acids. (b) PAGE analysis of MluI digestion products of a population of 5.8 × 106 DNA templates subjected to four iterated rounds of the cycle described in (a), resulting in the strong enrichment of a positive control template encoding a functionalized nucleic acid containing a carbonic anhydrase-binding pharmacophore.
In summary, we have developed a new system for the translation of DNA templates into sequence-defined highly functionalized nucleic acid polymers that uses T4 DNA ligase to catalyze the DNA-templated polymerization of functionalized trinucleotides. We incorporated eight different functional groups throughout a polymer product, with the possibility of expanding the substrate set up to 64. In addition to exhibiting a high degree of sequence specificity, polymerization was remarkably efficient and could generate a polymer of 50 consecutive substrates (150 nucleotides), corresponding to a polymer of a molecular weight of approximately 60 kDa. The functionalized nucleic acid polymers were amenable to primer extension by deep vent (exo-) to regenerate the encoding template with high fidelity. Iterative cycles of translation, selection, template regeneration, and PCR amplification enabled the enrichment of a single library member encoding a carbonic anhydrase II inhibitor from a library of 5.8 × 106 highly functionalized DNAs. The ability to sequence specifically introduce a wide array of functionality within an evolvable nucleic acid polymer should increase their structural and functional capabilities and therefore may help bridge the gap between nucleic acid polymers and proteins.
Acknowledgments
This work was supported by the Howard Hughes Medical Institute and the NIH/NIGMS (R01GM065865). R. H. is a NSERC postdoctoral fellow. J. N. is partially supported by an Eli Lilly Organic Chemistry Graduate Fellowship.
Supporting Information Available
Experimental procedures, molecular characterization data, and supporting experiments and results. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Vinkenborg J. L.; Karnowski N.; Famulok M. Nat. Chem. Biol. 2011, 7, 519–527. [DOI] [PubMed] [Google Scholar]; b Silverman S. K. Angew. Chem., Int. Ed. 2010, 49, 7180–7201. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Mayer G. Angew. Chem. Int. Ed. Ed. 2009, 48, 2672–2689. [DOI] [PubMed] [Google Scholar]; d Famulok M.; Hartig J. S.; Mayer G. Chem. Rev. 2007, 107, 3715–3743. [DOI] [PubMed] [Google Scholar]; e Joyce G. F. Annu. Rev. Biochem. 2004, 73, 791–836. [DOI] [PubMed] [Google Scholar]
- Wilson D. S.; Szostak J. W. Annu. Rev. Biochem. 1999, 68, 611–647. [DOI] [PubMed] [Google Scholar]
- a Pinheiro V. B.; Taylor a. I.; Cozens C.; Abramov M.; Renders M.; Zhang S.; Chaput J. C.; Wengel J.; Peak-Chew S.-Y.; McLaughlin S. H.; Herdewijn P.; Holliger P. Science 2012, 336, 341–344. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lam C. H.; Hipolito C. J.; Hollenstein M.; Perrin D. M. Org. Biomol. Chem. 2011, 9, 6949–6954. [DOI] [PubMed] [Google Scholar]; c Obeid S.; Baccaro A.; Welte W.; Diederichs K.; Marx A. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 21327–21331. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Vaught J. D.; Bock C.; Carter J.; Fitzwater T.; Otis M.; Schneider D.; Rolando J.; Waugh S.; Wilcox S. K.; Eaton B. E. J. Am. Chem. Soc. 2010, 132, 4141–4151. [DOI] [PubMed] [Google Scholar]; e Ramsay N.; Jemth A.-S.; Brown A.; Crampton N.; Dear. P.; Holliger P. J. Am. Chem. Soc. 2010, 132, 5096–5104. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Hollenstein M.; Hipolito C. J.; Lam C. H.; Perrin D. M. Nucleic Acids Res. 2009, 37, 1638–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Loakes D.; Holliger P. Chem. Commun 2009, 45, 4619–4631. [DOI] [PubMed] [Google Scholar]; h Shoji A.; Kuwahara M.; Ozaki H.; Sawai H. J. Am. Chem. Soc. 2007, 129, 1456–1464. [DOI] [PubMed] [Google Scholar]; i Jäger S.; Rasched G.; Kornreich-Leshman H.; Engeser M.; Thum O.; Famulok M. J. Am. Chem. Soc. 2005, 127, 15071–15082. [DOI] [PubMed] [Google Scholar]; j Jäger S.; Famulok M. Angew. Chem., Int. Ed. 2004, 43, 3337–3340. [DOI] [PubMed] [Google Scholar]
- Expanding the genetic code can also increase functionality present within DNA, for recent reports see:Malyshev D. A.; Dhami K.; Quach H. T.; Lavergne T.; Ordoukhanian P.; Torkamani A.; Romesberg F. E. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 12005–12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wu D. Y.; Wallace R. B. Gene 1989, 76, 245–254. [DOI] [PubMed] [Google Scholar]; b Taylor W. H.; Hagerman P. J. J. Mol. Biol. 1990, 212, 363–376. [DOI] [PubMed] [Google Scholar]; c Rossi R.; Montecucco A.; Ciarrocchi G.; Biamonti G. Nucleic Acids Res. 1997, 25, 2106–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Arabshahi A.; Frey P. A. J. Biol. Chem. 1999, 274, 8586–8588. [DOI] [PubMed] [Google Scholar]; e Cherepanov A. V.; de Vries S. Biophys. J. 2001, 81, 3545–3559. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Cherepanov A. V.; Doroshenko E. V.; Matysik J.; de Vries S.; de Groot H. J. M. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 8563–8568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Szybalski W. Gene 1990, 90, 117–178. [Google Scholar]; b Kotler L. E.; Zevin-Sonkin D.; Sobolev I. A.; Beskin A. D. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 4241–4245. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Dunn J. J.; Butler-Loffredo L.-L.; Studier W. Anal. Biochem. 1995, 228, 91–100. [DOI] [PubMed] [Google Scholar]; d Kaczorowski T.; Szybalski W. Gene 1998, 223, 83–91. [DOI] [PubMed] [Google Scholar]
- a Mendel-Hartvig M.; Kumar A.; Landergren U. Nucleic Acids Res. 2004, 32, e2. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Liang X.; Fujioka K.; Asanuma H. Chem.—Eur. J. 2011, 17, 10388–10396. [DOI] [PubMed] [Google Scholar]
- Nilsson S. V.; Magnusson G. Nucleic Acids Res. 1982, 10, 1425–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman S. B.; Minton A. P. Annu. Rev. Biophys. Biomol. Struct. 1993, 22, 27–65. [DOI] [PubMed] [Google Scholar]
- Hermanson G. T.Bioconjugate Techniques, 2nd ed.; Academic Press: Amsterdam, 2008. [Google Scholar]
- a Sanger F.; Nicklen S.; Coulson R. Proc. Natl. Acad. Sci. U.S.A. 1977, 74, 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Brudno Y.; Birnbaum M. E.; Kleiner R. E.; Liu D. R. Nat. Chem. Biol. 2010, 6, 148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGregor L. M.; Gorin D. J.; Dumelin C. E.; Liu D. R. J. Am. Chem. Soc. 2010, 132, 15522–15524. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.