

Abstract
Well-differentiated pancreatic neuroendocrine tumors (PanNETs) comprise ~1–3% of pancreatic neoplasms. Although long considered as reasonably benign lesions, PanNETs have considerable malignant potential, with a 5-year survival of ~65% and a 10-year survival of 45% for resected lesions. As PanNETs have a low incidence, they have been understudied, with few advances made until the completion of their exomic sequencing in the past year. In this Review, we summarize some of the latest insights into the genetics of PanNETs, and their probable implications in the context of prognosis and therapy. In particular, we discuss two genes (DAXX and ATRX) that have collectively been identified as mutated in >40% of PanNETs, and the biological and prognostic implications of these novel mutations. The identification of recurrent somatic mutations within the mTOR signaling pathway and the therapeutic implications for personalized therapy in patients with PanNETs are also discussed. Finally, this Review outlines state-of-the-art advances in the biology of PanNETs that are of emerging translational importance.
Introduction
Well-differentiated pancreatic neuroendocrine tumors (PanNETs) comprise ~1–3% of pancreatic neoplasms and are the second most commonly encountered primary malignancy of the pancreas.1 The estimated incidence of PanNETs in Europe and the USA is <1 per 100,000 individuals per year; however, the number of PanNETs diagnosed has increased considerably over the past four decades.1,2 Improvements in diagnostic imaging and increased use of imaging techniques are partly responsible for the increased detection of these neoplasms. PanNETs are sometimes erroneously considered as fairly benign or indolent tumors, but the 5-year survival following resection of a PanNET is ~65%, and the 10-year survival is 45%.2 Moreover, the fact that the majority (~70%) of patients present with metastatic disease underscores the malignant potential of PanNETs.1 For patients who have localized disease, surgical resection with curative intent remains the most effective therapeutic option. For patients with advanced disease, either streptozocin monotherapy, or its combination with fluorouracil or doxorubicin, were the only approved regimens in the US for more than three decades.3–5 However, analyses in the past 15 years suggest that these regimes are not as efficacious as previously thought.6,7
As a neoplastic entity, PanNETs have remained largely understudied, both at the molecular level and in the clinic. This deficiency can be mostly attributed to the low incidence of PanNETs, but also to the fact that these neoplasms can have heterogeneous clinical manifestations. From a scientific perspective, the scant availability of preclinical models (that is, cell lines and genetically engineered mouse models) has limited studies into the biology of PanNETs; as a result, progress in terms of early diagnosis and targeted therapies has been slow.
This Review provides the salient clinicopathological features of PanNETs, including the new WHO classification system, to ensure uniform assessment of disease, prognosis and treatment. We also discuss the latest advances in the biology of PanNETs, including the sequencing of the PanNET exome that has revealed recurrent, and hitherto unreported, somatic mutations in a new cancer-associated pathway. The implications of mutational analysis for personalized therapy are also discussed, as is the approval of new targeted agents tailored specifically towards PanNETs.
Classification and grading
According to the 2010 ‘WHO Classification of Tumours of the Digestive System’, PanNETs (also known as ‘islet cell tumors’) are defined as well-differentiated neoplasms ≥0.5 cm in size, with predominantly neuroendocrine differentiation and low proliferation indices (Figure 1).8–10 This Review does not discuss pancreatic neuroendocrine carcinomas (PanNECs), which are poorly differentiated high-grade neoplasms defined by the presence of >20 mitoses per 10 high power fields (HPFs). PanNECs exhibit different (that is, more aggressive) biological behavior and are genetically distinct from PanNETs, and as such should be considered as a different entity within the pancreatic neuroendocrine neoplasms.11
Figure 1.
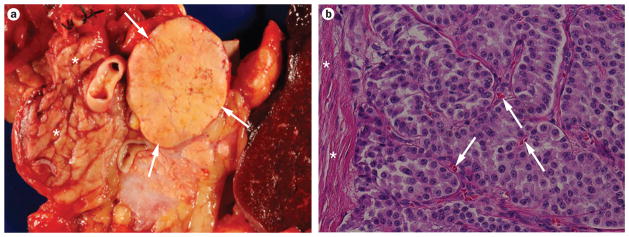
Gross appearance and histologic features of well-differentiated pancreatic neuroendocrine tumors (PanNETs). a | A well-demarcated PanNET (white arrows) in the tail of the pancreas is seen surrounded by normal parenchyma (white asterisks). The sample is comprised of a distal pancreatectomy with en bloc splenectomy. b | Microscopic appearance of a well-differentiated PanNET, in which neoplastic cells are organized in a nested fashion and demonstrate minimal pleomorphism or mitotic activity (low-grade or G1 lesion). A feature of PanNETs that differentiates them from pancreatic adenocarcinomas is the presence of abundant intratumoral microvessels (white arrows). In contrast to the infiltrating nature of adenocarcinomas, PanNETs are generally well demarcated from the surrounding non-neoplastic pancreas by fibrous tissue bands (white asterisks). Hematoxylin and eosin staining, 200× original magnification.
PanNETs are dichotomized into functioning (syndromic) or nonfunctioning (nonsyndromic) tumors. Functioning tumors are neoplasms that produce clinical symptoms as a result of the hypersecretion of one or more specific hormones, whereas nonfunctioning PanNETs generally present with mass effects. Of note, the mere identification of hormonal products in an otherwise clinically silent PanNET by special assays, such as immunohistochemistry, should not result in it being given a ‘functioning’ designation.
Of the functioning PanNETs, 30–45% are functioning insulin-producing PanNETs (insulinomas), which cause symptoms associated with excessive insulin production.12 These symptoms are linked to hypoglycemia and are reversible with glucose administration (Whipple’s triad).13 Functioning gastrin-producing tumors (gastrinomas) constitute 16–30% of functioning PanNETs and can result in Zollinger–Ellison syndrome, the symptoms of which include refractory peptic ulcerations and secretory diarrhea.14 In addition to the prototype clinical symptoms of these hormone-producing PanNETs, a profiling study published in 2011 also suggests that distinct underlying expression of transcription factors, probably reflecting a different cell or cells of origin, could be used to identify PanNETs.15
Other, infrequent (<10%) functional PanNETs include VIPomas and glucagonomas. VIPomas secrete vasoactive intestinal polypeptide (VIP), which gives rise to symptoms of watery diarrhea, hypokalemia and achlorhydria (WDHA syndrome, also known as Verner–Morrison syndrome).16 The most typical presentation of glucagonomas is a form of dermatitis known as migratory necrolytic erythema, in addition to diabetes (glucose intolerance), depression and deep venous thrombosis—symptoms that are referred to as the ‘4Ds’ syndrome.17 Less than 5% of PanNETs are somatostatinomas; as a result of the general inhibitory effect of somatostatin on both gastrointestinal exocrine and endocrine glands, these neoplasms produce several nonspecific and apparently unrelated symptoms. These symptoms include steatorrhea (or diarrhea), recent-onset diabetes mellitus, cholelithiasis, anemia, weight loss and features of hypochlorhydria.18 This compendium of clinical manifestations has been referred to as the ‘somatostatinoma syndrome’; however, as a large retrospective study showed, the strict definition of the syndrome is rarely present in its entirety in most patients who have an underlying somatostatin-secreting PanNET.19
In the current WHO classification, PanNETs are graded histologically on the basis of their proliferation rate, as assessed by either mitotic counts or nuclear Ki-67 immunolabeling.10 Such grading is an essential component in the classification of these tumors, as this parameter is a significant independent prognostic factor. Thus, low-grade or G1 PanNETs are those with a mitotic count of 0–1 per 10 HPFs or a nuclear Ki-67 labeling index of 0–2%. Intermediate-grade or G2 PanNETs are those with 2–20 mitoses per 10 HPFs or a nuclear Ki-67 labeling index of 3–20%. The highest grade (G3) neuroendocrine neoplasms (mitotic counts >20 per 10 HPFs or >20% nuclear Ki-67 labeling index) are, as previously stated, classified as PanNECs, and such lesions uniformly have a poor clinical outcome. The histological grading (especially as determined by the Ki-67 labeling index) is an independent predictor of disease progression in patients with resected PanNETs, with a hazard ratio (HR) of 1.52 when comparing G2 with G1 lesions, and an HR of 3.43 for G3 compared with G1 lesions.20 As such, the Ki-67 labeling index should now be provided as a standard component of the pathology report on any resected PanNET.
Cancer-predisposing syndromes
Although the vast majority of PanNETs occur sporadically, up to 10% of cases occur in patients who have a cancer-predisposition syndrome. These syndromes include multiple endocrine neoplasia type 1 (MEN1), von Hippel–Lindau disease (VHL), neurofibromatosis type 1 (NF1), and tuberous sclerosis complex (TSC), which are characterized by mutations in the tumor suppressor genes MEN1, VHL, NF1 and TSC1 or TSC2, respectively (Table 1).21–24 PanNETs can be the first manifestation of these syndromes.
Table 1.
Genetic syndromes associated with PanNETs
| Syndrome | Affected gene | Prevalence in PanNETs (%) |
|---|---|---|
| Multiple endocrine neoplasia type 1 | MEN1 | 20–70 |
| Von Hippel–Lindau | VHL | ≤20 |
| Neuro bromatosis type 1 | NF1 | ≤10 |
| Tuberous sclerosis complex | TSC2 | Rare (~1) |
Abbreviation: PanNETs, pancreatic neuroendocrine tumors.
All individuals who have MEN1 syndrome will develop some kind of neuroendocrine lesion of the pancreas during their lifetime, predominantly ‘micro-adenomas’ (defined as adenomas <0.5 cm in diameter) that are typically multifocal and, per definition, nonfunctioning.25 Between 20% and 70% of patients (the prevalence increases with age) who have MEN1 will develop symptomatic PanNETs.22,26,27 Approximately half of patients with MEN1 will develop Zollinger–Ellison syndrome because of an underlying gastrinoma, which usually arises in the duodenum, and roughly one-fifth of patients with MEN1 have clinical symptoms attributable to an insulinoma.28 Although patients with MEN1 often develop neuroendocrine lesions in other organs, PanNETs are the most common underlying cause of disease-associated mortality in these patients.29
Pancreatic involvement is also not uncommon in patients with VHL syndrome; however, only about one-fifth of these involvements are PanNETs, with the remaining being cystic lesions, specifically serous cystadenomas of the pancreas.21,22,30 PanNETs in patients with VHL syndrome are almost exclusively nonfunctioning, and are typically detected at routine follow-up for tumors associated with the syndrome that are more prevalent than PanNETs, such as hemangioblastomas, renal cell carcinomas or adrenal pheochromocytomas.31,32 Of note, the PanNETs in patients with VHL syndrome are often mixed serous cystic–neuroendocrine neoplasms.
In contrast to MEN1 and VHL syndromes, PanNETs are uncommon in patients with NF1 (also known as von Recklinghausen disease), and are reported in <10% of patients.33,34 Similarly, PanNETs are also uncommon in patients with TSC (~1%), with most reported cases occurring in patients with TSC type 2.22,35
Genetic alterations in PanNETs
The first generation
Before the PanNET exome was sequenced, much of the knowledge of the genetics of PanNETs stemmed from discoveries made studying the aforementioned familial syndromes, and applying these discoveries to sporadic lesions. This paradigm is best illustrated by the MEN1 syndrome, and the identification of the MEN1 gene.36,37 The PanNETs that arise in patients with MEN1 have a germline loss-of-function mutation in one allele of the MEN1 gene on chromosome 11q13.1, coupled with a second somatic mutation in the other allele, either through loss of heterozygosity or a smaller ‘intragenic mutation’, which inactivate both copies of the MEN1 gene.36 The protein product of MEN1, menin, is an essential component of the MLL/SET1-like histone methyltransferase complex and regulates chromatin remodeling, functioning as a cell-type specific activator or repressor of gene transcription.38,39 The contextual specificity of menin as an activator or repressor of transcription leads to a curious paradox, wherein menin has an oncogenic role in promoting MLL-dependent leukemias, but is a tumor suppressor in cells of neuroendocrine lineage.40 In pancreatic islet cells, menin activates the transcription of the genes encoding cyclin-dependent kinase (CDK) inhibitors CDK inhibitor 2C (CDKN2C) and CDK inhibitor 1B (CDKN1B), through histone H3 lysine 4 methylation, thus regulating cell-cycle progression.41,42 Loss of menin function in islet cells results in downregulation of CDK inhibitor 2C and CDK inhibitor 1B expression, and unchecked S-phase progression.
The importance of MEN1 in the pathogenesis of PanNETs is further strengthened by evidence from genetically engineered mouse models, in which the mouse homolog Men1 is either hemizygously deleted in the germline or conditionally inactivated in the β cells of the pancreas, which results in the formation of a range of lesions that most closely resemble microadenomas and insulinomas.43–45 Since the initial discovery of MEN1 mutations in familial PanNETs, a large body of evidence has shown that this gene is also somatically mutated in 22–34% of sporadically occurring PanNETs.46,47 Genetic analysis of a large series of 100 sporadic PanNETs found that 25% had somatic mutations in the MEN1 gene.48
PTEN is a tumor suppressor gene that is located on chromosome 10q23, which encodes a dual lipid and protein phosphatase upstream of the oncogenic PI-3-kinase (PI3K)–Akt–mammalian target of rapamycin (mTOR) pathway.49 In tumors with loss-of-function mutations of PTEN, unrestrained activation of PI3K–Akt–mTOR signaling results in cell growth and proliferation, as well as other features associated with neoplastic transformation (Figure 2). On the basis of comparative genomic hybridization data that revealed the loss of chromosome arm 10q in 25% of PanNETs,50 Perren et al. analyzed the PTEN gene in a series of sporadic PanNETs, and identified loss of heterozygosity at 10q23 in one-third of tumors, although somatic mutations were uncommon (~3%).51
Figure 2.

The mTOR signaling pathway. Mutations in the gene that encodes PI3K are activating (*), whereas those in the genes that encode PTEN and TSC2 are inactivating (‡) in PanNETs. Rapalogs are first-generation inhibitors of mTOR complex 1 (mTORC1) and include everolimus. Abbreviations: 4E-BP1, eIF4E-binding protein 1; AMPK, AMP-activated kinase; EGFR, epidermal growth factor receptor; IGFR, insulin-like growth factor receptor; IRS1, insulin receptor substrate 1; GβL, G protein β subunit-like; mTOR, mammalian target of rapamycin; PI3K, phosphatidylinositol 3-kinase; PTEN, phosphatase and tensin homolog deleted at chromosome 10; Raptor, regulatory associated protein of mTOR; Rheb, Ras homolog enriched in brain; Rictor, rapamycin-insensitive companion of mTOR; S6K1, p70 S6 kinase 1; Sin1, stress-activated protein kinase interaction protein 1; STK11, serine/threonine-protein kinase 11; TSC, tuberous sclerosis complex.
As stated previously, germline mutations in TSC2 are another uncommon hereditary cause of PanNETs, and the protein product encoded by TSC2 also regulates mTOR signaling downstream of Akt. In light of the perceived importance of PI3K–Akt–mTOR signaling to PanNET pathogenesis, Missiaglia et al. determined the expression profiles of the proteins encoded by the PTEN and TSC2 genes in ~70 sporadic primary tumors, and found downregulation of either transcript in ~80% of cases; the loss of PTEN and TSC2 expression was correlated with reduced disease-free and overall survival.52 As detailed below, these findings provide a rational basis for the first successful targeted therapy in patients with PanNETs, with the intent of blocking mTOR signaling.
In addition to mutational events in cancer-associated pathways, numerous abnormalities in expression have been described in PanNETs; the detailed description of these abnormalities is beyond the scope of this article, but they have been covered elsewhere.53,54 A survey of the literature points to nodal pathways that are recurrently altered in these lesions. For example, upregulation of cyclin D1, which promotes the G1/S cell-cycle transition by binding to CDKs, is observed in nearly half of all PanNETs.55 As a corollary of this finding, global expression profiling of PanNETs compared with normal pancreatic islet cells has identified downregulation of CDK inhibitor 1, which blocks G1 progression, in approximately half of patients.56 Thus, aberrant control of the cell cycle seems to be a key pathway involved in PanNET pathogenesis, and from a clinical standpoint this observation is reflected in the impact of proliferation rates (Ki-67 labeling index) on disease prognosis (detailed previously).
The next generation
An advance in elucidating the biology of PanNETs was made with the publication of the PanNET exome by Jiao and colleagues in 2011 (Table 2).57 In this study, the complete exomes of 10 primary PanNETs were sequenced (discovery phase), followed by screening for mutations in the most commonly altered genes in an additional 58 tumors (validation phase). As the same group had previously sequenced the pancreatic ductal adenocarcinoma (PDAC) exome, they were able to compare the genetic changes in exocrine carcinomas with those in neuroendocrine tumors.58 Notably, mutations that define the genetic landscape of PDACs, such as frequent alterations of the KRAS oncogene and loss-of-function mutations or homozygous deletions of CDKN2A and SMAD4, were not encountered in PanNETs. At a prevalence of 5%, the rate of mutations in TP53 is significantly lower than the ~70% observed in PDAC. In addition, PDACs generally seem to have a greater degree of genetic complexity than PanNETs, with a median of 66 somatic mutations versus 16 in PanNETs. Needless to add, these genetic features probably contribute to the observed differences in the natural history between the two tumor types.
Table 2.
Genetic alterations in PanNETs and potential targeted therapies
| Gene | Mutation frequency (%)* | Protein function | Targeted therapy |
|---|---|---|---|
| MEN1 | 44 | Histone remodeling | NA |
| DAXX | 25 | Chromatin assembly | NA |
| ATRX | 18 | Chromatin assembly | NA |
| TSC2 | 9 | GTPase-activating protein | Everolimus, sirolimus, temsirolimus |
| PTEN | 7 | Dual-specificity protein phosphatase | Everolimus, sirolimus, temsirolimus |
| PIK3CA | 1 | Phosphoinositide 3-kinase | Everolimus, sirolimus, temsirolimus |
| TP53 | 5 | Cell-cycle arrest | NA |
Abbreviations: NA, not available; PanNETS, pancreatic neuroendocrine tumors.
Percentages taken from Jiao et al.57
Not surprisingly, MEN1 was found to be the most frequently mutated gene in PanNETs, as it was altered in 44% of PanNETs.57 In addition to MEN1, several genes in the PI3K–Akt–mTOR pathway were also found to be mutated in PanNETs—alterations of PTEN, TSC2 or PIK3CA were found in ~16% of the tumors. Of these, mutations in PTEN and TSC2 are loss-of-function mutations, whereas the PIK3CA mutations are present at a previously described oncogenic ‘hotspot’ residue that constitutively activates the kinase domain of the encoded protein.59 The finding of mutations in the mTOR pathway in PanNETs has considerable clinical therapeutic implications, as further discussed below.
The most striking discovery from the exome sequencing was the identification of recurrent somatic mutations in two genes that had previously not been associated with cancer—DAXX and ATRX. These genes were mutated in 25% and 18% of PanNETs, respectively.57 Alterations of ATRX or DAXX were mutually exclusive (that is, they did not both occur in the same tumor), which suggests that the encoded proteins function in the same pathway. Furthermore, changes in the nucleotide sequence often resulted in truncating (nonsense) mutations, a feature typically associated with tumor suppressor genes. Patients with PanNETs that contained altered ATRX or DAXX genes had appreciably longer survival than did patients with wild-type tumors. Moreover, patients with PanNETs that had mutant ATRX or DAXX plus mutant MEN1 were alive at 10 years following resection. As the numbers of patients with each subset of mutations are fairly low, these findings need to be substantiated in a large series; however, these findings do indicate that there is an intriguing genetic association with prognosis.
As this cancer-associated pathway is still novel, little information exists on how loss of ATRX or DAXX function precisely effects tumor initiation and/or progression in PanNETs. In terms of their normal functions, both of the proteins encoded by these genes participate in heterochromatin assembly, particularly in the incorporation of the histone variant H3.3 at the telomeric ends of chromosomes.60–62 Thus, not surprisingly, tumors with wild-type ATRX or DAXX demonstrate intact nuclear immunolabeling (presumably the compartment that is most important for protein function), whereas this nuclear localization is typically lost in PanNETs that have mutations in ATRX or DAXX (Figure 3a–d).
Figure 3.

Loss of ATRX or DAXX function in pancreatic neuroendocrine tumors (PanNETs) correlates with the alternative lengthening of telomeres (ALT) phenotype. a | Immunolabeling for ATRX demonstrates strong nuclear labeling of ATRX protein in a well-differentiated PanNET. b | Immunolabeling for DAXX of the same PanNET demonstrates strong nuclear labeling of tumor nuclei. c | The same PanNET is negative for the ALT phenotype. Telomere florescence in situ hybridization (FISH) signals (red) are markedly dimmer in neoplastic cells (asterisk) than in the surrounding stromal cells. Centromere-specific FISH probe (green) serves as a positive control for hybridization. d | ATRX immunolabeling in a PanNET with ATRX mutation showing loss of nuclear protein expression. e | Telomere-specific FISH of the same PanNET displaying large, ultrabright telomere FISH signals indicative of ALT (white arrows). All images 400× original magnification.
As the ATRX and DAXX proteins are important in heterochromatin maintenance at telomeres, a well-described quantitative telomere-specific fluorescence in situ hybridization (FISH) technique that labels the telomere hexameric repeats63,64 was used by Heaphy and colleagues to examine the status of telomeres in a large series of genetically characterized PanNETs.65,66 Remarkably, a perfect correlation was observed between loss of ATRX or DAXX function (as assessed by immunolabeling for nuclear protein) and the presence of a telomerase-independent telomere maintenance mechanism known as alternative lengthening of telomeres (ALT).65 The ALT phenotype can be readily identified by the presence of ultrabright telomere-specific signals and the presence of so-called ALT-associated PML bodies on telomere-specific FISH (Figure 3e), and is present in only ~3% of all human neoplasms.66 Notably, the strong correlation between ALT and loss of nuclear immunolabeling for either protein persisted when extrapolated to other solid neoplasms, such as glioblastoma multiformae, which reiterates a general role for ATRX and DAXX in telomere maintenance.65 The association of ATRX and DAXX inactivation with the ALT phenotype might explain previous observations in other tumor types that connected the ALT phenotype with improved prognosis.67,68
In summary, the sequencing of the PanNET exome has defined the key genetic pathways that are altered in these tumors, and these pathways have potential prognostic and therapeutic implications. For example, if corroborated by large studies, a simple immunolabeling test for either the ATRX or the DAXX protein on resected PanNET specimens could provide useful prognostic insights. Numerous questions remain unanswered, however, such as identifying the fundamental mechanisms by which alterations in ATRX and DAXX produce the ALT phenotype and promote PanNET pathogenesis. Whether the resulting ALT phenotype is the basis for the improved survival in patients (probably by preventing the onset of widespread chromosomal instability and poor prognosis that accompanies these features)69 or is simply an epiphenomenon is still to be ascertained.
Functional studies that use appropriate in vitro or conditional mouse models might help to elucidate the telomere-independent functions of ATRX and DAXX that are required for tumor suppression. The development of such models might also identify potential targets that can be used to design therapies that produce a synthetic lethal effect in neoplastic cells with loss of ATRX or DAXX function, a finding that could have therapeutic implications beyond PanNETs. The utility of such pre-clinical models in PanNET research remains to be established, but parallels in the PDAC field are encouraging, as the development of genetically and biologically relevant preclinical disease models of PDAC has led to tangible improvements in our understanding of the underlying disease process.70–72
Therapeutic advances
In patients with resectable PanNETs, surgery with curative intent (that is, R0 or margins that are microscopically free of tumor) remains the treatment of choice.73 Unfortunately, as the majority of patients with PanNETs either present with metastatic disease or have disease recurrence within 2 years of surgery, effective systemic therapies are also needed.20
Various somatostatin analogues, such as octreotide, lanreotide and pasireotide, have therapeutic benefits in patients with advanced PanNETs.74 For example, octreotide lengthens the time to progression in patients with advanced PanNETs; the progression time in patients who received this agent was more than twice as long as the progression time in individuals administered a placebo (14.3 months versus 6 months, respectively).75 The ‘theranostic’ rationale for the use of somatostatin analogues is that the majority of PanNETs express somatostatin receptors on their cell surface.76 This concept has been taken a step further by labeling somatostatin analogues with radionuclides such as indium (111I), yttrium (90Y) or lutetium (177Lu), which can then be utilized for so-called peptide receptor radionuclide therapy (PRRT). Studies on PRRT in advanced PanNETs are limited, but emerging data suggest that this strategy results in favorable response rates and a delay to progression.77,78 In the future, prospective randomized trials will have to confirm the benefit of PRRT compared with current standard-of-care treatments for patients with advanced PanNETs.79
Much of the focus on treatment over the past half century has been on the use of conventional cytotoxic agents such as streptozocin3–5,80,81 or temozolomide.82–84 Streptozocin, an alkylating agent that is essentially an antibiotic with islet-cell-specific toxicity, was approved by the FDA in the early 1980s after the confirmation of positive results when administered with fluorouracil.4,80 An analogue of streptozocin—chlorozotocin—has comparable therapeutic activity to the combination of streptozocin with fluorouracil. Subsequently, a combination of streptozocin plus doxorubicin demonstrated improved activity compared with the original fluorouracil-based regimen.5 Even though the results from some of these trials were positive, others failed to demonstrate unequivocal improvement in progression-free survival or overall survival, lending an element of skepticism to relying on conventional cytotoxic agents alone.6,7,85
The emergence of two related agents, temozolamide and dacarbazine, has resulted in the efficacy of alkylating agents in patients with advanced PanNETs being reexamined.86 The cytotoxic effects of temozolomide result from DNA methylation (alkylation) at the O6 position of guanine residues, which ultimately results in apoptosis and cell death.87 In patients with advanced PanNETs, several retrospective studies have demonstrated response rates to temozolomide-based treatments that vary between 33% and 74%.82–84 Of note, the DNA repair enzyme, O6-methylguanine DNA methyltransferase (MGMT), which reverts the alkylation at the O6 position on guanines, can confer resistance to temozolomide.87 Not surprisingly, the efficacy of temozolomide is greater in patients with PanNETs that lack MGMT expression (as a result of epigenetic silencing of the corresponding gene promoter) than in patients with those that express MGMT.88 Dacarbazine demonstrates more equivocal efficacy in patients with PanNETs than temozolomide, with reported response rates of 16–33%.76
The serine-threonine kinase mTOR is at the center of an oncogenic pathway that is involved in cell growth, proliferation and anabolic metabolism. As previously discussed, Jiao et al. found somatic mutations in the genes encoding proteins of the PI3K–Akt–mTOR pathway in 16% of PanNETs, all of which are predicted to result in aberrant mTOR signaling.52,57 Hence, it would be expected that inhibiting mTOR signaling in PanNETs, at least in a subset of these tumors, would be effective in attenuating tumor progression.
A number of inhibitors of mTOR signaling have been developed, including the prototype rapamycin and several water-soluble analogues of rapamycin (‘rapa-logs’) that demonstrate improved pharmacokinetics.89 Within this class of agents, everolimus has shown promise as a therapeutic agent in patients with advanced PanNETs. On the basis of the results of two encouraging phase II trials,90,91 Yao and colleagues conducted a prospective randomized trial to evaluate the efficacy of everolimus in 410 patients with advanced PanNETs (RADIANT-3).92 The patients in this phase III trial were randomly assigned to receive either 10 mg daily everolimus or placebo. Patients receiving everolimus had a significant improvement in median progression-free survival (11.0 months) compared with patients in the placebo arm (4.6 months); however, no differences in overall survival were noted at the interim analysis. The HR for disease progression or death from any cause was 0.35 in the everolimus arm, underscoring the efficacy of the therapy. Although only 16% of PanNETs have mutations in genes encoding proteins in the mTOR pathway, the sense from this trial was that >16% of the patients responded to everolimus, suggesting that there might be additional mechanisms by which this pathway is activated in PanNETs.
On the basis of the success of the RADIANT-3 trial, the FDA recommended approval for the use of everolimus in patients with unresectable, locally advanced or metastatic PanNETs in May 2011. Follow-up analyses on the RADIANT-3 cohorts (including unpublished data presented at a 2011 annual oncology meeting) have elucidated additional information on the use of everolimus in this setting. For example, the efficacy of everolimus is retained in patients who have received prior chemotherapy, or have been previously or concomitantly treated with the somatostatin analogue, octreotide, which is typically beneficial in patients with midgut carcinoids.93,94 Finally, data on the safety of everolimus were evaluated with an increased length of follow-up. Stomatitis and diarrhea were among the most common adverse events; in general, everolimus was very well tolerated at the recommended oral dose of 10 mg per day.95
Additional combinatorial regimens of everolimus that are currently being evaluated should provide even greater efficacy, and an improvement in the overall survival.96 The need for combinatorial regimens is reiterated by the all too frequent observation, including in preclinical models of PanNETs, that monotherapy with rapalogs often induces compensatory upregulation of parallel growth factor signaling pathways and eventual therapeutic resistance.97,98 Despite its limitations, everolimus has emerged as one of the most promising molecularly targeted therapies in the armamentarium against PanNETs in more than a generation.
Other approaches to targeted therapies for PanNETs are also on the horizon. One of the distinguishing features of well-differentiated PanNETs, particularly compared with PDACs, is the presence of abundant intratumoral vasculature (Figure 1b). The PanNET milieu is rich in a variety of growth factors that support this tumor-associated angiogenesis, including vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF)—the latter binds to the cognate receptor (PDGFR) that is expressed on vascular pericytes.99,100 Notably, in addition to their endothelial and stromal expression, VEGFR and PDGFR are also present on the neoplastic cells themselves, which suggests that autocrine loops in PanNET progression exist.101,102
In a series of elegant preclinical studies using a viral oncogene-driven mouse model of PanNETs,103 Hanahan and colleagues have demonstrated the efficacy of anti-angiogenic therapy, specifically in the context of dual targeting of the activities of both VEGFR and PDGFR.104–106 Many of these preclinical studies utilized the multi-tyrosine kinase inhibitor sunitinib, which is a potent antagonist of VEGFR and PDGFR signaling. These preclinical advances have, in turn, led to phase I and II trials in patients who have advanced PanNETs, with encouraging results.107,108 These studies culminated in a pivotal randomized placebo-controlled phase III trial of sunitinib in patients with advanced PanNETs.109 In this trial, patients receiving the optimal oral dose of 37.5 mg sunitinib a day demonstrated a median progression-free survival of 11.4 months versus 5.5 months in those who received placebo. At the data cutoff point, the HR for death in the treatment arm was 0.41, although median overall survival could not be estimated in the two arms as a result of the fairly high number of censored events.
In a follow-up to the sunitinib trial, Raymond et al. reported updated progression-free survival data of 12.6 months in the treatment group versus 5.8 months in the placebo group.110 By contrast, the differences in overall survival have not reached statistical significance in the trial update. Nevertheless, the results were encouraging and the FDA approved sunitinib as yet another option for patients with unresectable, locally advanced or metastatic PanNETs. The cautionary statements vis-à-vis acquired resistance and compensatory bypass mechanisms alluded to in the context of everolimus monotherapy also hold true for sunitinib. As amply demonstrated in preclinical models, antiangiogenesis therapy can lead to paradoxical enhancement of invasive and metastatic disease.105 Thus, it is probable that the ongoing clinical trials evaluating combinatorial regimens will emerge as the eventual protocol or protocols of choice in patients with advanced PanNETs.
Conclusions
Even though PanNETs are the second-most common malignancy of the pancreas, they have long been understudied as a result of their low incidence and heterogeneous clinical presentation. The overwhelming majority of patients with PanNETs present with advanced disease, underscoring their inherent malignant potential. The sequencing of the PanNET exome has defined the key genetic pathways that are altered in these tumors. In particular, recurrent somatic mutations have been identified in two genes, ATRX and DAXX, whose encoded products participate in hetero-chromatin assembly, with either ATRX or DAXX being mutated in 43% of PanNETs. Understanding how these genes contribute to the pathogenesis and natural history of PanNETs will probably become an area of intense research over the next decade. Whereas cytotoxic agents have been at the core of systemic therapy for PanNETs for over 30 years, two molecularly targeted agents —everolimus and sunitinib—have been approved by the FDA, providing clinicians and patients alike with the first new therapeutic choices in more than a generation. In some respects, the next decade will probably be the golden years for PanNET research, which for too long has been the poor cousin of the better studied PDACs. The juxtaposition of molecular aberrations with targeted therapies has the potential to bring in a much-needed era of personalized medicine for patients with advanced PanNETs.
Key points.
Pancreatic neuroendocrine tumors (PanNETs) are fully malignant neoplasms, with the majority (~70%) of patients presenting with advanced disease and a 5-year survival of ~65%
In 2010, the WHO implemented a new classification for PanNETs that stratifies these tumors by stage and histological grade (the latter defined by mitotic counts or Ki-67 labeling index)
Sequencing of the PanNET exome has identified somatic mutations in MEN1, DAXX, ATRX and mTOR pathway genes in 44%, 25%, 18% and ~16% of PanNETs, respectively
ATRX and DAXX are PanNET tumor suppressor genes that are involved in chromatin remodeling—mutations in these genes are correlated with the alternative lengthening of telomeres phenotype
Two molecularly targeted therapies—sunitinib (VEGF inhibitor) and everolimus (mTOR inhibitor)—have been independently shown to improve progression-free survival in patients with metastatic PanNETs, and are approved by the FDA
Review criteria.
This Review is based on the professional experience of the authors in addition to an extensive literature search in PubMed. The PubMed search string contained the following terms “pancreas” OR “pancreatic” AND “endocrine” OR “neuroendocrine” OR “carcinoid” OR “islet cell” AND “tumor” OR “tumors” OR “neoplasm” OR “neoplasms” OR “microadenoma” OR “microadenomas” OR “carcinoma” OR “carcinomas”. The search string was limited to “[Title/Abstract]” and language or publication date did not serve as a search criterion. All used citations were available in full-text except for updates (that is, only an abstract available) presented at the American Society of Clinical Oncology Meeting. Additional sources of information consisted of topic-related literature collected by the authors and the identification of additional citations by cross-checking references.
Acknowledgments
The authors would like to acknowledge the support of the Sol Goldman Pancreatic Cancer Research Center and the Caring for Carcinoid Foundation. The authors are grateful to Drs Alan Meeker and Christopher Heaphy (Johns Hopkins University) for assistance with Figure 3 of this Review and helpful suggestions.
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
R. F. de Wilde and A. Maitra researched data for the article. All authors contributed equally to discussion of the content, writing the manuscript and reviewing/editing the manuscript before submission.
References
- 1.Yao JC, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26:3063–3072. doi: 10.1200/JCO.2007.15.4377. [DOI] [PubMed] [Google Scholar]
- 2.Halfdanarson TR, Rabe KG, Rubin J, Petersen GM. Pancreatic neuroendocrine tumors (PNETs): incidence, prognosis and recent trend toward improved survival. Ann Oncol. 2008;19:1727–1733. doi: 10.1093/annonc/mdn351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kouvaraki MA, et al. Fluorouracil, doxorubicin, and streptozocin in the treatment of patients with locally advanced and metastatic pancreatic endocrine carcinomas. J Clin Oncol. 2004;22:4762–4771. doi: 10.1200/JCO.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 4.Moertel CG, Hanley JA, Johnson LA. Streptozocin alone compared with streptozocin plus fluorouracil in the treatment of advanced islet-cell carcinoma. N Engl J Med. 1980;303:1189–1194. doi: 10.1056/NEJM198011203032101. [DOI] [PubMed] [Google Scholar]
- 5.Moertel CG, Lefkopoulo M, Lipsitz S, Hahn RG, Klaassen D. Streptozocin-doxorubicin, streptozocin-fluorouracil or chlorozotocin in the treatment of advanced islet-cell carcinoma. N Engl J Med. 1992;326:519–523. doi: 10.1056/NEJM199202203260804. [DOI] [PubMed] [Google Scholar]
- 6.Cheng PN, Saltz LB. Failure to confirm major objective antitumor activity for streptozocin and doxorubicin in the treatment of patients with advanced islet cell carcinoma. Cancer. 1999;86:944–948. [PubMed] [Google Scholar]
- 7.McCollum AD, et al. Lack of efficacy of streptozocin and doxorubicin in patients with advanced pancreatic endocrine tumors. Am J Clin Oncol. 2004;27:485–488. doi: 10.1097/01.coc.0000135343.06038.eb. [DOI] [PubMed] [Google Scholar]
- 8.Klöppel G. Classification and pathology of gastroenteropancreatic neuroendocrine neoplasms. Endocr Relat Cancer. 2011;18 (Suppl 1):S1–S16. doi: 10.1530/ERC-11-0013. [DOI] [PubMed] [Google Scholar]
- 9.Rindi G, et al. In: WHO Classification of Tumours of the Digestive System. Bosman FT, Carneiro F, Hruban RH, Theise N, editors. IARC; Lyon: 2010. pp. 13–14. [Google Scholar]
- 10.Klimstra DS, et al. In: WHO Classification of Tumours of the Digestive System. Bosman FT, Carneiro F, Hruban RH, Theise N, editors. IARC; Lyon: 2010. pp. 322–326. [Google Scholar]
- 11.Yachida S, et al. Small cell and large cell neuroendocrine carcinomas of the pancreas are genetically similar and distinct from well-differentiated pancreatic neuroendocrine tumors. Am J Surg Pathol. 2012;36:173–184. doi: 10.1097/PAS.0b013e3182417d36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Halfdanarson TR, Rubin J, Farnell MB, Grant CS, Petersen GM. Pancreatic endocrine neoplasms: epidemiology and prognosis of pancreatic endocrine tumors. Endocr Relat Cancer. 2008;15:409–427. doi: 10.1677/ERC-07-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whipple AO. The surgical therapy of hyperinsulinism. J Int Chir. 1938;3:237–276. [Google Scholar]
- 14.Zollinger RM, Ellison EH. Primary peptic ulcerations of the jejunum associated with islet cell tumors of the pancreas. Ann Surg. 1955;142:709–723. [PMC free article] [PubMed] [Google Scholar]
- 15.Hermann G, Konukiewitz B, Schmitt A, Perren A, Klöppel G. Hormonally defined pancreatic and duodenal neuroendocrine tumors differ in their transcription factor signatures: expression of ISL1, PDX1, NGN3, and CDX2. Virchows Arch. 2011;459:147–154. doi: 10.1007/s00428-011-1118-6. [DOI] [PubMed] [Google Scholar]
- 16.Verner JV, Morrison AB. Islet cell tumor and a syndrome of refractory watery diarrhea and hypokalemia. Am J Med. 1958;25:374–380. doi: 10.1016/0002-9343(58)90075-5. [DOI] [PubMed] [Google Scholar]
- 17.Kindmark H, et al. Endocrine pancreatic tumors with glucagon hypersecretion: a retrospective study of 23 cases during 20 years. Med Oncol. 2007;24:330–337. doi: 10.1007/s12032-007-0011-2. [DOI] [PubMed] [Google Scholar]
- 18.Larsson LI, et al. Pancreatic somatostatinoma. Clinical features and physiological implications. Lancet. 1977;1:666–668. doi: 10.1016/s0140-6736(77)92113-4. [DOI] [PubMed] [Google Scholar]
- 19.Garbrecht N, et al. Somatostatin-producing neuroendocrine tumors of the duodenum and pancreas: incidence, types, biological behavior, association with inherited syndromes, and functional activity. Endocr Relat Cancer. 2008;15:229–241. doi: 10.1677/ERC-07-0157. [DOI] [PubMed] [Google Scholar]
- 20.Panzuto F, et al. Metastatic and locally advanced pancreatic endocrine carcinomas: analysis of factors associated with disease progression. J Clin Oncol. 2011;29:2372–2377. doi: 10.1200/JCO.2010.33.0688. [DOI] [PubMed] [Google Scholar]
- 21.Alexakis N, et al. Hereditary pancreatic endocrine tumours. Pancreatology. 2004;4:417–433. doi: 10.1159/000079616. [DOI] [PubMed] [Google Scholar]
- 22.Jensen RT, Berna MJ, Bingham DB, Norton JA. Inherited pancreatic endocrine tumor syndromes: advances in molecular pathogenesis, diagnosis, management, and controversies. Cancer. 2008;113 (7 Suppl):1807–1843. doi: 10.1002/cncr.23648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anlauf M, et al. Microadenomatosis of the endocrine pancreas in patients with and without the multiple endocrine neoplasia type 1 syndrome. Am J Surg Pathol. 2006;30:560–574. doi: 10.1097/01.pas.0000194044.01104.25. [DOI] [PubMed] [Google Scholar]
- 24.Périgny M, et al. Pancreatic endocrine microadenomatosis in patients with von Hippel-Lindau disease: characterization by VHL/HIF pathway proteins expression. Am J Surg Pathol. 2009;33:739–748. doi: 10.1097/PAS.0b013e3181967992. [DOI] [PubMed] [Google Scholar]
- 25.Perren A, et al. Multiple endocrine neoplasia type 1 (MEN1): loss of one MEN1 allele in tumors and monohormonal endocrine cell clusters but not in islet hyperplasia of the pancreas. J Clin Endocrinol Metab. 2007;92:1118–1128. doi: 10.1210/jc.2006-1944. [DOI] [PubMed] [Google Scholar]
- 26.Pipeleers-Marichal M, et al. Gastrinomas in the duodenums of patients with multiple endocrine neoplasia type 1 and the Zollinger-Ellison syndrome. N Engl J Med. 1990;322:723–727. doi: 10.1056/NEJM199003153221103. [DOI] [PubMed] [Google Scholar]
- 27.Triponez F, et al. Epidemiology data on 108 MEN 1 patients from the GTE with isolated nonfunctioning tumors of the pancreas. Ann Surg. 2006;243:265–672. doi: 10.1097/01.sla.0000197715.96762.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carty SE, et al. The variable penetrance and spectrum of manifestations of multiple endocrine neoplasia type 1. Surgery. 1998;124:1106–1113. doi: 10.1067/msy.1998.93107. [DOI] [PubMed] [Google Scholar]
- 29.Dean PG, et al. Are patients with multiple endocrine neoplasia type I prone to premature death? World J Surg. 2000;24:1437–1441. doi: 10.1007/s002680010237. [DOI] [PubMed] [Google Scholar]
- 30.Schmitt AM, et al. VHL inactivation is an important pathway for the development of malignant sporadic pancreatic endocrine tumors. Endocr Relat Cancer. 2009;16:1219–1227. doi: 10.1677/ERC-08-0297. [DOI] [PubMed] [Google Scholar]
- 31.Hammel PR, et al. Pancreatic involvement in von Hippel-Lindau disease. The Groupe Francophone d’Etude de la Maladie de von Hippel-Lindau. Gastroenterology. 2000;119:1087–1095. doi: 10.1053/gast.2000.18143. [DOI] [PubMed] [Google Scholar]
- 32.Lonser RR, et al. von Hippel-Lindau disease. Lancet. 2003;361:2059–2067. doi: 10.1016/S0140-6736(03)13643-4. [DOI] [PubMed] [Google Scholar]
- 33.Mao C, Shah A, Hanson DJ, Howard JM. Von Recklinghausen’s disease associated with duodenal somatostatinoma: contrast of duodenal versus pancreatic somatostatinomas. J Surg Oncol. 1995;59:67–73. doi: 10.1002/jso.2930590116. [DOI] [PubMed] [Google Scholar]
- 34.Fujisawa T, et al. Malignant endocrine tumor of the pancreas associated with von Recklinghausen’s disease. J Gastroenterol. 2002;37:59–67. doi: 10.1007/s535-002-8135-x. [DOI] [PubMed] [Google Scholar]
- 35.Ilgren EB, Westmoreland D. Tuberous sclerosis: unusual associations in four cases. J Clin Pathol. 1984;37:272–278. doi: 10.1136/jcp.37.3.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chandrasekharappa SC, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276:404–407. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- 37.Marx S, et al. Multiple endocrine neoplasia type 1: clinical and genetic topics. Ann Intern Med. 1998;129:484–494. doi: 10.7326/0003-4819-129-6-199809150-00011. [DOI] [PubMed] [Google Scholar]
- 38.Agarwal SK, et al. Menin interacts with the AP1 transcription factor JunD and represses JunD-activated transcription. Cell. 1999;96:143–152. doi: 10.1016/s0092-8674(00)80967-8. [DOI] [PubMed] [Google Scholar]
- 39.Hughes CM, et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell. 2004;13:587–597. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- 40.Yokoyama A, Cleary ML. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell. 2008;14:36–46. doi: 10.1016/j.ccr.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karnik SK, et al. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc Natl Acad Sci USA. 2005;102:14659–14664. doi: 10.1073/pnas.0503484102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schnepp RW, et al. Mutation of tumor suppressor gene Men1 acutely enhances proliferation of pancreatic islet cells. Cancer Res. 2006;66:5707–5715. doi: 10.1158/0008-5472.CAN-05-4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bertolino P, Tong WM, Galendo D, Wang ZQ, Zhang CX. Heterozygous Men1 mutant mice develop a range of endocrine tumors mimicking multiple endocrine neoplasia type 1. Mol Endocrinol. 2003;17:1880–1892. doi: 10.1210/me.2003-0154. [DOI] [PubMed] [Google Scholar]
- 44.Crabtree JS, et al. A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc Natl Acad Sci USA. 2001;98:1118–1123. doi: 10.1073/pnas.98.3.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crabtree JS, et al. Of mice and MEN1: Insulinomas in a conditional mouse knockout. Mol Cell Biol. 2003;23:6075–6085. doi: 10.1128/MCB.23.17.6075-6085.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Toliat MR, Berger W, Ropers HH, Neuhaus P, Wiedenmann B. Mutations in the MEN I gene in sporadic neuroendocrine tumours of gastroenteropancreatic system. Lancet. 1997;350:1223. doi: 10.1016/S0140-6736(05)63453-8. [DOI] [PubMed] [Google Scholar]
- 47.Moore PS, et al. Role of disease-causing genes in sporadic pancreatic endocrine tumors: MEN1 and VHL. Genes Chromosomes Cancer. 2001;32:177–181. doi: 10.1002/gcc.1180. [DOI] [PubMed] [Google Scholar]
- 48.Corbo V, et al. MEN1 in pancreatic endocrine tumors: analysis of gene and protein status in 169 sporadic neoplasms reveals alterations in the vast majority of cases. Endocr Relat Cancer. 2010;17:771–783. doi: 10.1677/ERC-10-0028. [DOI] [PubMed] [Google Scholar]
- 49.Hay N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell. 2005;8:179–183. doi: 10.1016/j.ccr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 50.Speel EJ, et al. Genetic differences in endocrine pancreatic tumor subtypes detected by comparative genomic hybridization. Am J Pathol. 1999;155:1787–1794. doi: 10.1016/S0002-9440(10)65495-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perren A, et al. Mutation and expression analyses reveal differential subcellular compartmentalization of PTEN in endocrine pancreatic tumors compared to normal islet cells. Am J Pathol. 2000;157:1097–1103. doi: 10.1016/S0002-9440(10)64624-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Missiaglia E, et al. Pancreatic endocrine tumors: expression profiling evidences a role for AKT-mTOR pathway. J Clin Oncol. 2010;28:245–255. doi: 10.1200/JCO.2008.21.5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gumbs AA, et al. Review of the clinical, histological, and molecular aspects of pancreatic endocrine neoplasms. J Surg Oncol. 2002;81:45–53. doi: 10.1002/jso.10142. [DOI] [PubMed] [Google Scholar]
- 54.Oberg K. Genetics and molecular pathology of neuroendocrine gastrointestinal and pancreatic tumors (gastroenteropancreatic neuroendocrine tumors) Curr Opin Endocrinol Diabetes Obes. 2009;16:72–78. doi: 10.1097/med.0b013e328320d845. [DOI] [PubMed] [Google Scholar]
- 55.Chung DC, et al. Overexpression of cyclin D1 occurs frequently in human pancreatic endocrine tumors. J Clin Endocrinol Metab. 2000;85:4373–4378. doi: 10.1210/jcem.85.11.6937. [DOI] [PubMed] [Google Scholar]
- 56.Maitra A, et al. Global expression analysis of well-differentiated pancreatic endocrine neoplasms using oligonucleotide microarrays. Clin Cancer Res. 2003;9:5988–5995. [PubMed] [Google Scholar]
- 57.Jiao Y, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331:1199–1203. doi: 10.1126/science.1200609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jones S, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Samuels Y, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 60.Elsaesser SJ, Allis CD. HIRA and Daxx constitute two independent histone H3.3-containing predeposition complexes. Cold Spring Harb Symp Quant Biol. 2010;75:27–34. doi: 10.1101/sqb.2010.75.008. [DOI] [PubMed] [Google Scholar]
- 61.Goldberg AD, et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010;140:678–691. doi: 10.1016/j.cell.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lewis PW, Elsaesser SJ, Noh KM, Stadler SC, Allis CD. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc Natl Acad Sci USA. 2010;107:14075–14080. doi: 10.1073/pnas.1008850107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meeker AK, et al. Telomere shortening is an early somatic DNA alteration in human prostate tumorigenesis. Cancer Res. 2002;62:6405–6409. [PubMed] [Google Scholar]
- 64.van Heek NT, et al. Telomere shortening is nearly universal in pancreatic intraepithelial neoplasia. Am J Pathol. 2002;161:1541–1547. doi: 10.1016/S0002-9440(10)64432-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heaphy CM, et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011;333:425. doi: 10.1126/science.1207313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Heaphy CM, et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am J Pathol. 2011;179:1608–1615. doi: 10.1016/j.ajpath.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hakin-Smith V, et al. Alternative lengthening of telomeres and survival in patients with glioblastoma multiforme. Lancet. 2003;361:836–838. doi: 10.1016/s0140-6736(03)12681-5. [DOI] [PubMed] [Google Scholar]
- 68.Ulaner GA, et al. Absence of a telomere maintenance mechanism as a favorable prognostic factor in patients with osteosarcoma. Cancer Res. 2003;63:1759–1763. [PubMed] [Google Scholar]
- 69.Thompson SL, Bakhoum SF, Compton DA. Mechanisms of chromosomal instability. Curr Biol. 2010;20:R285–R295. doi: 10.1016/j.cub.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bardeesy N, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci USA. 2006;103:5947–5952. doi: 10.1073/pnas.0601273103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hingorani SR, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 72.Hingorani SR, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 73.Fendrich V, Bartsch DK. Surgical treatment of gastrointestinal neuroendocrine tumors. Langenbecks Arch Surg. 2011;396:299–311. doi: 10.1007/s00423-011-0741-7. [DOI] [PubMed] [Google Scholar]
- 74.Eriksson B. New drugs in neuroendocrine tumors: rising of new therapeutic philosophies? Curr Opin Oncol. 2010;22:381–386. doi: 10.1097/CCO.0b013e32833adee2. [DOI] [PubMed] [Google Scholar]
- 75.Rinke A, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27:4656–4663. doi: 10.1200/JCO.2009.22.8510. [DOI] [PubMed] [Google Scholar]
- 76.Faivre S, Sablin MP, Dreyer C, Raymond E. Novel anticancer agents in clinical trials for well-differentiated neuroendocrine tumors. Endocrinol Metab Clin North Am. 2010;39:811–826. doi: 10.1016/j.ecl.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 77.Bushnell DL, Jr, et al. 90Y-edotreotide for metastatic carcinoid refractory to octreotide. J Clin Oncol. 2010;28:1652–1659. doi: 10.1200/JCO.2009.22.8585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kwekkeboom DJ, et al. Treatment with the radiolabeled somatostatin analog [177 Lu-DOTA 0, Tyr3]octreotate: toxicity, efficacy, and survival. J Clin Oncol. 2008;26:2124–2130. doi: 10.1200/JCO.2007.15.2553. [DOI] [PubMed] [Google Scholar]
- 79.Rindi G, Wiedenmann B. Neuroendocrine neoplasms of the gut and pancreas: new insights. Nat Rev Endocrinol. 2012;8:54–64. doi: 10.1038/nrendo.2011.120. [DOI] [PubMed] [Google Scholar]
- 80.Broder LE, Carter SK. Pancreatic islet cell carcinoma. II Results of therapy with streptozotocin in 52 patients. Ann Intern Med. 1973;79:108–118. doi: 10.7326/0003-4819-79-1-108. [DOI] [PubMed] [Google Scholar]
- 81.Chernicoff D, Bukowski RM, Groppe CW, Jr, Hewlett JS. Combination chemotherapy for islet cell carcinoma and metastatic carcinoid tumors with 5-fluorouracil and streptozotocin. Cancer Treat Rep. 1979;63:795–796. [PubMed] [Google Scholar]
- 82.Kulke MH, et al. Phase II study of temozolomide and thalidomide in patients with metastatic neuroendocrine tumors. J Clin Oncol. 2006;24:401–406. doi: 10.1200/JCO.2005.03.6046. [DOI] [PubMed] [Google Scholar]
- 83.Ekeblad S, et al. Temozolomide as monotherapy is effective in treatment of advanced malignant neuroendocrine tumors. Clin Cancer Res. 2007;13:2986–2991. doi: 10.1158/1078-0432.CCR-06-2053. [DOI] [PubMed] [Google Scholar]
- 84.Strosberg JR, et al. First-line chemotherapy with capecitabine and temozolomide in patients with metastatic pancreatic endocrine carcinomas. Cancer. 2011;117:268–275. doi: 10.1002/cncr.25425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Eriksson B, et al. Medical treatment and long-term survival in a prospective study of 84 patients with endocrine pancreatic tumors. Cancer. 1990;65:1883–1890. doi: 10.1002/1097-0142(19900501)65:9<1883::aid-cncr2820650902>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 86.Alexandraki KI, Kaltsas G. Gastroenteropancreatic neuroendocrine tumors: new insights in the diagnosis and therapy. Endocrine. 2012;41:40–52. doi: 10.1007/s12020-011-9562-2. [DOI] [PubMed] [Google Scholar]
- 87.Liu L, Gerson SL. Targeted modulation of MGMT: clinical implications. Clin Cancer Res. 2006;12:328–331. doi: 10.1158/1078-0432.CCR-05-2543. [DOI] [PubMed] [Google Scholar]
- 88.Kulke MH, et al. O6-methylguanine DNA methyltransferase deficiency and response to temozolomide-based therapy in patients with neuroendocrine tumors. Clin Cancer Res. 2009;15:338–345. doi: 10.1158/1078-0432.CCR-08-1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang X, Sun SY. Enhancing mTOR-targeted cancer therapy. Expert Opin Ther Targets. 2009;13:1193–1203. doi: 10.1517/14728220903225008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yao JC, et al. Daily oral everolimus activity in patients with metastatic pancreatic neuroendocrine tumors after failure of cytotoxic chemotherapy: a phase II trial. J Clin Oncol. 2010;28:69–76. doi: 10.1200/JCO.2009.24.2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yao JC, et al. Efficacy of RAD001 (everolimus) and octreotide LAR in advanced low- to intermediate-grade neuroendocrine tumors: results of a phase II study. J Clin Oncol. 2008;26:4311–4318. doi: 10.1200/JCO.2008.16.7858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yao JC, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:514–523. doi: 10.1056/NEJMoa1009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pommier RF, et al. Impact of prior chemotherapy on progression-free survival in patients (pts) with advanced pancreatic neuroendocrine tumors (pNET): Results from the RADIANT-3 trial [abstract] J Clin Oncol. 2011;29 (Suppl):4103. [Google Scholar]
- 94.Shah MH, et al. Everolimus in patients with advanced pancreatic neuroendocrine tumors (pNET): Impact of somatostatin analog use on progression-free survival in the RADIANT-3 trial [abstract] J Clin Oncol. 2011;29 (Suppl):4010. [Google Scholar]
- 95.Strosberg JR, Lincy J, Winkler RE, Wolin EM. Everolimus in patients with advanced pancreatic neuroendocrine tumors (pNET): Updated results of a randomized, double-blind, placebo-controlled, multicenter, phase III trial (RADIANT-3) [abstract] J Clin Oncol. 2011;29 (Suppl):4009. [Google Scholar]
- 96.ClinicalTrials.gov [online] 2011 http://www.clinicaltrials.gov/ct2/home.
- 97.Chiu CW, Nozawa H, Hanahan D. Survival benefit with proapoptotic molecular and pathologic responses from dual targeting of mammalian target of rapamycin and epidermal growth factor receptor in a preclinical model of pancreatic neuroendocrine carcinogenesis. J Clin Oncol. 2010;28:4425–4433. doi: 10.1200/JCO.2010.28.0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.O’Reilly KE, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Joyce JA, et al. Stage-specific vascular markers revealed by phage display in a mouse model of pancreatic islet tumorigenesis. Cancer Cell. 2003;4:393–403. doi: 10.1016/s1535-6108(03)00271-x. [DOI] [PubMed] [Google Scholar]
- 100.Lindahl P, Johansson BR, Levéen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–245. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- 101.Fjällskog ML, Hessman O, Eriksson B, Janson ET. Upregulated expression of PDGF receptor beta in endocrine pancreatic tumors and metastases compared to normal endocrine pancreas. Acta Oncol. 2007;46:741–746. doi: 10.1080/02841860601048388. [DOI] [PubMed] [Google Scholar]
- 102.Hansel DE, et al. Liver metastases arising from well-differentiated pancreatic endocrine neoplasms demonstrate increased VEGF-C expression. Mod Pathol. 2003;16:652–659. doi: 10.1097/01.MP.0000077416.68489.50. [DOI] [PubMed] [Google Scholar]
- 103.Hanahan D. Heritable formation of pancreatic beta-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature. 1985;315:115–122. doi: 10.1038/315115a0. [DOI] [PubMed] [Google Scholar]
- 104.Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. 2003;111:1287–1295. doi: 10.1172/JCI17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pàez-Ribes M, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pietras K, Hanahan D. A multitargeted, metronomic, and maximum-tolerated dose “chemo-switch” regimen is antiangiogenic, producing objective responses and survival benefit in a mouse model of cancer. J Clin Oncol. 2005;23:939–952. doi: 10.1200/JCO.2005.07.093. [DOI] [PubMed] [Google Scholar]
- 107.Faivre S, et al. Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J Clin Oncol. 2006;24:25–35. doi: 10.1200/JCO.2005.02.2194. [DOI] [PubMed] [Google Scholar]
- 108.Kulke MH, et al. Activity of sunitinib in patients with advanced neuroendocrine tumors. J Clin Oncol. 2008;26:3403–3410. doi: 10.1200/JCO.2007.15.9020. [DOI] [PubMed] [Google Scholar]
- 109.Raymond E, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:501–513. doi: 10.1056/NEJMoa1003825. [DOI] [PubMed] [Google Scholar]
- 110.Raymond E, et al. Updated overall survival (OS) and progression-free survival (PFS) by blinded independent central review (BICR) of sunitinib (SU) versus placebo (PBO) for patients (Pts) with advanced unresectable pancreatic neuroendocrine tumors (NET) J Clin Oncol. 2011;29 (Suppl):4008. [Google Scholar]