

Abstract
A soluble, short chain phosphatidylserine, 1,2-dicaproyl-sn-glycero-3-phospho-L-serine (C6PS), binds to discrete sites on FXa, FVa, and prothrombin to alter their conformations, to promote FXa dimerization (Kd ~ 14 nM), and to enhance both the catalytic activity of FXa and the cofactor activity of FVa. In the presence of calcium, C6PS binds to two sites on FXa, one in the epidermal growth factor like (EGF) domain and one in the catalytic domain; the latter interaction is sensitive to Na+ binding and probably represents a protein recognition site. Here we ask whether dimerization of FXa and its binding to FVa in the presence of C6PS are competitive processes. We monitored FXa activity at 5, 20 and 50 nM FXa while titrating with FVa in the presence of 400 µM C6PS and 3 or 5 mM Ca2+ to show that the apparent Kd of FVa-FXa interaction increased with increasing FXa concentration at 5 mM Ca2+, but the Kd was only slightly affected at 3 mM Ca2+. A mixture of 50 nM FXa and 50 nM FVa in the presence of 400 µM C6PS yielded both Xa homodimers and Xa ·Va heterodimers but no FXa dimers bound to FVa. A mutant FXa (R165A) that has reduced prothrombinase activity showed both reduced dimerization (Kd~147 nM) and reduced FVa binding (apparent Kd, = 58, 92 and 128 nM, respectively for 5, 20 and 50 nM R165A FXa). Native gel electrophoresis showed that the GLA-EGFNC fragment of FXa (lacking the catalytic domain) neither dimerized nor formed a complex with FVa in the presence of 400 µM C6PS and 5 mM Ca2+. Our results demonstrate that the dimerization site and FVa binding site are both located in the catalytic domain of FXa and that these sites are linked thermodynamically.
Introduction
Thrombin production, located at the convergence of the three legs of the traditional blood coagulation cascade, is arguably the central reaction of blood coagulation, and thrombin is the central metabolite of blood coagulation (1). A complex between two soluble clotting factors, FXa and FVa, catalyzes the conversion of prothrombin to thrombin. This prothrombin-activating complex (or prothrombinase) assembles and functions in vivo on activated platelet membranes (2, 3). Phosphatidylserine (PS), which, along with phosphatidylethanolamine, is exposed on activated platelet membranes (4), appears to be the platelet lipid required for efficient blood coagulation (5). In order to examine the molecular details of regulation of FXa and FVa by PS, we introduced a soluble (CMC ~ 650–800 µM depending on exact conditions) form of PS (1,2-dicaproyl-sn-glycero-3-phospho-L-serine, C6PS) that has allowed us to locate the PS regulatory sites on FXa (6) and FVa (7) and to establish the regions of these molecules affected by occupancy of these sites by PS. While C6PS does not occur in vivo, it has proven to be a very important tool in our efforts to reveal the molecular details of PS regulation of these two coagulation factors and of blood coagulation in general.
FXa has an N-terminal GLA module essential for membrane binding and two highly Cys-rich domains (epidermal-growth-factor-like cassettes, EGFN and EGFC) that link the Gla module to the C-terminal catalytic domain containing the catalytic triad (H/D/S) and FVa interaction sites (8–11). The EGF cassette pair binds a molecule of C6PS, which is soluble up to ~700–800 µM under our conditions to form in solution, an inactive dimer (12, 13) or a 60-fold activated monomer (14, 15). Binding of C6PS to the EGF pair is specific for L-phosphatidylserine (16), triggers binding of a second molecule of C6PS to a Ca2+- and Na+-dependent site in the catalytic domain and alters the active site (6, 14), and elicits changes in FXa activity (15) only when the GLA and EGF domains are linked by C6PS binding (6).
FVa is a heterodimer (17) composed of a heavy chain (FVa-HC; A1 & A2 domains; Mr = 105,000 in human) and a light chain (FVa-LC; A3, C1, & C2 domains; Mr = 74,000 or 71,000) (18, 19). Heterogeneity of the light chain is seen in both bovine and human FVa (18–20) and is due to alternative glycosylation at Asn2181 in the C2 domain (21). The species that is glycosylated at Asn2181 is termed factor Va1 while the species lacking glycosylation at this position is factor Va2. FVa2 differs from FVa1 in its ability to bind tightly to FXa in the presence of phosphatidylserine (PS)-containing membranes or a short-chain PS (C6PS) (22, 23). For this reason, all of our studies are performed with FVa2 that, for simplicity, will be referred to as FVa. FVa binds 4 molecules of C6PS, two molecules to the FVa-HC moiety and two molecules to the FVa-LC moiety (24). PS binding to the C2 site in FVa-LC is required for tight membrane association (25), while binding to a site in the C1 domain is required for tight association between FXa and FVa on a membrane or in solution (7). We have not identified a function for the C6PS-binding sites in FVa-HC.
The FVa binding site in FXa is thought to be located in the catalytic domain: residues 162–169 (8, 9); residues 185–189 (26); and residues 230–245 (27, 28) and is thermodynamically linked to a Na+ binding site and the active site (29) (numbering is in the chymotrypsin system). C6PS binding to a regulatory sites in the EGFNC pair (ideally attached to the Gla (GLA) domain) is also linked to the Na+ binding and active sites (6). C6PS binding to the regulatory site triggers formation of an inactive FXa dimer in which Lysine 90 in the catalytic domain becomes buried (13). Because C6PS binding to the regulatory site is linked to both the FXa dimer- and the Na+/FVa binding regions, we speculated that these binding regions might be directly linked within the catalytic domain. Here we test this hypothesis and show that FVa binding and dimerization of FXa are competitive processes. Next, we show that the GLA-EGFNC fragment of FXa (lacking the catalytic domain) does not contribute to dimerization or to interaction with FVa, in the presence of C6PS, thus locating both interactions exclusively to the catalytic domain. Finally, we show that a FXa mutation in the catalytic domain that interferes with FVa binding (R165A, (8)) also interferes with both FXa dimer formation, thus locating the linkage between these binding regions to the catalytic domain. Because these regions are far separated in current molecular models of FXa, our results imply either that a linkage path exists between them or that PS triggers formation of mutually exclusive FXa-binding or FVa-binding conformations of the FXa catalytic domain.
Materials & Methods
Materials
The sodium salt of 1, 2-dicaproyl-sn-glycero-3-phospho-L-serine (C6PS) was purchased from Avanti Polar Lipids (Birmingham, AL). Human prothrombin and FXa were obtained from Hematologic Technologies Inc. (Essex Junction, VT). FXa was assayed prior to each experiment using an active-site titration standard (30) to measure its activity. The R165A mutant of FXa (chymotrypsin numbering (11)) was expressed and purified as described in (8) and the GLA-EGFNC fragment of FXa was provided by Dr. Stenflo and prepared as described in (31). Purified human factor V was prepared from fresh frozen human plasma (American Red Cross Center, Durham, NC) and then activated to FVa as described (18, 32). The glycosylated form of human FVa (referred to here as simply FVa) was isolated as described previously (21, 23). The activity of FVa was assayed using either 25/75 PS/DOPC vesicles (33)or C6PS (25). Dansylarginine-N- (3-ethyl-1, 5-pentanediyl) amide (DAPA) was obtained from Haematologic Technologies Inc. (Essex Junction, VT). All other chemicals were ACS reagent grade; all solvents were HPLC grade.
Short-chain phospholipid sample preparation
Chloroform was removed from measured amounts of C6PS stock using a stream of nitrogen, and the lipid sample was dissolved in cyclohexane and lyophilized overnight. The lyophilized lipid was then suspended in an appropriate amount of buffer to reach a desired concentration and used within 1–2 days.
Critical Micelle Concentration (CMC) Determination
Changes in pyrene florescence were used to document the CMC of C6PS (34). Pyrene fluorescence has been shown to detect micelles with aggregation numbers as small as 10, as validated by comparison to CMC determination by surface tension and conductivity measurements (34, 35).
Native polyacrylamide gel electrophoresis
Separation of protein mixtures and protein standards on native gels of varying percentages allows the determination of both charge and mass of the sample proteins. The graphic analysis used is known as the Ferguson Plot (36). The details of the procedures are described elsewhere (12, 23, 30, 36). Five marker proteins (myosin, BSA, Trypsin inhibitor, α-lactalbumin, chicken albumin, carbonic anhydrase) were subjected to native polyacrylamide gel electrophoresis at varying degrees of gel cross-linking, and a Ferguson plot (retardation coefficient versus molecular weight) was constructed based on their mobilities. FXa, FVa, or their mixture (50 nM) in 50 mM Tris, 150 mM NaCl, 5 mM CaCl2, pH 7.5, 0.6 % PEG (buffer A) were incubated at 37°C for 10 minutes in the presence and absence of 400 µM C6PS. These samples were run together with the marker proteins in 5% or 8% polyacrylamide gels in a BioRad Mini-Protean II™ minigel apparatus (Biorad Corp., Hercules, CA) and stained with colloidal Coomassie blue (37). The molecular masses of Xa, Va, and their complex were obtained by comparison with the marker proteins.
Prothrombin Activation in the Presence of C6PS
The rate of prothrombin activation was derived from the time-dependent change of DAPA fluorescence as it bound to the activation products (30). Stopped-flow measurements were performed at 37°C using an SLM-Aminco Milliflow stopped-flow reactor (Spectronic Instruments, Inc., Rochester, NY) attached to the Spex® FluoroLog-3 spectrofluorometer (Jobin Yvon Inc., Edison, NJ), as described previously (38). Reactions were initiated by rapidly mixing equal volumes (400 µL) of the contents of the two driving syringes. One syringe contained prothrombin and DAPA in Buffer A (substrate:DAPA ratio 1:5), and the other syringe contained pre-assembled prothrombinase (FXa, FVa and C6PS) in the same buffer. The FVa concentration was varied from 0 to 200 nM.
Results
Dimerization of FXa Affects The Activity of The Prothrombinase Complex at 5 mM Ca+2
The initial rates of thrombin formation by the prothrombinase complex formed at 5, 20 and 50 nM FXa in the presence of 400 µM C6PS and 5 mM Ca2 are plotted in Figure 1B as a function of FVa concentration. Under these conditions, C6PS is present as a monomer (CMC be ~650 µM) that binds to FXa to trigger dimer formation (12) and to FVa to trigger its tight association with FXa in solution (30). Prothrombinase activity decreased with increasing FXa concentration at a constant FVa concentration, i.e., FXa inhibited prothrombinase activity. The red hyperbolic curves (single-site binding model for FXa-FVa interaction) extrapolated to similar saturating activities (kcat/KM ≈ IR/[Xatot]/[S](1000 nM) = 11 ± 3×107 M−1s−1, 12 ± 1×107 M−1s−1 and 10 ± 1×107 M−1s−1, for 5, 20 and 50 nM FXa, respectively), meaning that inhibition was overcome at saturating FVa concentration. The of FXa-FVa interaction, as estimated from a single-site binding model (solid red lines in Figure 1B), increased with FXa concentration: 2.2 ±0.6, 20 ±4.2 and 43 ± 13.3 nM for 5, 20 and 50 nM FXa, respectively. These results suggest that FXa interferes with prothrombinase complex formation. Because we know that human FXa forms a dimer in solution in the presence of C6PS (12, 13), it is reasonable to think that this inhibition is due to formation of inactive FXa dimers, thus, that dimer formation competes with prothrombinase formation. Using a previously reported Kd for FXa dimerization (13), we calculated the potential percent of FXa present as monomer at 5 mM Ca2+ to be 46, 62, and 83 %, at 50, 20, and 5 nM Xatot, respectively (Figure 1B inset), whereas at 3 mM Ca2+ the potential percent of FXa present as monomer is 94, 97.5, and 99 %, at 50, 20, and 5 nM Xatot, respectively (Figure 2B inset) under the assumption that all FXa is free to dimerize (i.e., none is tied up in FVa-FXa complex). Because FXa dimerization is highly sensitive to Ca2+ concentration (12, 13), and 5 mM Ca2+ is the optimum concentration for FXa dimerization (12, 13), we observed a lower amount of FXa monomer at 5 mM Ca2+. In the presence of FXa dimers, FVa is required to compete with FXa to form the prothrombinse complex. The inset to Figure 1B shows that the apparent Kd of FXa-FVa interaction decreased as the percent of FXa potentially present as monomer increased. This result is consistent with FXa dimerization and binding to FVa being competitive processes. Nonetheless, this result is an insufficient test of competition.
Figure 1. Initial Rate of Prothrombin Activation by FXa-FVa at increasing Concentrations of FVa under Conditions Favoring Dimers.
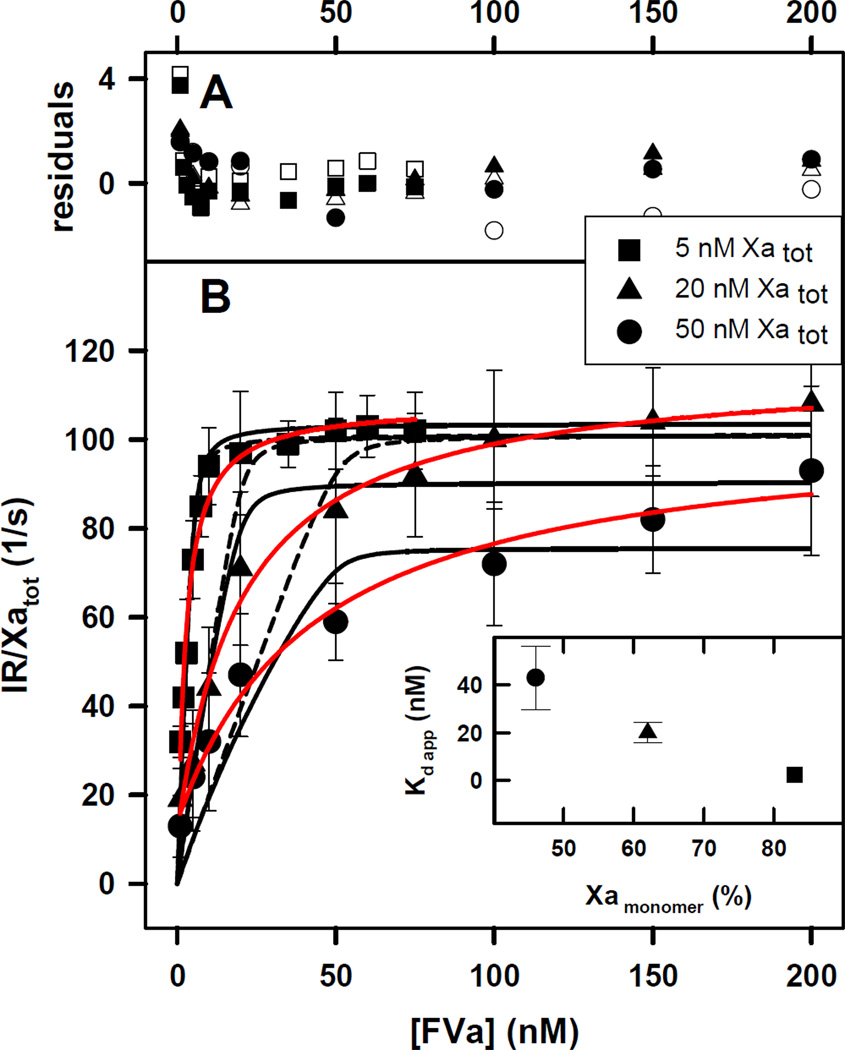
The FXa-FVa interaction was monitored as an increase in the ability of FXa at 5 (squares), 20 (triangles) and 50 (circles) nM concentrations to activate 1 µM prothrombin while titrating with FVa in the presence of 400 µM C6PS at 5 mM Ca2+. These conditions favor formation of both the FXa dimer and the FXa-FVa complex.
A) the deviation of experimental data from a value predicted by Model 1 (open symbols) and Model 5a (closed symbols) relative to the average standard deviation (Rexp – Rfit)/σmean.
B) The black curves drawn through the symbols indicate the global fit of the data to a simple competition model as described in the Supplement (dashed black line) or to the (XaVa)2 model (Model 5a in Supplement; solid black line. Both models assume that the FXa dimer and XaVa dimer are inactive. Solid red lines represent a fit with hyperbola. Inset: The apparent Kds of FXa-FVa dissociation were 2.2 ±0.6, 20±4.2, and 43 ± 13.3 nM for 5, 20 and 50 nM FXa, respectively. The inset shows a plot of apparent Kd versus % of monomer of FXa obtained using Equation 2 in Methods.
Figure 2. Initial Rate of Prothrombin Activation by FXa-FVa at increasing concentrations of FVa under Conditions Favoring Monomers.
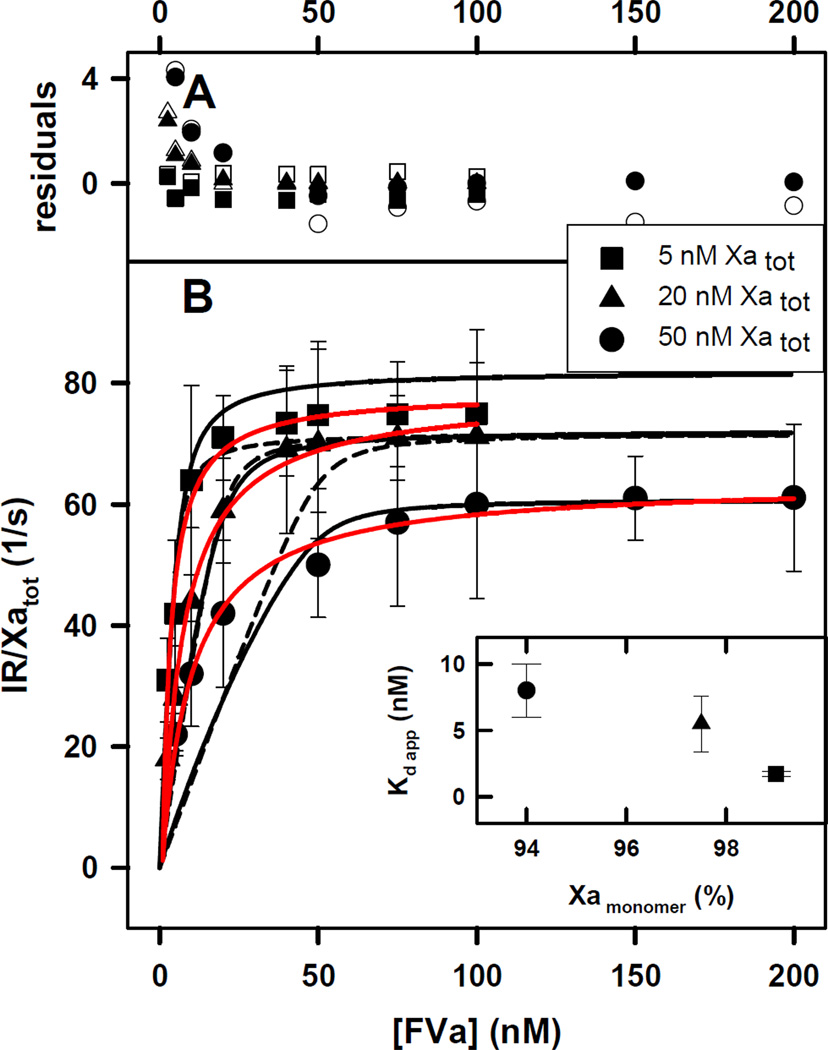
Conditions and symbols are as in Figure 1 except that Ca2+ concentration was 3 mM.
A) the deviation of experimental data from a value predicted by the Model 1 (open symbols) and Model 5a (closed symbols) relative to the average standard deviation (Rexp–Rfit)/σmean.
B) The black curves drawn through the symbols indicate the global fit of the data to a simple dimer competition model as described in Appendix A (dashed black line) or to an aggregate model as described in Appendix B (solid black line), both models predict that a dimer or aggregates are inactive. Solid red lines represent a fit with hyperbola.
B inset) The apparent equilibrium constants (Kd) for FXa-FVa dissociation were 1.7 ± 0.4, 5.8 ± 0.8, and 8.6 ± 1.2 nM for 5, 20 and 50 nM FXa, respectively. The inset shows a plot of apparent Kd versus % of monomer obtained using Equation 2 in Methods.
To test the competition hypothesis more quantitatively, we used MatLab to test the ability of a dimer competition model to account for our observations. Data at all three FXa concentrations were fit globally as described in detail in Supplement Section A, under Model 1, and the results are shown in Figure 1B as dashed black curves (▲ 5nM FXa, ■ 20 nM FXa, and ● for 50nM FXa). The kcat/KM that yielded the best fit (9.8 ± 0.9 ×107 M−1s−1; Table 1) was in reasonable agreement with a previous estimate (22 ×107 M−1s−1) (30). Activity measurements at 37 °C have estimated the Kd of complex formation as ~3 nM (23), while measurements with active-site labeled human FXa (DEGR-Xa) have estimated this as ~ 0.6 nM (7), in good agreement with the value obtained from our fit of 0.32 (Table 1). Nonetheless, it is evident that the simple competition model significantly underestimated the degree of observed inhibition of the complex formation at low to intermediate FVa concentrations and 50 nM FXa, and thus reached an asymptote well before the experimental data. Hyperbolic fits to our data (red lines), also suggest this asymptotic behavior. The reduced chi square (χ̅2) of the fit was large (1.38, Table 1) while its significance was low (0.1). While the simple dimer competition model confirmed that Xa dimer formation could result in inhibition of FXa-FVa complex formation, it fell short in its ability to account quantitatively for our data, leading us to consider alternative models that might provide better quantitative descriptions of our data.
Table 1.
Goodness of fit reduced chi-squared and best fit parameters obtained by simultaneously fitting all three data sets (5, 20, and 50 nM Xa tot) by either the simple Xa dimer – prothrombinase competition model (Model 1, Supplement) or the (XaVa)2 plus Xa dimer model (Model 5a, Supplement).
| Figure 1 – 5 mM Calcium | Kd (nM) |
kcat/KM (107 M−1s−1) |
|||||
| parameter model/α |
chi2 |
|
|
|
|||
| 1) simple Xa2 XaVa comp./0.1 | 1.38 | 0.32 +0.73 −0.31 | / | 9.8 ± 0.9 | |||
|
5a) (XaVa)2 aggregate from XaVa/0.29 2XaVa↔(XaVa)2 |
1.14 | 0.43 ± 0.3 | 150 +80 −50 | 11 ± 0.6 | |||
| Figure 2 – 3 mM Ca | Kd (nM) |
kcat/KM (107 M−1s−1) |
|||||
| parameter model |
chi2 |
|
|
|
|||
| 1) simple Xa2 XaVa comp. /0.09 | 1.41 | 0.76 +2 −0.74 | / | 7.2 ± 0.8 | |||
|
5a) (XaVa)2 aggregate from XaVa/0.17 2XaVa↔(XaVa)2 |
1.25 | 1.5 +2 −1.1 | 166 +230 −80 | 8.7 ± 0.9 | |||
Fixed parameters at 5 mM Ca for both models: Kd Xa2 = 14 nM, and kcat/KM Xa = 1.36x 104 M−1s−1 as measured previously (Majumder et al., 2003).
Fixed parameters at 3 mM Ca for both models: Kd Xa2 = 600 nM, and kcat/KM Xa = 1.36x 104 M−1s−1 as determined in this paper.
Because dimer formation underestimated the extent of inhibition, we considered the possibility that FXa might form a higher order aggregate, e.g., a tetramer (Model 2 in Section B of Supplement). Model 2 provided no improvement in data description despite requiring an additional adjustable parameter . As a control, we tried an alternative form of the “FXa tetramer” model (Model 2a in Section C of Supplement) and found, as anticipated if our analysis methods were robust, that the results of our fitting were independent of the thermodynamic path assumed in forming a tetramer. Inability of the “FXa tetramer” model to fit the data better than the simple dimer competition model (model 1) allows us to reject the “higher-order Xa aggregate” (as embodied to a first approximation in a tetramer) model to explain inhibition observed at higher concentrations of FXa. Next, we considered whether aggregation of FVa (Model 3 in Section D of Supplement) and subsequent formation of an aggregated FXaFVa complex (model 4) might contribute to the less than satisfactory quantitative description we had obtained using the dimer competition model (model 1). Both models provided better fit to the data, however the parameter values were quite unreasonable relative to other reports ( relative to the reported 0.6 nM (7) or to common sense ( means that FVa binds to FVa 100 times more tightly than it does to FXa). Because we also didn’t see any evidence of FVa dimer in native gels of mixtures of FVa and FXa in the presence of C6PS (Figure 3), we must conclude that aggregation of FVa cannot offer an explanation for the poor quantitative agreement between the FXa dimer competition model (model 1) and our data.
Figure 3.
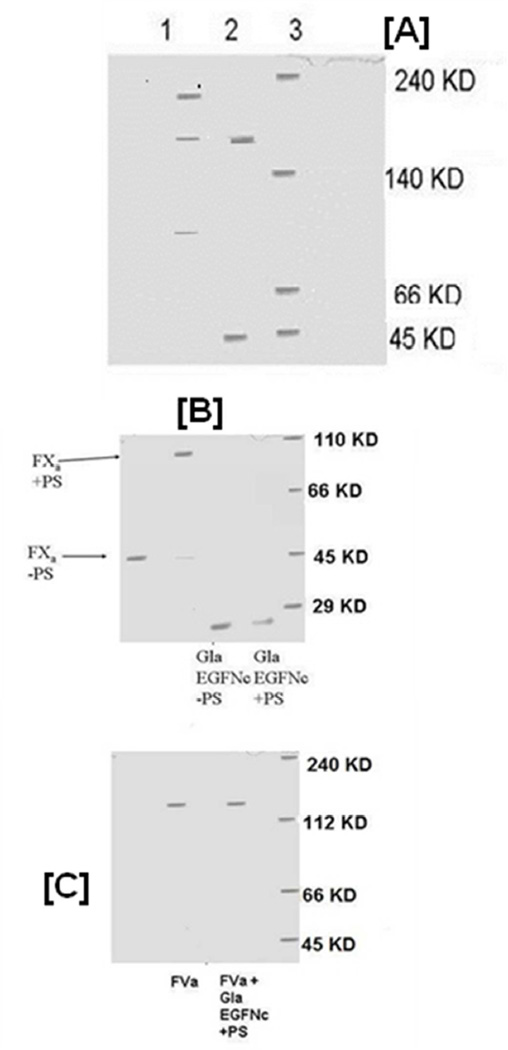
3A. Native Gel Electrophoresis of a FXa and FVa Mixture in the Presence and Absence of C6PS. A 5% polyacrylamide gel of a mixture 50 nM FXa and 50 nM FVa in the presence of 400 µM C6PS and 5 mM Ca2+ (lane 1) shows coexistence of homodimers (FXa ·FXa; 90 KD), heterodimers (FXa · FVa; 224 KD), and FVa (178 KD), showing that FXa monomer interacts with both FVa and with FXa, thus consistent with the hypothesis that FXa and FVa compete to bind FXa. Lane 2 shows that the same reaction mixture in the absence of C6PS contains only monomers of both FVa and FXa, showing that this competition is regulated by phosphatidylserine (in this case C6PS). Lane 3 shows molecular mass markers.
3B: Native Gel Electrophoresis of FXa and its GLA-EGFNC Fragment. 8% native polyacrylamide gel electrophoresis of FXa and GLA-EGFNC fragment of FXa in the presence and absence of µM 400 C6PS and 5 mM Ca2+ shows that the GLA-EGFNC fragment does not dimerize in the presence of C6PS and 5 mM Ca2+. Figure 3C: 5% native polyacrylamide gel electrophoresis of 50 nM FVa with and without 50 nM GLA-αEGFNC domain in the presence of 400 µM C6PS, 5 mM Ca2+. The results show that the GLA-EGFNC fragment of FXa does not form a complex with FVa.
Finally, we considered whether dimerization of the XaVa complex might help account for our data. The description of our data by this model is better than for any other model considered (i.e., lowest for physically reasonable parameters; Table S1). The parameters for this model (; (Kcat / KM)XaVa) were also in reasonable agreement with literature estimates (30), while the predicted (150 nM) is consistent with our failure to observe this species in native gels (Figure 3). However, Figure 1B reveals that the predicted curves do not approach a common asymptotic (Kcat / KM)XaVa at saturating FVa, which hyperbola through our data appears to do (see red hyperbolic curves in Figure 1B). Thus, allowing for a dimer of the FXa-FVa complex considerably improves description of our high FXa data considerably at low and intermediate FVa concentrations but still falls somewhat short of accounting for the apparent asymptotic behavior of our data at very high FVa concentration and 5 mM Ca2+, which favors FXa dimerization.
Dimerization of FXa Affects The Activity of The Prothrombinase Complex at 3 mM Ca+2
We showed previously that FXa dimerization is sensitive to Ca2+ concentration, with at 3 mM Ca2+ being roughly 40-fold larger than that at 5 mM Ca2+ (12, 13). Thus, to further test whether FXa dimerization might be a reasonable explanation for the variation with FXa concentration of for FXaFVa complex, we repeated the experiments of Figure 1B at 3 mM Ca2+. Surprisingly, Figure 2B still shows inhibition of prothrombinase assembly at increasing concentrations of FXa, but the inset to Figure 2B shows a 10-fold increase in FXa concentration produced a 3–4-fold increase in at 3 mM Ca2+ (1.7 ±0.2, 5.5 ± 2 and 8 ± 2 nM for 5, 20 and 50 nM FXa, respectively) compared to the 20–25-fold increase at 5 mM Ca2+ (Figure 1B inset), suggesting that FXa dimer formation may be less important at 3 mM Ca2+, as expected.
To explore quantitatively the reasons for this unanticipated result, we again used the procedures described in the Supplement to test the compatibility of our data with the two most successful models we identified for 5 mM Ca2+ (Model 1: the dimer competition model and Model 5a: the XaVa dimerization model). Because thermodynamic constants for XaVa complex formation and human FXa dimer formation are not reported at 3 mM Ca2+, we measured these constants at 37°C using previously documented methods that use active site labeled human DEGR-FXa (12, 30) to obtain and . This value of was fixed in our calculations, while was adjustable to remain consistent with our computations for 5 mM Ca2. The parameters providing this fit are summarized in Table 1. The best-fit value for was somewhat lower than we measured but still well within the error range of these two estimates. The effect of Ca2+ on seen in the context of the dimer competition model is thus small. Same was also true for (kcat / KM)XaVa (9.8 ±0.8 vs 7.2 ±0.8 ×107 M−1s−1 for 5 and 3 mM Ca2+, respectively). Apparently, within the context of this model, only FXa dimer formation was significantly affected by a change in Ca2+ concentration from 5 to 3 mM.
As for 5mM Ca2+, Model 5a (XaVa dimerization) also offered a better description of the data at 3 mM Ca2+ than did the dimer competition model (model 1). Better description occurred because the data at 3 mM Ca2+ showed a difference in asymptotic behavior between 5 and 50 nM FXa, a feature predicted by the XaVa dimerization model. The parameters defined by this fit are also given in Table 1, where we see that the value for (kcat / KM)XaVa for the FXa-FVa complex within the context of this model is also not significantly Ca2+-dependent (11.0 ± 0.9 and 8.7 ± 0.7 ×107 M−1s−1 for 5 and 3 mM Ca2+, respectively). In addition, was also not significantly Ca2+-dependent (0.43± 0.2 vs. 0–2nM for 5 and 3 mM Ca2+, respectively) nor was FXa-FVa complex dimer formation significantly Ca2+-dependent (150 vs 166 nM for 5 and 3 mM Ca2+, respectively). Thus, within the context of both models, Ca2+ significantly affected only FXa dimer formation ( for 5 and 3 mM Ca2+, respectively) in this concentration range.
Coexistence of FXa–FVa Complex with Dimer at 5mM Ca2+
Native polyacrylamide gel electrophoresis, in the presence of C6PS, detects both human FXa-FVa complex and human FXa dimers of the expected molecular masses (measured 224±3.7 kDa for FXa-FVa (30)) and ~ 90 kDa for (FXa)2 (13). In order for FXa dimer formation and FXa-FVa complex formation to be competitive processes, the species FVa, FXa, FXa-FVa, and (FXa)2 should all be able to coexist in equilibrium under a condition for which both FXa dimers and prothrombinase complex can form (400 µM C6PS, 5 mM Ca2+, 50 nM FXa, and 50 nM FVa). Figure 3A (lane 1) shows FVa (~178KD), FXa2 (~90 KD) and FXa-FVa complex (~224 KD) bands, thus confirming that FVa monomer FXa dimer and FXa-FVa complex can all coexist under appropriate conditions. Previous hydrodynamic studies showed that the solution prothrombinase formed in the presence of C6PS is a 1:1 complex at low FXa concentration (30). These results show that the C6PS-induced complex is 1:1 even at a FXa concentration for which FXa dimers form. A complex of the FXa dimer with FVa (FXa·FXa·FVa) should enter the gel (270 KD) and locate to the top of the gel, but it was not detected. This result is consistent with the hypothesis that only monomer FXa binds to FVa and that both FXa and FVa compete to bind FXa. Note that the FXa monomer does not appear in the gel, because, under these conditions, most FXa is incorporated into Xa2 or (FXa-FVa). We note also that (FXa-FVa)2 (~ 450 kDa) would not have entered this 5% gel, but we did not detect any staining at the running gel/loading gel interface even at 200 nM FVa.
Thus, if this species is present, it must be present in a small quantity. Not only by simulation but also directly from experimental data at [FXa]=50 nM, the IR reaches more than 50% of the final IR at [FVa]=50 nM, which indicates that more than 50% of all species should be in FXa-FVa form. Is it possible that this occurs due to concentration gradient within the gel? More massive species travel in the front while less massive species remain in the back, if FXaVa is disassembling, and it’s reassembly is hindered because FXa is no longer present at the initial concentration.
A R165A FXa mutation in the FVa Binding Region Interferes with Dimerization
Arg 165 in the human FXa catalytic domain is part of a basic region involved in FVa binding (8, 9, 26, 27). We also obtained values for R165A FXa-FVa interaction from plots of initial rates of prothrombin activation (400 µM C6PS and 5 mM Ca2+) catalyzed by 5, 20 and 50 nM R165A mutant human FXa in the presence of increasing concentrations of FVa (Table 2). These apparent Kd values along with the values of the wild type FXa-FVa (Figure 1B) association are presented in Table2. The results show that the apparent affinity of the R165A mutant FXa with FVa was lowered by 3–28 fold relative to native FXa, depending on the concentrations of mutant and wild type FXa. This finding is consistent with a previous report of a 6-fold effect of this mutation on the interaction with FXa (39).
Table 2.
Apparent Dissociation Constants for Interactions of Wild-Type and R165A Mutant FXa with FVa
| FXa, nM | Kd,app FXa-FVa, nM | |
|---|---|---|
| Wild Type FXa | Mutant (R165A) FXa |
|
| 5 | 2.2 ± 0.6 | 58 ± 7 |
| 20 | 20 ± 4.2 | 92 ± 12 |
| 50 | 43 ± 13.3 | 128 ± 27 |
| Kddimer, nM | ||
|---|---|---|
| Wild Type FXa | Mutant (R165A) FXa |
|
| 16 ± 4 | 147 ± 25 | |
Dissociation constants obtained from the data plotted in Figure 1B and from similar experiments with R165A rHFXa show that the R165A mutation in FXa inhibits both the interaction with FVa and FXa dimer formation. The value of was obtained from fitting the data in the figure. We previously reported a value of 14 ± 1 nM obtained from more extensive sets of data in the absence of FVa (13).
The initial rates of prothrombin activation by different concentrations of wild type and R165A FXa in the presence of 400 µM C6PS versus FXa concentration are plotted in Figure 4. The curves were fit to a dimerization model (12) as described in Methods. The for R165A FXa was 9-fold larger than for wild type FXa (Table 2). We conclude that the R165A mutation alters dimerization as well as binding to FVa in the presence of C6PS.
Figure 4. Prothrombin activation by increasing concentration of wild type and mutant (R165A) FXa in the presence of C6PS and 5 mM Ca2+.
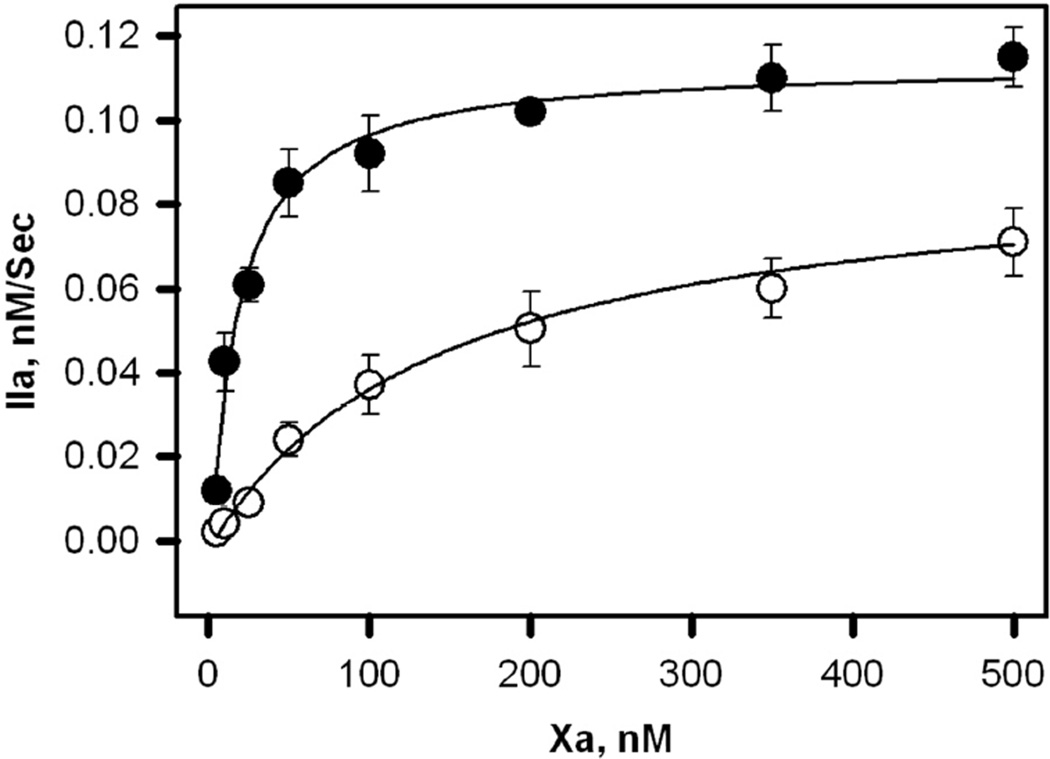
The initial rates of prothrombin activation by FXa in the presence of FVa and 400 µM C6PS at different of FXa concentrations were plotted as a function of FXa. The appearance of thrombin was determined by the rate of hydrolysis of DAPA (as described in Methods) at 37°C. The reaction was carried out at increasing concentrations of wild type FXa (closed circles) and mutant R165A FXa (open circles). The reaction mixture contained 1 µM prothrombin, FXa in 50 mM Tris, 175 nM NaCl, 0.6% poly(ethylene glycol), 5 mM Ca+2 and 400 µM C6PS. The dimerization Kds obtained in the presence of wild type and mutant FXa are 16 and 147 nM respectively.
Role of catalytic domain of FXa in dimerization of FXa and its interaction with FVa
FXa is generally thought to interact with Va through sites in the FXa catalytic domain (8, 10, 27, 40). We wanted to test whether there might be other regions of FXa required for FVa interaction and dimerization of FXa in the presence of 400 µM C6PS. Figure 3B shows an 8% native polyacrylamide gel of a GLA-EGFNC fragment of FXa (lacking the catalytic domain) along with intact FXa in the absence and presence of C6PS, showing that FXa dimerizes but that GLA-EGFNC does not dimerize in the presence of C6PS. Figure 3C shows a 5% native polyacrylamide gel of FVa in the absence and presence of C6PS and GLA-EGFNC fragment, showing that FVa cannot form a complex with the GLA-EGFNC fragment of FXa. Because we have shown previously that native polyacrylamide gel electrophoresis can detect both FXa dimer and FXa-FVa complexes (12, 30), these results demonstrate that, although C6PS binds to a regulatory site in the EGFNC pair (6), interactions between this region of FXa do not contribute directly either to FXa dimerization or to binding to FVa.
Discussion
Our results support the following conclusions:
1] FXa dimer formation and prothrombinase complex formation are thermodynamically competing processes in solution that are both triggered by binding of C6PS to a regulatory site in the EGFNC pair.
Because of this competition, estimates of the for prothrombinase complex formation will depend on the reaction conditions (FXa, FVa, Ca2+ concentrations), at least if activity is used to monitor prothrombinase formation. This dependence is demonstrated in Figures 1 and 2 but is also surmised from the dimer competition model (Model 1 in Supplement) we used to interpret our results. Because DEGR-FXa dimerizes weakly in solution (Koklic, 2009 #169}, estimates made of using active-site-labeled FXa (0.6 ± 0.09 nM (30)) are lower than estimates made using activity to follow complex assembly (2.8 ± 0.3 nM (23). Estimates of using either active-site-labeled FXa or using activity may not differ for complex assembly on a membrane because DEGR-FXa dimerizes reasonably well on a membrane surface (41).
2] Other interactions appear to complicate further complex formation in solution.
This conclusion is implied by the observation that the dimer competition model (Model 1 in Supplement) was unable to account for our data at either 5 mM Ca2+ (where dimerization should be significant) or 3 mM Ca2+ (where dimer formation should be minimal). In both instances, less FXa-FVa complex was predicted to form than was suggested by observed prothrombin-activation rates. We considered what other species might be limiting formation of prothrombinase complex. Inclusion of higher order aggregates of FXa (tetramer) produced no reduction in and produced an estimate of the tetramer dissociation constants that resulted in this species not being present (Table S1 in Supplement). The possibility that FVa formed a dimer reduced the but predicted that the FVa dimer should be the dominant species, in contradiction with results (Fig. 3A). Next, the assumption that a dimer of the FXa-FVa complex might form produced an improved description of observations in Fig. 1 and a significant reduction in as well as a considerable increase in the goodness of fit (α, Table 1). FXa-FVa dimer formation (Model 5a in Supplement) also improved our description of the data collected at 3 mM Ca2+ (Table 1 and Fig. 2) and is concluded to provide some insight into why the simple dimer competition model does not account well for our data. Within the context of this model, only the FXa dimerization constant is significantly dependent on Ca2+ concentration; neither nor were dependent on Ca2+ concentration, suggesting that formation of (XaVa)2 involves a binding region other than that involved in Xa2 formation. Since FVa seems not to form a dimer (Model 3 in Supplement), the most likely interaction region to support formation of the (XaVa)2 species would be the dimer interaction region. If so, we would have to surmise that binding of FVa to the FVa binding region of FXa, which is distant from the FXa dimerization region (13), must alter the dimerization region so that a FXa-FXa interaction becomes more likely at low Ca2+ concentration. If so, our hypothesis that these two binding regions are linked, is more credible.
Despite the success of the XaVa dimer model (model 5a) relative to the Xa dimmer competition model (model 1), the former model at 5 mM Ca2+ still had shortcomings in its ability to predict observed asymptotic behavior (Fig. 1). Reference to simulations presented in Figure S3 reveals that the concentration of the prothrombinase complex does not approach its maximum possible value at high FVa and FXa concentrations because a stable amount of the FXa-FVa dimer species ((XaVa)2) is predicted to form under these circumstances. At 3 mM Ca2+, when very little FXa2 species is expected, incomplete formation of the prothrombinase complex at high FXa and FVa concentrations is both predicted (Fig S3) and observed (Fig. 2). This result led us to suspect that the value of that we used might be in error. To test this suspicion, we fixed best-fit parameters (, (Kcat / KM)XaVa) and varied without producing a better description of asymptotic behavior. In this regard, we note the large parameter uncertainty for (Table 1) that likely reflects the need to rectify different behaviors of the data at high and intermediate FVa concentrations. The reasons for this discrepancy between the data and XaVa dimer model (model 5a) at high FVa concentrations could be 1] uneven distribution of data points, 2] large uncertainties in the rate data at high FVa concentrations or 3] inadequacy of the model. Indeed, use of an average 12% error for all rate data led to a somewhat larger 1.3×108 M−1s−1 A (kcat/KM)XaVa, a somewhat improved appearance of the fit at high FVa and parameters still within the range of uncertainties shown in Table 1, but also a somewhat larger . Of course, we must consider that a XaVa dimer may not explain our observations, but, if not this model, then what model? We know that FXa forms a dimer in solution under the influence of C6PS (12, 13), and the data in Figures 1 and 2 make it clear that both FXa and FVa concentrations must be taken into account to model the formation of the active FXa-FVa complex in solution at high FXa and FVa concentrations. We have eliminated a model that posits a FVa dimer (Model 3 in Supplement) and models that posit higher order aggregates of FXa (Models 2 and 2a in Supplement). While we could still imagine other species (e.g., FXa·FXa·FVa), each such species added to the model adds a new adjustable parameter, meaning that any resulting improvement in is less likely to be significant. We conclude that some higher order aggregated species are required to explain the dependence of soluble prothrombinase formation on both FXa and FVa concentrations and that the most likely species at this point is a dimer of the FXa·FVa prothrombinase complex.
3] FVa and FXa binding regions are distinct linked sites within the catalytic domain.
Binding of C6PS to the regulatory site in the EGFNC region (6) of human FXa is linked to binding sites in the catalytic domain responsible for interaction with FVa and for dimmer formation (13, 30). Because the EGFN domain is near the membrane-binding Gla domain, linkage of these sites must occur via the FXa light chain. Because FXa dimer formation and prothrombinase complex formation are not mediated by C6PS-triggered interactions between FXa light chains or between FXa light chain and FVa (Fig. 3C), the rules of a thermodynamic cycle require linkage between the catalytic domain binding regions. Here we provide additional evidence for this linkage. We know that K90 in the catalytic domain becomes buried when C6PS triggers FXa dimer formation in the presence of 5 mM Ca2+ and is on the opposite face of a FXa structural model (42) from the most likely FVa binding regions (either or both of a 185–189 loop (26) or a 162–169 helix containing R165 (8, 9, 27)). Both of these regions were identified by mutational analysis. There are two general views of linkage (43). One asserts that binding of one ligand favors one of many conformational states of a protein that favors binding of another ligand. In the second view, inter-residue or inter-domain interactions along a path convey information between regions of a protein. Mutational analysis has been proposed to identify such paths (44). Some note that these views are not mutually exclusive but that “path” residues are residues that are crucial for protein conformational state equilibration. We show here that a mutation (R165A) that interferes with FXa interaction with FVa (8) also interferes with dimer formation (Fig 4). If R165 is in the FVa binding region, this observation supports linkage between the FVa binding and FXa binding regions. It is noteworthy that another Lys (K230) becomes exposed upon dimer formation (13) and is spatially close to R165 in the proposed FXa structural model. These observations suggest that K230 and R165 may be residues that are part of a linkage “path” or key to switching between dimer forming or FVa-binding conformational states, a possibility that also supports linkage between these two binding regions. Further supporting our hypothesis is the observation that K90 is contained in two peptides that mimic regions of FXa sequence that interfere with or block FXa binding to FVa (10, 40). The fact that K90 is far from the FVa-binding region supports our hypothesis that these two sites are linked and that binding of synthetic peptides to the FXa dimer interface interferes with FXa binding to FVa.
4] Linkage between the dimer interface and FVa binding regions of FXa may have physiological significance in blood coagulation.
Both FXa dimer formation and assembly of the prothrombinase complex are triggered by C6PS or by binding of these proteins to PS-containing membranes (13, 30, 41, 45). Thus, the competition between FXa dimer formation and prothrombinase assembly could well occur on a membrane in vivo. If so, its sensitivity to Ca2+ concentration could be significant during the early stages of platelet plug formation when calcium concentration in the local wounded area drops much below normal plasma concentration and increases back to normal levels within three hours after the injury (46). Thus, we speculate that FXa dimerization is a mechanism that limits prothrombinase formation when blood clotting is undesirable. We are investigating this possibility using model membranes.
Supplementary Material
Acknowledgments
This work was supported by grants GM32707 (BRL) and HL 101917 (ARR) from the National Institutes of Health.
Abbreviations
- DAPA
dansylarginine-N- (3-ethyl-1, 5-pentanediyl) amide
- S765
N-α-benzyloxycarbonyl-D-arginyl-L-glycyl-L-arginine-p-nitroaniline dihydrochloride
- C6PS
1,2-dicaproyl-sn-glycero-3-phospho-L-serine
- PS
phosphatidylserine
- FXa
factor Xa
- FVa
factor Va
- IIa
thrombin
- GLA
N-terminus γ-carboxyglutamic acid-rich region
- EGFC
epidermal growth factor nearest the C terminus
- PEG
polyethylene glycol
- DEGR-FXa
[5-(dimethylamino)1-naphthalenesulfonyl]-glutamylglycylarginyl chloromethyl ketone
Footnotes
Supporting Information Available. This material documents the ability of different binding models to account for observed binding data as well as the method used to obtain fit parameters and estimate their uncertainties. It is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Fenton Jn. Thrombin. Ann NY Acad Sci. 1986;485:5–15. doi: 10.1111/j.1749-6632.1986.tb34563.x. [DOI] [PubMed] [Google Scholar]
- 2.Jackson CM, Nemerson Y. Blood coagulation. Annu Rev Biochem. 1980;49:765–811. doi: 10.1146/annurev.bi.49.070180.004001. [DOI] [PubMed] [Google Scholar]
- 3.Mann KG, Jenny RJ, Krishnaswamy S. Cofactor proteins in the assembly and expression of blood clotting enzyme complexes. Annu Rev Biochem. 1988;57:915–956. doi: 10.1146/annurev.bi.57.070188.004411. [DOI] [PubMed] [Google Scholar]
- 4.Bevers EM, Comfurius P, Zwaal RF. Changes in membrane phospholipid distribution during platelet activation. Biochim Biophys Acta. 1983;736:57–66. doi: 10.1016/0005-2736(83)90169-4. [DOI] [PubMed] [Google Scholar]
- 5.Jones ME, Lentz BR, Dombrose FA, Sandberg H. Comparison of the abilities of synthetic and platelet-derived membranes to enhance thrombin formation. Thromb Res. 1985;39:711–724. doi: 10.1016/0049-3848(85)90255-5. [DOI] [PubMed] [Google Scholar]
- 6.Srivastava A, Wang J-F, Majumder R, Stenflo J, Rezaie AR, Esmon CT, Lentz BR. Locations of phosphatidylserine binding sites on Factor Xa. Journal of Biological Chemistry. 2002;277:1855–1863. doi: 10.1074/jbc.M105697200. [DOI] [PubMed] [Google Scholar]
- 7.Majumder R, Quinn-Allen MA, Kane WH, Lentz BR. A phosphatidylserine binding site in factor Va C1 domain regulates both assembly and activity of the prothrombinase complex. Blood. 2008;112:2795–2802. doi: 10.1182/blood-2008-02-138941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rezaie AR. Identification of basic residues in the heparin-binding exosite of factor Xa critical for heparin and factor Va binding [In Process Citation] J Biol Chem. 2000;275:3320–3327. doi: 10.1074/jbc.275.5.3320. [DOI] [PubMed] [Google Scholar]
- 9.Rudolph AE, Porche-Sorbet R, Miletich JP. Substitution of asparagine for arginine 347 of recombinant factor Xa markedly reduces factor Va binding. Biochemistry. 2000;39:2861–2867. doi: 10.1021/bi992379b. [DOI] [PubMed] [Google Scholar]
- 10.Sabharwal AK, Padmanabhan K, Tulinsky A, Mathur A, Gorka J, Bajaj SP. Interaction of calcium with native and decarboxylated human factor X. Effect of proteolysis in the autolysis loop on catalytic efficiency and factor Va binding. Journal of Biological Chemistry. 1997;272:22037–22045. doi: 10.1074/jbc.272.35.22037. [DOI] [PubMed] [Google Scholar]
- 11.Padmanabhan K, Padmanabhan KP, Tulinsky A, Park CH, Bode W, Huber R, Blankenship DT, Cardin AD, Kisiel W. Structure of human des(1–45) factor Xa at 2.2 A resolution. J Mol Biol. 1993;232:947–966. doi: 10.1006/jmbi.1993.1441. [DOI] [PubMed] [Google Scholar]
- 12.Majumder R, Wang J, Lentz BR. Effects of Water Soluble Phosphotidylserine on Bovine Factor X(a): Functional and Structural Changes Plus Dimerization. Biophys J. 2003;84:1238–1251. doi: 10.1016/S0006-3495(03)74939-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chattopadhyay R, Iacob R, Majumder R, Sen S, Tomer K, Lentz BR. Functional and Structural Characterization of Factor Xa Dimer In Solution. Biophysical Journal. 2009;96:974–986. doi: 10.1016/j.bpj.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koppaka V, Wang J, Banerjee M, Lentz BR. Soluble phospholipids enhance factor Xa-catalyzed prothrombin activation in solution. Biochemistry. 1996;35:7482–7491. doi: 10.1021/bi952063d. [DOI] [PubMed] [Google Scholar]
- 15.Banerjee M, Majumder R, Weinreb G, Wang J, Lentz BR. Role of Procoagulant Lipids in Human Prothrombin Activation. 2. Soluble Phosphatidylserine Upregulates and Directs Factor X(a) to Appropriate Peptide Bonds in Prothrombin. Biochemistry. 2002;41:950–957. doi: 10.1021/bi0116902. [DOI] [PubMed] [Google Scholar]
- 16.Banerjee M, Drummond DC, Srivastava A, Daleke D, Lentz BR. Specificity of soluble phospholipid binding sites on human factor Xa. Biochemistry. 2002;41:7751–7762. doi: 10.1021/bi020017p. [DOI] [PubMed] [Google Scholar]
- 17.Esmon CT. The subunit structure of thrombin-activated factor V. Isolation of activated factor V separation of subunits, and reconstitution of biological activity. J Biol Chem. 1979;254:964–973. [PubMed] [Google Scholar]
- 18.Kane WH, Majerus PW. Purification and characterization of human coagulation factor V. J Biol Chem. 1981;256:1002–1007. [PubMed] [Google Scholar]
- 19.Dahlback B, Stenflo J. The activation of prothrombin by platelet-bound factor Xa. European Journal of Biochemistry. 1980;104:549–557. doi: 10.1111/j.1432-1033.1980.tb04458.x. [DOI] [PubMed] [Google Scholar]
- 20.Ortel TL, Quinn-Allen MA, Keller FG, Peterson JA, Larocca D, Kane WH. Localization of functionally important epitopes within the second C- type domain of coagulation factor V using recombinant chimeras. J Biol Chem. 1994;269:15898–15905. [PubMed] [Google Scholar]
- 21.Kim SW, Ortel TL, Quinn-Allen MA, Yoo L, Worfolk L, Zhai X, Lentz BR, Kane WH. Partial glycosylation at asparagines-2181 of the second C-type domain of human factor V modulates assembly of the prothrombinase complex. Biochemistry. 1999;38:11448–11454. doi: 10.1021/bi991275y. [DOI] [PubMed] [Google Scholar]
- 22.Koppaka V, Talbot WF, Zhai X, Lentz BR. Roles of factor Va heavy and light chains in protein and lipid rearrangements associated with the formation of a bovine factor Va- membrane complex. Biophys J. 1997;73:2638–2652. doi: 10.1016/S0006-3495(97)78293-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Majumder R, Weinreb G, Zhai X, Lentz BR. Soluble Phosphatidylserine Triggers Assembly in Solution of a Prothrombin-activating Complex in the Absence of a Membrane Surface. J. Biol. Chem. 2002;277:29765–29773. doi: 10.1074/jbc.M200893200. [DOI] [PubMed] [Google Scholar]
- 24.Zhai X, Srivastava A, Drummond DC, Daleke D, Lentz BR. Phosphatidylserine Binding Alters the Conformation and Specifically Enhances the Cofactor Activity of Bovine Factor V(a) Biochemistry. 2002;41:5675–5684. doi: 10.1021/bi011844d. [DOI] [PubMed] [Google Scholar]
- 25.Majumder R, Quinn-Allen MA, Kane WH, Lentz BR. The phosphatidylserine binding site of the factor Va C2 domain accounts for membrane binding but does not contribute to the assembly or activity of a human factor Xa-factor Va complex. Biochemistry. 2005;44:711–718. doi: 10.1021/bi047962t. [DOI] [PubMed] [Google Scholar]
- 26.Rezaie AR, Kittur FS. The critical role of the 185–189-loop in the factor Xa interaction with Na+ and factor Va in the prothrombinase complex. J Biol Chem. 2004;279:48262–48269. doi: 10.1074/jbc.M409964200. [DOI] [PubMed] [Google Scholar]
- 27.Rudolph AE, Porche-Sorbet R, Miletich JP. Definition of a factor Va binding site in factor Xa. J Biol Chem. 2001;276:5123–5128. doi: 10.1074/jbc.M006961200. [DOI] [PubMed] [Google Scholar]
- 28.Yegneswaran S, Mesters RM, Griffin JH. Identification of distinct sequences in human blood coagulation factor Xa and prothrombin essential for substrate and cofactor recognition in the prothrombinase complex. Journal of Biological Chemistry. 2003;278:33312–33318. doi: 10.1074/jbc.M305906200. [DOI] [PubMed] [Google Scholar]
- 29.Camire RM. Prothrombinase Assembly and S1 Site Occupation Restore the Catalytic Activity of FXa Impaired by Mutation at the Sodium-binding Site. J Biol Chem. 2002;277:37863–37870. doi: 10.1074/jbc.M203692200. [DOI] [PubMed] [Google Scholar]
- 30.Majumder R, Weinreb G, Lentz BR. Efficient thrombin generation requires molecular phosphatidylserine, not a membrane surface. Biochemistry. 2005;44:16998–17006. doi: 10.1021/bi051469f. [DOI] [PubMed] [Google Scholar]
- 31.Valcarce C, Persson E, Astermark J, Ohlin AK, Stenflo J. Isolation of intact modules from noncatalytic parts of vitamin K-dependent coagulation factors IX and X and protein C. Methods Enzymol. 1993;222:416–435. doi: 10.1016/0076-6879(93)22027-d. [DOI] [PubMed] [Google Scholar]
- 32.Cutsforth GA, Koppaka V, Krishnaswamy S, Wu JR, Mann KG, Lentz BR. Insights into the complex association of bovine factor Va with acidic-lipid-containing synthetic membranes. Biophys J. 1996;70:2938–2949. doi: 10.1016/S0006-3495(96)79864-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krishnaswamy S, Russell GD, Mann KG. The reassociation of factor Va from its isolated subunits. J Biol Chem. 1989;264:3160–3168. [PubMed] [Google Scholar]
- 34.Haque ME, Das AR, Moulik SP. Behaviors of Sodium Deoxycholate (Nadc) and Polyoxyethylene Tert-Octylphenyl Ether (Triton X-100) at the Air/Water Interface and in the Bulk. Journal of Physical Chemistry. 1995;99:14032–14038. [Google Scholar]
- 35.Haque ME, Das AR, Moulik SP. Mixed micelles of sodium deoxycholate and polyoxyethylene sorbitan monooleate (Tween 80) Journal of Colloid and Interface Science. 1999;217:1–7. doi: 10.1006/jcis.1999.6267. [DOI] [PubMed] [Google Scholar]
- 36.Alexander E, Jr, Mitchell OC, Ferguson KG, Leinbach LB. Excretory Urography Urography by-Product of Cerebral Angiography. J S C Med Assoc. 1964;60:237–239. [PubMed] [Google Scholar]
- 37.Mitra P, Pal AK, Basu D, Hati RN. A staining procedure using Coomassie brilliant blue G-250 in phosphoric acid for detection of protein bands with high resolution in polyacrylamide gel and nitrocellulose membrane. Anal Biochem. 1994;223:327–329. doi: 10.1006/abio.1994.1594. [DOI] [PubMed] [Google Scholar]
- 38.Weinreb GE, Mukhopadhyay K, Majumder R, Lentz BR. Cooperative roles of factor V(a) and phosphatidylserine-containing membranes as cofactors in prothrombin activation. J Biol Chem. 2003;278:5679–5684. doi: 10.1074/jbc.M208423200. [DOI] [PubMed] [Google Scholar]
- 39.Rezaie AR, He X. Sodium binding site of factor Xa: role of sodium in the prothrombinase complex. Biochemistry. 2000;39:1817–1825. doi: 10.1021/bi992006a. [DOI] [PubMed] [Google Scholar]
- 40.Chattopadhyay A, James HL, Fair DS. Molecular recognition sites on factor Xa which participate in the prothrombinase complex. Journal of Biological Chemistry. 1992;267:12323–12329. [PubMed] [Google Scholar]
- 41.Koklic T, Majumder R, Weinreb GE, Lentz BR. Factor XA binding to phosphatidylserine-containing membranes produces an inactive membrane-bound dimer. Biophys J. 2009;97:2232–2241. doi: 10.1016/j.bpj.2009.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Venkateswarlu D, Perera L, Darden T, Pedersen LG. Structure and dynamics of zymogen human blood coagulation factor x. Biophys J. 2002;82:1190–1206. doi: 10.1016/S0006-3495(02)75476-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whitley MJ, Lee AL. Frameworks for understanding long-range intraprotein communication. Curr Protein Pept Sci. 2009;10:116–127. doi: 10.2174/138920309787847563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Horovitz A, Fersht AR. Strategy for analysing the co-operativity of intramolecular interactions in peptides and proteins. J Mol Biol. 1990;214:613–617. doi: 10.1016/0022-2836(90)90275-Q. [DOI] [PubMed] [Google Scholar]
- 45.Krishnaswamy S. Prothrombinase complex assembly. Contributions of proteinprotein and protein-membrane interactions toward complex formation. Journal of Biological Chemistry. 1990;265:3708–3718. [PubMed] [Google Scholar]
- 46.Stokes BT, Fox P, Hollinden G. Extracellular calcium activity in the injured spinal cord. Experimental Neurology. 1983;80:561–572. doi: 10.1016/0014-4886(83)90307-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.